On the importance of synthetic organic chemistry in drug discovery: reflections on the discovery of antidiabetic agent ertugliflozin†‡§
Vincent
Mascitti
*,
Benjamin A.
Thuma
,
Aaron C.
Smith
,
Ralph P.
Robinson
,
Thomas
Brandt
,
Amit S.
Kalgutkar
,
Tristan S.
Maurer
,
Brian
Samas
and
Raman
Sharma
Pfizer Global Research & Development, Groton Laboratories, Eastern Point Road, Groton, Connecticut 06340, USA. E-mail: vincent.mascitti@pfizer.com; Fax: +1 845 474 5876; Tel: +1 860 686 3455
First published on 16th July 2012
Abstract
The discovery of antidiabetic agent ertugliflozin is described. The compound belongs to a new class of SGLT2 inhibitors bearing a dioxa-bicyclo[3.2.1]octane motif. This article describes the critical role that organic synthesis played in both influencing our medicinal chemistry strategy and speeding up the progression of our program.
1. Introduction
In recent years, the pharmaceutical industry has been at the center of profound changes aimed at improving the efficiency of the drug discovery process.1 Within research organizations like Pfizer, this has led to a critical refocus on creative organic synthesis as a cornerstone of the medicinal chemistry discipline.2 Indeed, by helping to translate hypotheses (derived from observations at the macroscopic level) into molecular architectures (microscopic level), synthetic organic chemistry is crucial for rapid navigation of increasingly complex drugable chemical space. It is also critical in determining discovery cycle times and development speeds associated with drug candidates.3 The recent discovery of the dioxa-bicyclo[3.2.1]octane class of sodium glucose co-transporter 2 (SGLT2)¶inhibitors is a timely example (Fig. 1).4 In this article we will highlight how organic synthesis was instrumental in the progression of Pfizer's SGLT2 inhibitor research program which resulted in the identification of ertugliflozin (PF-04971729, 1, Fig. 1) as an antidiabetic agent.5![Dioxa-bicyclo[3.2.1]octane SGLT2 inhibitor ertugliflozin.](/image/article/2013/MD/c2md20163a/c2md20163a-f1.gif) | ||
| Fig. 1 Dioxa-bicyclo[3.2.1]octane SGLT2 inhibitor ertugliflozin. | ||
2. SGLT2 inhibitors
Sodium glucose co-transporter 2 is a glucose transport protein that is exclusively expressed in renal proximal tubules and is responsible for the majority of glucose re-uptake from the pro-urine. Blockade of SGLT2 induces glucosuria (excretion of glucose in the urine). As a result, reduction of blood glucose concentration can be achieved in a glucose-dependent and insulin-independent manner, thus minimizing the risk of hypoglycemia and potentially favoring preservation of pancreatic beta cells.6–10 In addition, SGLT2 inhibition is associated with clinically significant blood pressure reduction and weight loss. Because of this combination of beneficial effects on plasma glucose concentration, weight loss and blood pressure, inhibition of SGLT2 has emerged as a very promising approach for the pathophysiologic treatment of Type 2 diabetes mellitus (T2DM),11 a disease spreading at an alarming rate worldwide.12Starting from the weakly active dual SGLT1/SGLT2 inhibitor natural product phlorizin (2, Fig. 2),13 clinical candidates from two classes of selective SGLT2 inhibitors emerged: O- and C-aryl glucosides.14 Compounds such as sergliflozin (3, Fig. 2), from the O-aryl glucoside class, suffered degradation by glucosidase enzymes found in the gut. Although this problem could be partially avoided by administration of the compounds as esterprodrug derivatives,15 it appears now that all clinical candidates from this class have been abandoned, presumably due to suboptimal pharmacokinetic (PK) profiles. The PK liabilities associated with O-aryl glucosides were elegantly solved by medicinal chemists at Bristol-Myers Squibb, with the discovery of potent compounds in the C-aryl glucoside class, an advance driven in part by characterization of an unexpected product from an attempted O-glucosylation reaction.16 Exploration of the structure activity relationships around the aglycone moiety led to the identification of the antidiabetic agent dapagliflozin (Forxiga®; 4, Fig. 2).17 Interestingly, a survey of the SGLT2 competitive landscape revealed that, until very recently, most of the disclosed clinical candidates from this class resulted from modifications of the aglycone, the carbohydrate part remaining as an unmodified glucose motif (5–9; Fig. 2).18 A dual SGLT2/1 inhibitor, LX4211, has also been recently disclosed (10, Fig. 2).19
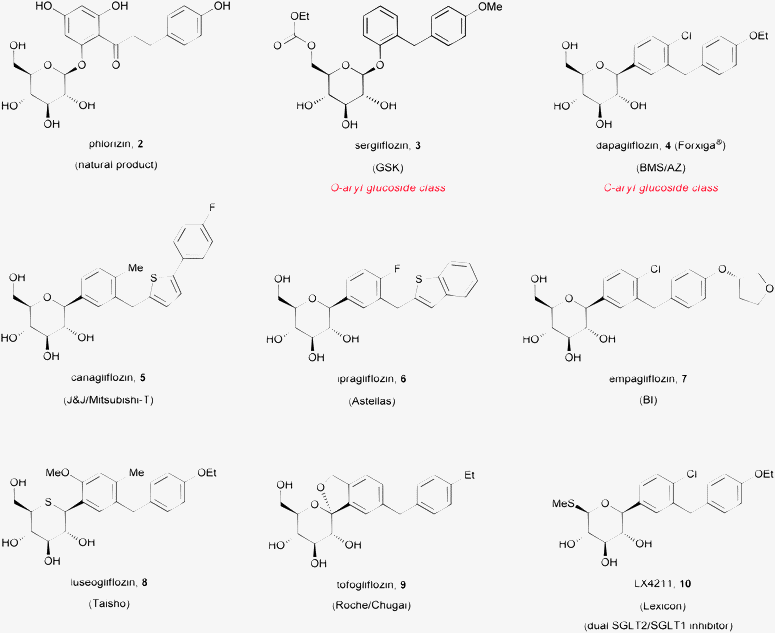 | ||
| Fig. 2 Structure of some SGLT2 inhibitor clinical candidates. | ||
3. C-Aryl glucosides: aglycone modifications enabled through synthesis
Developments in organic synthesis have played an ever-increasing role in advancing SGLT2 projects and shaping the SGLT2 competitive landscape. Most notably, advances in the synthesis of β-C-aryl glucosides have been critical to the discovery and progression of this class of inhibitors.17b The general synthetic sequence is shown in Scheme 1. Thus, the appropriate aglycone motif is added in the form of an organometallic reagent to a gluconolactone derivative to produce a ketal intermediate which upon treatment under reductive conditions followed by deprotection produces the desired C-aryl glucoside. Following the disclosure of dapagliflozin, this same sequence has been adapted to the synthesis of numerous other potential SGLT2 inhibitors having modifications on the aglycone moiety. Some of these compounds have reached clinical trials, e.g., canagliflozin (5),20 ipragliflozin (6),21 empagliflozin (7),22 and luseogliflozin (8).23 The same approach was also used by the medicinal chemists from Chugai for the synthesis of the conformationally constrained O-spiroketal C-aryl glucoside tofogliflozin (9, Fig. 2). In this case, spiroketalization was achieved by intramolecular trapping of the putative oxonium ion formed upon exposure of the ketal intermediate to acidic conditions (Scheme 1).24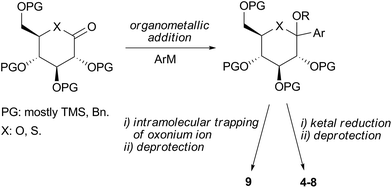 | ||
| Scheme 1 General synthesis of C-aryl glucoside SGLT2 inhibitors. | ||
However, fueled by the need to secure proprietary chemical space, some of the structural changes associated with aglycone modification in the C-aryl glucoside class have not necessarily been in line with maintaining optimal drug-like physicochemical properties. Thus, in some cases, associated increases in molecular weight and lipophilicity have led to suboptimal PK properties (e.g., high clearance, short half-life) requiring increased dose. Also, structural features associated with potential reactive metabolite formation (e.g., thiophene) have sometimes been introduced.25 For a chronic disease such as T2DM, this may lead to higher risk of idiosyncratic toxicity. Additionally, some compounds from the C-aryl glucoside class have tested micronucleus positive in vitro.26
4. Discovery of ertugliflozin: dioxa-bicyclo[3.2.1]octane class enabled through synthesis
The above observations prompted the Pfizer SGLT2 group to take on the synthetically more difficult challenge of sugar modification. Based upon systematic modification and deletion of the hydroxyl groups, the C-5 position was identified as being tolerant of structural change. This observation led to the discovery of the C-5-spirocyclic C-glycoside series, exemplified by spirocyclopropane 11 (Fig. 3).27,28 This compound was readily synthesized in four steps by cyclopropanation of the exocyclic enol ether derived from the corresponding C-aryl glucoside.27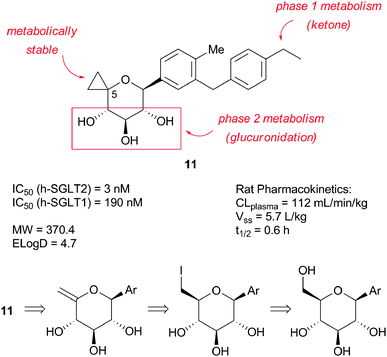 | ||
| Fig. 3 C-5-spirocyclopropane C-glycoside lead 11. | ||
In spite of acceptable potency at human SGLT2, this compound had unacceptable PK properties and only moderate selectivity against human SGLT1 (∼60 fold). In vitro qualitative metabolite identification in rat and human hepatocytes helped understand the cause of the PK findings at the molecular level. Studies revealed that, although the spirocyclopropane ring was metabolically robust, the compound was oxidized to a long lived acetophenone-like metabolite and was glucuronidated (regioselectivity not determined). These considerations led us to focus our attention on spirooxetane compounds such as 12 (Fig. 4).
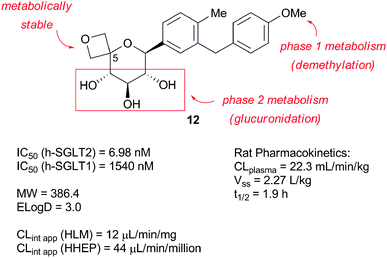 | ||
| Fig. 4 C-5-spirooxetane C-glycoside lead 12. | ||
We reasoned that the spirooxetane motif would help reduce the overall lipophilicity (as measured by ELogD) of the molecule and thereby improve CLplasma. Besides, the introduction of a methoxy substituent in lieu of the original ethyl substituent in 11, would prevent the formation of the observed long lived ketonemetabolite. Compound 12 was synthesized according to the linear sequence described in Scheme 2.29 Of particular note, we utilized an aldol-Cannizzaro sequence to produce diol13 and introduce the tetrasubstitution at C-5 (carbohydrate nomenclature) required in the final analogue.30 Formation of monotosylate intermediate 14, followed by intramolecular displacement completed the formation of the oxetane motif.31 Unfortunately, compound 12 tested positive in the in vitro micronucleus assay. Besides, in spite of improved selectivity and PK properties, this compound still proved to have suboptimal projected human PK (short half life <5 h). Based on our SGLT2 pharmacokinetic-pharmacodynamic (PKPD) relationship model,32 it was anticipated that this short half life would translate into a projected high dose in humans (>100 mg once daily) to achieve near maximal theoretical urinary glucose excretion (UGE) at steady state.4 Indeed, the PKPD model revealed that the projected dose increased exponentially as the projected t1/2 diminished.4
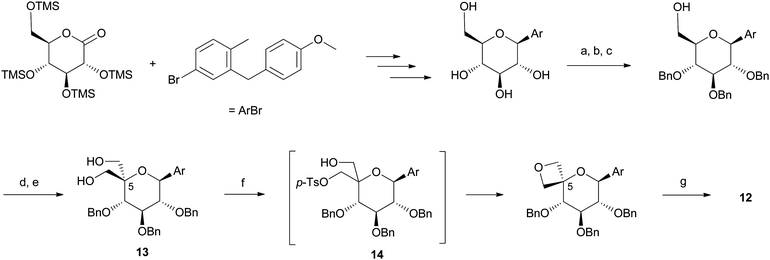 | ||
| Scheme 2 Multigram-scale synthesis of spirooxetane C-glycoside 12. Reagents and conditions: (a) PhCH(OMe)2 (1.3 equiv.), p-TsOH (cat.), DMF, 60 °C; (b) BnBr (3 equiv.), NaH (3 equiv.), DMF, 0 to 23 °C, (82%, 2 steps); (c) LiAlH4 (5 equiv.), AlCl3 (4 equiv.), Et2O/CH2Cl2, reflux, (90%); (d) (COCl)2, DMSO, Et3N, CH2Cl2, −78 to −20 °C; (e) NaOH (1.5 equiv.), H2CO, H2O/1,4-dioxane, 23 °C, (45%, 2 steps); (f) n-BuLi (1 equiv.), then p-TsCl, then n-BuLi (another 1 equiv.), THF, 0 to 65 °C, (65%); (g) Pd black (3 equiv.), HCO2H (80 equiv.), EtOH/THF, 23 °C, (95%). | ||
Thus, in order to identify a compound of lower projected dose, we sought to increase the projected human t1/2 by reducing clearance. However, the long linear sequence used to produce each spirooxetane analogue (11 steps from persilylated gluconolactone; Scheme 2) and lack of modularity (aglycone introduced at the first step) precluded the rapid exploration of this series. Nonetheless, studies analyzing the elimination mechanism(s) of 12, revealed interesting observations that allowed us to move the project forward, in conjunction with opportunistic organic synthesis. First, CLplasma in rats was largely mediated via hepatic metabolism (CLhepatic), the renal clearance (CLrenal) being generally low and passive; second, in vitro stability studies on 12 in human hepatic tissue revealed a higher apparent intrinsic clearance (CLint app) in HHEP relative to HLM, suggestive of non-cytochrome P450 mediated metabolic elimination; third, qualitative in vitrometabolite identification in HHEP confirmed the formation of glucuronide conjugates of 12.
Access to intermediates such as 13, paved the way to the rapid exploration of other C-5 modifications and led to a key observation. Indeed, replacement of the oxetane ring in 12 by an azetidine, synthesized in 3 steps from diol intermediate 15 (Fig. 5) by bistriflation followed by displacement in presence of excess benzyl amine and deprotection, produced 16 (IC50(h-SGLT2) = 5100 nM), a compound of similar lipophilicity to that of 12 but with markedly reduced CLint app in HHEP (Fig. 5). Mindful of this observation, permitted by rapid synthesis of 16, we proposed that the increased human phase 2 metabolism of 12 was due to a lack of an H-bond donor or polar group at the C-5 position.4 This hypothesis appeared to be corroborated by the fact that C-aryl glucosides of similar lipophilicity, such as 17 and 18, have a reduced CLint app (∼2.0 to 4.0 μL per min per million cells) in HHEP (Fig. 5).
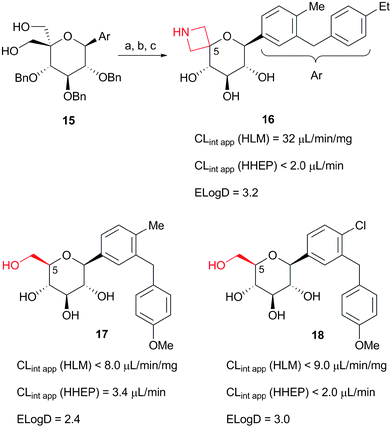 | ||
| Fig. 5 In vitro CLint app considerations. CLint app (HHEP) values are expressed per million cells. Reagents and conditions: (a) Tf2O (5 equiv.), pyridine (30 equiv.), CH2Cl2, −20 °C; (b) i-Pr2NEt (3 equiv.), BnNH2, 80 °C, (75%); (c) Pd black (7 equiv.), HCO2H, EtOH/THF (2/1 vol.), 23 °C, (63%). | ||
Lessons learned from the C-5-spirocyclic C-glycosides prompted us to deprioritize this series and led to the exploration of the dioxa-bicyclo[3.2.1]octane class (Fig. 1).4 It was hypothesized that the bridged ketal system would confer rigidity that could potentially positively impact potency and selectivity for SGLT2. Moreover, it would also allow introduction of a hydroxymethylene H-bond donor group (in place of the spirocycle), which was anticipated to help reduce the rate of human phase 2 metabolism (relative to that of 12) and further improve potency. Compounds from this class were prepared in an analogue-friendly fashion starting from advanced intermediate Weinreb amide19, synthesized in 10 steps from D-glucose (Scheme 3).33 Intermediate 19, which presents the required carbon framework in the proper oxidation state, was primed for the three-step analoguing sequence as shown in Scheme 3. Nucleophilic addition of the appropriate organometallic reagent to 19 in THF produced the corresponding cyclic lactol20. Acid promoted one-pot PMB removal followed by stereoselective intramolecular trapping of the putative oxonium ion intermediate gave compound 21 containing the desired dioxa-bicyclo[3.2.1]octane ring system.34 Finally, hydrogenolysis of the benzyl protecting groups under transfer hydrogenation conditions yielded analogues 22 and 23 as a mixture of diastereomers at C-4, which were separated by HPLC. The epimerization at C-4 may be rationalized via the formation of a putative enol or enol ether intermediate in the analoguing sequence. In contrast to the linear synthesis described in Scheme 2, this analoguing sequence allowed us to: (i) rapidly explore chemical space, (ii) establish structure–activity relationships, and (iii) identify 1, a compound of optimal safety and PK/PD profiles (Fig. 6).
![Three-step analoguing sequence to dioxa-bicyclo[3.2.1]octane SGLT2 inhibitors. Reagents and conditions: (a) ArLi, THF, −78 to 0–23 °C; (b) TFA, anisole, CH2Cl2, 23 °C; (c) Pd black, HCO2H, EtOH/THF, 23 °C, then HPLC separation.](/image/article/2013/MD/c2md20163a/c2md20163a-s3.gif) | ||
| Scheme 3 Three-step analoguing sequence to dioxa-bicyclo[3.2.1]octane SGLT2 inhibitors. Reagents and conditions: (a) ArLi, THF, −78 to 0–23 °C; (b) TFA, anisole, CH2Cl2, 23 °C; (c) Pd black, HCO2H, EtOH/THF, 23 °C, then HPLC separation. | ||
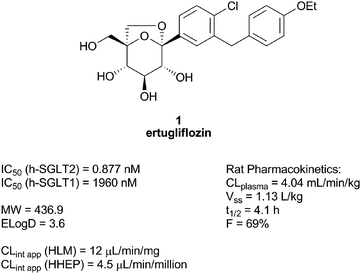 | ||
| Fig. 6 Some properties of ertugliflozin. | ||
Compound 1, had a low turnover in vitro in human hepatic tissues, thereby validating our hypothesis, and a low clearance in our preclinical species (rat and dog).35 This translated in a projected human PK (half life ∼12 h) compatible with a projected low dose once a day (ED50 ∼ 4 mg at steady state in healthy volunteers).4 Furthermore, 1 tested negative in the in vitro micronucleus test. Based on its excellent preclinical profile, compound 1 was advanced to clinical trials as the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 L-pyroglutamic acid co-crystal (mp = 142.5 °C) and is currently in phase 2 (Fig. 7).
:1 L-pyroglutamic acid co-crystal (mp = 142.5 °C) and is currently in phase 2 (Fig. 7).
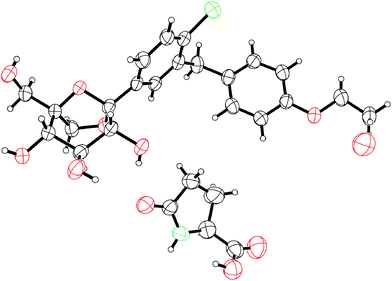 | ||
| Fig. 7 ORTEP representation of the X-ray crystal structure of a co-crystal of ertugliflozin. (molecule of water also shown). | ||
5. Ertugliflozin: development support enabled through synthesis
The identification of 1 as a promising clinical candidate presented a new exciting challenge. Indeed, the versatile synthetic sequence described in Scheme 3 was a useful discovery tool that enabled rapid analogue preparation and identification of compounds with optimal PK/PD profiles; however, the route was not well suited for the synthesis of specific compounds on larger scale. For example, it provided 1 in 0.3% overall yield over 13 linear steps starting from D-glucose including final HPLC separation from its epimer at C-4. These considerations prompted the development of a new stereoselective route that would provide efficient access to 1. Practicality being of paramount importance at this stage, we focused our disconnective analysis using the Chiron approach pioneered by Hanessian.36 Indeed, in such an approach, minimum perturbation of chirality is at a premium during the bond disconnection process. Our retrosynthetic analysis37 led us to develop a synthesis which exploits the pseudo C2-symmetry found in the chiron D-mannose. The synthesis is outlined in Scheme 4.38 Highly diastereoselective dithiane anion (derived from 24) addition to aldehyde25 produced intermediate 26 having the required stereochemistry installed at C-4. Aldol condensation of intermediate lactol27 with formaldehyde under basic conditions gave intermediate 28 bearing the tetrasubstituted carbon found at C-1 in the final product. Deprotection and cyclization of advanced acyclic intermediate 29 under acidic conditions led to the formation of the dioxa-bicyclo[3.2.1]octane-2,3,4-triol motif with exclusive stereocontrol. Overall, this synthesis provided efficient access to ertugliflozin in 5 steps and 26% overall yield starting from readily available dithiane24 and aldehyde25.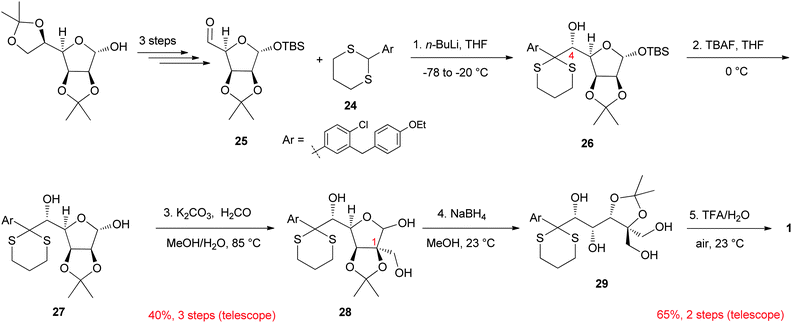 | ||
| Scheme 4 Stereoselective synthesis of ertugliflozin. | ||
Organic synthesis has had a profound impact well beyond the preclinical phase with the development of routes to support toxicological studies, clinical trials, commercialization, and other activities such as drug disposition studies requiring radiolabeled material. A recent example of synthesis impact was prompted by readout from a human mass balance that required the synthesis of the major glucuronide of 1 for structure confirmation. The route used is presented in Scheme 5. Conversion of 1 to the corresponding benzylidene acetal 30 followed by TMSOTf promoted glycosylation using trichloroacetimidate glucuronyl donor 31 led to a mixture of C-3 and C-4 glucuronides (∼4:1). Following acetylation, the regioisomers were separated to provide 32 in 55% yield. Deprotection of the benzylidene acetal under acidic conditions produced crystalline intermediate 33 in 94% yield. Single crystal X-ray diffraction analysis of 33 confirmed the 3-β-O-structure of the glucuronide formed (Fig. 8). Global deprotection under basic conditions led to the desired 3-β-O-glucoronide 34 in 59% yield. Additional results related to the human mass balance studies will be reported in a separate communication.
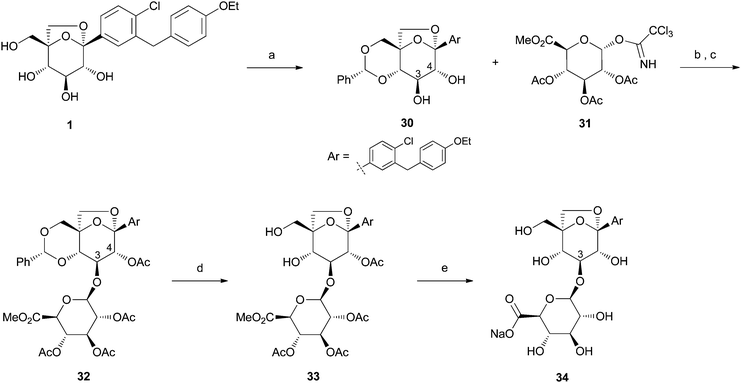 | ||
| Scheme 5 Synthesis of ertugliflozin-3-β-O-glucuronide 34. Reagents and conditions: (a) p-TsOH, PhCH(OMe)2, DMF, 80 °C, (84%); (b) 3 Å MS, CH2Cl2, TMSOTf, −20 to −10 °C; (c) DMAP, i-Pr2NEt, Ac2O, THF, 23 °C, (55%); (d) TFA, H2O, CH2Cl2, 23 °C, (94%); (e) NaOH, H2O, MeOH, 23 °C, (59%). | ||
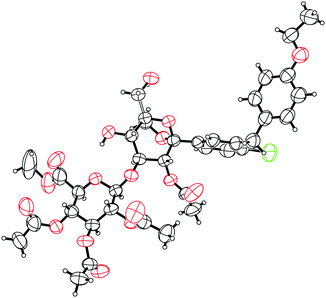 | ||
| Fig. 8 ORTEP representation of the X-ray crystal structure of 33. | ||
6. Conclusion
Based on Phase 1 and 2 clinical data, ertugliflozin proved safe and well tolerated in patients with T2DM. Importantly, significant improvements with respect to glycemic control,39 blood pressure lowering,40 and body weight reduction39 were observed.41 At a time where multiple conceptual avenues are explored to try and help improve the research and development engine of the pharmaceutical industry, the in-house discovery of the antidiabetic agent ertugliflozin offers a concrete and timely example of the importance of organic synthesis in the rapid progression and lifecycle of a successful research program. We dare to hope that lessons learned from this medicinal chemistry story will prove useful to the pharmaceutical industry community and, beyond medicinal chemistry, will contribute to the refocus of creative organic synthesis as a cornerstone of the drug discovery process.7. Experimental
General
Unless specified otherwise, starting materials are generally available from commercial sources. NMR spectra were recorded on a Varian Unity™ 400 (available from Varian Inc., Palo Alto, CA) at room temperature at 400/500 MHz for proton. Chemical shifts are expressed in parts per million (δ) relative to residual solvent as an internal reference (for methanol-d, δH = 3.29 ppm and δC = 47.8 ppm). The peak shapes are denoted as follows: s, singlet; d, doublet; dd, doublet of doublet; t, triplet; q, quartet; m, multiplet; bs or br.s., broad singlet; 2 s, two singlets; br.d., broad doublet. High resolution mass spectra (HRMS) were obtained on an Agilent™ Model 6210 time of flight. Where the intensity of single chlorine ions are described, the expected intensity ratio was observed (approximately 3:1 for 35Cl/37Cl-containing ions) and the intensity of only the lower mass ion is given. Column chromatography was performed with either Baker™ silica gel (40 μm; J.T. Baker, Phillipsburg, NJ) or Silica Gel 50 (EM Sciences™, Gibbstown, NJ) in glass columns or in Flash 40 Biotage™ columns (ISC, Inc., Shelton, CT). MPLC (medium pressure liquid chromatography) was performed using a Biotage™ SP or Isolera One purification systems or a Combiflash® Companion® from Teledyne™ Isco™; Biotage™ SNAP cartridge KPsil or Redisep Rf silica (from Teledyne™ Isco™) under low nitrogen pressure were used. HPLC was performed using a Shimadzu™ 10A LC-UV or an Agilent™ 1100 preparatory HPLC. The tested compounds were determined to be >95% pure by HPLC. Except where otherwise noted, all reactions were run under an inert atmosphere of nitrogen gas using anhydrous solvents. Also, except where otherwise noted, all reactions were run at room temperature (∼23 °C).
(2R,4aS,7S,8R,9R,9aS)-7-[4-Chloro-3-(4-ethoxybenzyl)phenyl]-2-phenyltetrahydro-4a,7-epoxy[1,3]dioxino[5,4-c]oxepine-8,9(4H,5H)-diol (30)
To a solution of (1S,2S,3S,4R,5S)-5-[4-chloro-3-(4-ethoxybenzyl)phenyl]-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol 1 (ref. 33 and 38) (9.67 g, 22.1 mmol) in N,N-dimethylformamide (10 mL) was added p-toluenesulfonic acid monohydrate (130 mg, 0.673 mmol) followed by benzaldehyde dimethyl acetal (4.6 mL, 29 mmol) and the mixture was heated at 80 °C for 16 hours. The reaction mixture was then cooled to room temperature, diluted with water (500 mL), and extracted three times with ethyl acetate (100 mL). The combined organic layers were washed with brine (100 mL), dried over sodium sulfate, filtered and concentrated under reduced pressure. The crude reaction mixture was purified using a Biotage Isolera One (SNAP 100 g silica gel column) eluting with a gradient of 0–20% methanol/dichloromethane. The product was triturated with heptane (100 mL) and concentrated under reduced pressure to give 9.80 g (84% yield) of the desired product.1H NMR (500 MHz, CD3OD) δ = 7.53–7.47 (m, 2H), 7.43 (s, 1H), 7.40–7.33 (m, 5H), 7.09 (m, J = 8.5 Hz, 2H), 6.81 (m, J = 8.5 Hz, 2H), 5.67 (s, 1H), 4.55 (d, J = 7.6 Hz, 1H), 4.18 (s, 2H), 4.04 (s, 2H), 3.99 (q, J = 7.0 Hz, 2H), 3.91 (dd, J = 1.0, 8.5 Hz, 1H), 3.85 (t, J = 7.8 Hz, 1H), 3.82 (dd, J = 1.2, 7.6 Hz, 1H), 3.64 (d, J = 7.8 Hz, 1H), 1.36 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CD3OD) δ = 159.0, 140.0, 139.0, 138.0, 131.0, 130.5, 130.2, 130.0, 129.2, 127.6, 127.1, 115.6, 110.3, 103.4, 82.8, 79.7, 76.3, 74.2, 72.5, 72.2, 64.6, 39.4, 15.3. HRMS (ESI): m/z calcd for [C29H29ClO7 + NH4]: 542.1940, found: 542.1927.
(2R,4aS,7S,8R,9S,9aS)-8-(Acetyloxy)-7-[4-chloro-3-(4-ethoxybenzyl)phenyl]-2-phenyltetrahydro-4a,7-epoxy[1,3]dioxino[5,4-c]oxepin-9(4H,5H)-yl methyl 2,3,4-tri-O-acetyl-beta-D-glucopyranosiduronate (32)
A round bottomed flask was charged with freshly activated 3 Å molecular sieves (500 mg). A solution of 30 (500 mg, 0.952 mmol) and methyl 2,3,4-tri-O-acetyl-1-O-(2,2,2-trichloroethanimidoyl)-alpha-D-glucopyranuronate 31 (500 mg, 1.04 mmol) in dichloromethane was then added to the flask. The resulting mixture was stirred for 30 minutes at room temperature, then cooled to −20 °C. Trimethylsilyltriflate (18 μL, 0.098 mmol) was added dropwise and the temperature maintained between −20 °C and −10 °C for 2 hours. The reaction was quenched with the addition of triethylamine (0.5 mL), filtered (washing with dichloromethane), and concentrated under reduced pressure. The resulting crude product was purified using a Biotage Isolera One (SNAP 50 g silica gel column) eluting with a gradient of 0–50% ethyl acetate/heptane to produce 450 mg (56% yield) of an inseparable mixture of C-3 and C-4 isomers.Note: This transformation was batched into two reactions and combined for purification. To a solution of the above mixture of isomers (420 mg, 0.499 mmol) in tetrahydrofuran (7.0 mL) was added dimethylaminopyridine (6.2 mg, 0.050 mmol) and N,N-diisopropylethylamine (175 μL, 0.998 mmol) followed by the addition of acetic anhydride (170 μL, 1.50 mmol). The resulting mixture was then stirred for 72 hours at room temperature and concentrated under reduced pressure. A second batch (734 mg of above mixture of isomers used, 0.873 mmol) was run under the same conditions; the crude material were combined and purified using a Biotage Isolera One (SNAP 50 g silica gel column) eluting with a gradient of 0–50% ethyl acetate/heptane to produce 1.12 g (92% yield) of an inseparable mixture of the acetylated C-3 and C-4 glucuronides as a white solid. Methanol was then added to this mixture (40 mL) and the slurry was heated until dissolution. The solution was allowed to cool to room temperature with stirring and stirred for an additional 2 hours at this temperature. The resulting white precipitate was filtered, washed with methanol (5 mL) and dried for 1 hour under high vacuum to give 668 mg (55% yield) of the pure C-3 isomer as a white solid. 1H NMR (500 MHz, CDCl3) δ = 7.50–7.44 (m, 2H), 7.43–7.32 (m, 4H), 7.32–7.27 (m, 2H), 7.07 (m, J = 8.5 Hz, 2H), 6.81 (m, J = 8.5 Hz, 2H), 5.61 (s, 1H), 5.29–5.22 (m, 1H), 5.21 (t, J = 9.5 Hz, 1H), 5.16 (t, J = 9.0 Hz, 1H), 5.00 (t, J = 8.7 Hz, 1H), 4.65 (t, J = 7.6 Hz, 2H), 4.23–4.16 (m, 2H), 4.16–4.12 (m, 2H), 4.10 (d, J = 15.4 Hz, 1H), 4.00 (q, J = 7.0 Hz, 2H), 3.95 (d, J = 15.4 Hz, 1H), 3.87 (d, J = 9.8 Hz, 1H), 3.84 (d, J = 8.3 Hz, 1H), 3.60 (s, 3H), 1.99 (s, 3H), 1.98 (s, 3H), 1.95 (s, 3H), 1.70 (s, 3H), 1.40 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ = 170.3, 169.4, 169.3, 168.5, 166.9, 157.6, 139.0, 136.8, 135.3, 134.1, 131.4, 129.9, 129.5, 129.3, 128.8, 128.4, 126.1, 125.4, 114.6, 107.6, 101.7, 101.1, 79.5, 78.6, 77.4, 75.6, 72.6, 72.4, 71.7, 71.1, 70.9, 69.5, 63.5, 52.9, 38.5, 20.7, 20.6, 20.5, 20.4, 15.0. HRMS (ESI): m/z calcd for [C44H47ClO17 + NH4]: 900.2840, found: 900.2820.
Methyl (2S,3S,4S,5R,6R)-3,4,5-tris(acetyloxy)-6-{[(1S,2S,3S,4R,5S)-4-(acetyloxy)-5-[4-chloro-3-(4-ethoxybenzyl)phenyl]-2-hydroxy-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]oct-3-yl]oxy}tetrahydro-2H-pyran-2-carboxylate (33)
Note: This transformation was batched into two reactions and combined for purification. To a solution of 32 (152 mg, 0.172 mmol) in dichloromethane (4.0 mL) was added water (5 μL, 0.278 mmol) followed by the addition of trifluoroacetic acid (40 μL, 0.52 mmol) and the reaction was allowed to stir for 16 hours at room temperature. After 16 hours, additional water (25 μL) and trifluoroacetic acid (50 μL) were added and the reaction allowed to stir for an additional 3 hours at room temperature. The reaction mixture was then poured into ethyl acetate (15 mL) and saturated aqueous sodium bicarbonate solution (20 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (15 mL). The combined organic layers were washed with brine (15 mL), dried over sodium sulfate, filtered, and concentrated under reduced pressure to provide 144 mg of crude product. A second batch (340 mg of 32 used, 0.385 mmol) was run under the same conditions; the crude material were combined and purified using a Biotage Isolera One (SNAP 50 g silica gel column) eluting with a gradient of 0–100% ethyl acetate/heptane. The resulting gum/solid was dissolved in a mixture of ethyl acetate (10 mL) and dichloromethane (2 mL), heptanes was added (20 mL) at which point the solution started to turn cloudy; the round bottom flask was swirled slowly thereby increasing the formation of precipitate and additional heptane (40 mL) was added. The mixture was concentrated under reduced pressure to give 417 mg (94% yield) of the desired product as a white solid. 1H NMR (500 MHz, CDCl3) δ = 7.35–7.31 (m, 2H), 7.28 (d, J = 2.4 Hz, 1H), 7.06 (m, J = 8.3 Hz, 2H), 6.79 (m, J = 8.8 Hz, 2H), 5.24 (t, J = 9.3 Hz, 1H), 5.22 (d, J = 7.8 Hz, 1H), 5.19 (t, J = 9.3 Hz, 1H), 4.98 (d, J = 7.8 Hz, 1H), 4.96 (d, J = 7.8 Hz, 1H), 4.59 (d, J = 7.8 Hz, 1H), 4.34 (d, J = 7.8 Hz, 1H), 4.18 (br.s., 1H), 4.10 (d, J = 9.8 Hz, 1H), 4.08 (d, J = 15.1 Hz, 1H), 4.04–4.00 (m, 1H), 3.99 (q, J = 6.8 Hz, 2H), 3.94 (d, J = 15.1 Hz, 1H), 3.91 (s, 2H), 3.80 (dd, J = 1.0, 7.8 Hz, 1H), 3.80 (d, J = 7.8 Hz, 1H), 3.77 (s, 3H), 2.05 (s, 3H), 2.01 (s, 3H), 1.96 (s, 3H), 1.71 (s, 3H), 1.39 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ = 170.2, 169.5, 169.3, 168.8, 167.1, 157.6, 139.0, 135.2, 134.5, 131.4, 129.8, 129.5, 128.9, 125.4, 114.6, 107.0, 100.8, 85.5, 84.4, 76.0, 72.2, 71.8, 71.5, 70.9, 68.9, 67.9, 63.5, 62.9, 53.5, 38.5, 20.7, 20.6, 20.5, 20.3, 15.0. HRMS (ESI): m/z calcd for [C37H43ClO17 + Na]: 817.2081, found: 817.2077.Melting point (filtered precipitate): 189.0 °C determined by using the Buchi Melting Point B-545 (60 °C set point, gradient 5.0 degrees per minute).
Single crystals were obtained by vapor diffusion over 72 hours of 33 (5.9 mg) in tetrahydrofuran (0.3 mL)/heptanes (5 mL). Melting point = 188.4–188.9 °C.
(2S,3S,4S,5R,6R)-6-{[(1S,2S,3S,4R,5S)-5-[4-Chloro-3-(4-ethoxybenzyl)phenyl]-2,4-dihydroxy-1-(hydroxymethyl)-6,8-dioxabicyclo[3.2.1]oct-3-yl]oxy}-3,4,5-trihydroxytetrahydro-2H-pyran-2-carboxylic acid (34)
To a solution of 33 (372 mg, 0.468 mmol) in methanol (6 mL) was added water (1.2 mL) followed by 3 M aqueous sodium hydroxide solution (1.8 mL). The resulting mixture was stirred at room temperature for 16 hours, and concentrated under reduced pressure. Ethyl acetate (30 mL) was added and the mixture was allowed to sit for 3 days before being filtered. The filtrate was concentrated under reduced pressure yielding 301 mg of the crude desired product. To the resulting material was added methanol (15 mL) and the mixture was heated until dissolution. The solution was then cooled slowly to room temperature and left at this temperature for 24 hours at which point the resulting mixture was filtered, the filter cake washed with methanol (8 mL), and dried under high vacuum for 2 hours to give 172 mg (59% yield) of the desired product as a white solid. 1H NMR (500 MHz, CD3OD) δ = 7.46 (d, J = 2.0 Hz, 1H), 7.39 (dd, J = 2.4, 8.3 Hz, 1H), 7.37 (d, J = 8.3 Hz, 1H), 7.09 (m, J = 8.8 Hz, 2H), 6.80 (m, J = 8.8 Hz, 2H), 4.60 (d, J = 7.8 Hz, 1H), 4.24 (d, J = 7.3 Hz, 1H), 4.04 (s, 2H), 3.99 (q, J = 6.8 Hz, 2H), 3.91 (d, J = 7.3 Hz, 1H), 3.89 (d, J = 12.2 Hz, 1H), 3.82 (t, J = 7.3 Hz, 1H), 3.79 (d, J = 7.3 Hz, 1H), 3.69 (d, J = 12.7 Hz, 1H), 3.63–3.57 (m, 2H), 3.48–3.40 (m, 2H), 3.30–3.27 (m, 1H), 1.36 (t, J = 6.8 Hz, 3H). 13C NMR (126 MHz, CD3OD) δ = 176.6, 158.9, 139.7, 138.3, 135.1, 132.8, 130.8, 130.5, 129.8, 127.1, 115.5, 109.3, 104.8, 88.0, 86.3, 78.4, 77.7, 75.4, 75.1, 73.5, 71.8, 68.1, 64.4, 62.1, 39.3, 15.2. HRMS (ESI): m/z calcd for [C28H33ClO13 + Na]: 635.1502, found: 635.1495.Notes and references
- (a) B. Munos, Lessons from 60 years of pharmaceutical innovation, Nat. Rev. Drug Discovery, 2009, 8, 959–968 CrossRef CAS; (b) S. M. Paul, D. S. Mytelka, C. T. Dunwiddie, C. C. Persinger, B. H. Munos, S. R. Lindborg and A. L. Schacht, How to improve R&D productivity: the pharmaceutical industry's grand challenge, Nat. Rev. Drug Discovery, 2010, 9, 203–214 CAS.
- (a) H. Wild, D. Heimbach and C. Huwe, Editorial: the importance of chemistry for the future of the pharma industry, Angew. Chem., Int. Ed., 2011, 50, 7452–7453 CrossRef CAS; (b) V. Mascitti, V. Snieckus and S. Haak, Editorial: a new vista in SYNFACTS, Synfacts, 2012, 8, I CrossRef.
- (a) F. Lovering, J. Bikker and C. Humblet, Escape from Flatland: Increasing Saturation as an Approach to Improvinc Clinical Success, J. Med. Chem., 2009, 52, 6752 CrossRef CAS; (b) A. Nadin, C. Hattotuwagama and I. Churcher, Lead-oriented synthesis: a new opportunity for synthetic chemistry, Angew. Chem., Int. Ed., 2012, 51, 1114 CrossRef CAS.
- For a prior communication of part of this work, see: V. Mascitti, T. S. Maurer, R. P. Robinson, J. Bian, C. M. Boustany-Kari, T. Brandt, B. M. Collman, A. S. Kalgutkar, M. K. Klenotic, M. T. Leininger, A. Lowe, R. J. Maguire, V. M. Masterson, Z. Miao, E. Mukaiyama, J. D. Patel, J. C. Pettersen, C. Préville, B. Samas, L. She, Z. Sobol, C. M. Steppan, B. D. Stevens, B. A. Thuma, M. Tugnait, D. Zeng and T. Zhu, Discovery of a clinical candidate from the structurally unique dioxa-bicyclo[3.2.1]octane class of sodium-dependent glucose cotransporter 2 inhibitors, J. Med. Chem., 2011, 54, 2952–2960 Search PubMed.
- For the first disclosure of the structure of 1 see: V. Mascitti, Discovery of a New Class of SGLT2 Inhibitors. Abstract of Papers. 240th National ACS Meeting; Boston, MA, United States, Aug 22–26, 2010, American Chemical Society, Washington, D.C., 2010, MEDI-21 Search PubMed.
- Y. Kanai, W. S. Lee, G. You, D. Brown and M. A. Hediger, The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose, J. Clin. Invest., 1994, 93, 397 CrossRef CAS.
- M. F. Albertoni Borghese and M. P. Majowicz, Inhibitors of sodium/glucose cotransporter, Drugs Future, 2009, 34, 297 Search PubMed.
- J. J. Neumiller, J. R. Jr White and R. K. Campbell, Sodium-glucose co-transport inhibitors. Progress and therapeutic potential in type 2 diabetes mellitus, Drugs, 2010, 70, 377 CrossRef CAS.
- E. C. Chao and R. R. Henry, SGLT2 inhibition - a novel strategy for diabetes treatment, Nat. Rev. Drug Discovery, 2010, 9, 551 CrossRef CAS.
- S. Nair and J. P. H. Wilding, Sodium glucose cotransporter 2 inhibitors as a new treatment for diabetes mellitus, J. Clin. Endocrinol. Metab., 2010, 95, 34 CrossRef CAS.
- R. A. DeFronzo, From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus, Diabetes, 2009, 58, 773 CrossRef CAS.
- International Diabetes Federation, Diabetes Atlas, International Diabetes Federation, Brussels, Belgium, 4th edn, 2009 Search PubMed.
- (a) J. R. L. Ehrenkranz, N. G. Lewis, C. R. Kahn and J. Roth, Phlorizin: a review, Diabetes/Metab. Res. Rev., 2005, 21, 31 Search PubMed; (b) J. R. White Jr, Apple trees to sodium glucose co-transporter inhibitors: a review of SGLT2 inhibition, Clin. Diabetes, 2010, 28, 5 Search PubMed.
- W. N. Washburn, Evolution of sodium glucose co-transporter 2 inhibitors as anti-diabetic agents, Expert Opin. Ther. Pat., 2009, 19, 1485–1499 Search PubMed.
- (a) E. K. Hussey, R. V. Clark, D. M. Amin, M. S. Kipnes, R. L. O'Connor-Semmes, E. C. O'Driscoll, J. Leong, S. C. Murray, R. L. Dobbins, D. Layko and D. J. R. Nunez, Single-dose pharmacokinetics and pharmacodynamics of sergliflozin etabonate, a novel inhibitor of glucose reabsorption, in healthy volunteers and patients with type 2 diabetes mellitus, J. Clin. Pharmacol., 2010, 50, 623 CrossRef CAS; (b) E. K. Hussey, R. L. Dobbins, R. R. Stoltz, N. L. Stockman, R. L. O'Connor-Semmes, A. Kapur, S. C. Murray, D. Layko and D. J. R. Nunez, Multiple-dose pharmacokinetics and pharmacodynamics of sergliflozin etabonate, a novel inhibitor of glucose reabsorption, in healthy overweight and obese subjects: a randomized double-blind study, J. Clin. Pharmacol., 2010, 50, 636 CrossRef CAS.
- W. N. Washburn, Development of the renal glucose reabsorption inhibitors: a new mechanism for the pharmacotherapy of diabetes mellitus type 2, J. Med. Chem., 2009, 52, 1785 CrossRef CAS.
- (a) W. Meng, B. A. Ellsworth, A. A. Nirschl, P. J. McCann, M. Patel, R. N. Girotra, G. Wu, P. M. Sher, E. P. Morrison, S. A. Biller, R. Zahler, P. P. Deshpande, A. Pullockaran, D. L. Hagan, N. Morgan, J. R. Taylor, M. T. Obermeier, W. G. Humphreys, A. Khanna, L. Discenza, J. G. Robertson, A. Wang, S. Han, J. R. Wetterau, E. B. Janovitz, O. P. Flint, J. M. Whaley and W. N. Washburn, Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes, J. Med. Chem., 2008, 51, 1145–1149 CrossRef CAS; (b) P. P. Deshpande, B. A. Ellsworth, F. G. Buono, A. Pullockaran, J. Singh, T. P. Kissick, M.-H. Huang, H. Lobinger, T. Denzel and R. H. Mueller, Remarkable β-selectivity in the synthesis of β-1-C-arylglucosides: stereoselective reduction of acetyl-protected methyl 1-C-arylglucosides without acetoxy-group participation, J. Org. Chem., 2007, 72, 9746–9749 CrossRef CAS.
- W. N. Washburn, Sodium glucose co-transporter 2 (SGLT2) inhibitors: novel antidiabetic agents, Expert Opin. Ther. Pat., 2012, 22, 483–494 Search PubMed , See also reference 14.
- See article related to first disclosure in: Chemical Engineering News, 2012, 90(16), 42, See also: Search PubMed (a) N. C. Goodwin, B. A. Harrison, S. Iimura, R. Mabon, Q. Song, W. Wu, J. Yan, H. Zhang and M. M. Zhao, Preparation of glycosyl sulfoxides as potential antidiabetic sodium glucose co-transporter 2 inhibitors, PCT Int. Appl. WO2009014970, 2009, Chem. Abstr., 2009, 150, 144780 Search PubMed; (b) N. C. Goodwin, R. Mabon, B. A. Harrison, M. K. Shadoan, Z. Y. Almstead, Y. Xie, J. Healy, L. M. Buhring, C. M. DaCosta, J. Bardenhagen, F. Mseeh, Q. Liu, A. Nouraldeen, A. G. E. Wilson, S. D. Kimball, D. R. Powell and D. B. Rawlins, Novel L-xylose derivatives as selective sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors for the treatment of type 2 diabetes, J. Med. Chem., 2009, 52, 6201–6204 CrossRef CAS.
- S. Nomura, S. Sakamaki, M. Hongu, E. Kawanishi, Y. Koga, T. Sakamoto, Y. Yamamoto, K. Ueta, H. Kimata, K. Nakayama and M. Tsuda-Tsukimoto, Discovery of canagliflozin, a novel C-glucoside with thiophene ring, as sodium-dependent glucose co-transporter 2 inhibitor for the treatment of type 2 diabetes mellitus, J. Med. Chem., 2010, 53, 6355–6360 CrossRef CAS.
- M. Imamura, T. Murakami, R. Shiraki, K. Ikegai, T. Sugane, F. Iwasaki, E. Kurosaki, H. Tomiyama, A. Noda, K. Kitta and Y. Kobayashi, Preparation of C-glycoside derivatives and salts thereof as Na glucose co-transporter inhibitor, PCT Int. Appl. WO2004080990, 2004, Chem. Abstr., 2004, 141, 296242 Search PubMed.
- D. Weber, S. Renner, T. Fiedler and S. Orlich, Method for the preparation of a crystalline form of 1-chloro-4-(beta-D-glucopyranos-1-yl)-2-(4-((S)-tetrahydrofuran-3-yloxy)benzyl)benzene, PCT Int. Appl. WO2011039107, 2011, Chem. Abstr., 2011, 154, 444966 Search PubMed.
- H. Kakinuma, T. Oi, Y. Hashimoto-Tsuchiya, M. Arai, Y. Kawakita, Y. Fukasawa, I. Iida, N. Hagima, H. Takeuchi, Y. Chino, J. Asami, L. Okumura-Kitajima, F. Io, D. Yamamoto, N. Miyata, T. Takahashi, S. Uchida and K. Yamamoto, (1S)-1,5-Anhydro-1-[5-(4- ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-D-glucitol (TS-071) is a potent, selective sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for type 2 diabetes treatment, J. Med. Chem., 2010, 53, 3247–3261 CrossRef CAS.
- (a) M. Murakata, T. Ikeda, N. Kimura, A. Kawase, M. Nagase, K. Yamamoto, N. Takata and S. Yoshizaki, Crystal of spiroketal derivative, and process for production thereof, PCT Int. Appl. WO2009154276, 2009, Chem. Abstr., 2009, 152, 75342 Search PubMed; (b) T. Sato, Discovery of O-spiroketal C-arylglucosides as novel and selective sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes, Abstracts of Papers, 240th National ACS Meeting, Boston, MA, Aug 22–26, 2010, American Chemical Society, Washington, DC, 2010, MEDI-202. Search PubMed For further examples of conformationally constrained spiro C-arylglucosides see: B. Lv, Y. Feng, J. Dong, M. Xu, B. Xu, W. Zhang, Z. Sheng, A. Welihinda, B. Seed and Y. Chen, Conformationally constrained spiro C-arylglucosides as potent and selective renal sodium-dependent glucose co-transporter 2 (SGLT2) inhibitors, ChemMedChem, 2010, 5, 827–831 Search PubMed.
- (a) A. S. Kalgutkar, I. Gardner, R. S. Obach, C. L. Shaffer, E. Callegari, K. R. Henne, A. E. Mutlib, D. K. Dalvie, J. S. Lee, Y. Nakai, J. P. O'Donnell, J. Boer and S. P. Harriman, A comprehensive listing of bioactivation pathways of organic functional groups, Curr. Drug Metab., 2005, 6, 161–225 Search PubMed; (b) A. S. Kalgutkar and J. R. Soglia, Minimising the potential for metabolic activation in drug discovery, Expert Opin. Drug Metab. Toxicol., 2005, 1, 91–142 Search PubMed.
- (a) In Vitro Mammalian Cell Micronucleus Test (MNvit), OECD Guideline for Testing of Chemicals No. 487, OECD, Paris, 2010, http://www.oecd.org/env/testguidelines Search PubMed; (b) W. Washburn and W. Meng, C-aryl glucoside SGLT2 inhibitors and method for the treatment of diabetes and related diseases, US Pat., US 7,589,193, 2009, Chem. Abstr., 2006, 144, 305154 Search PubMed.
- R. P. Robinson, V. Mascitti, C. M. Boustany-Kari, C. L. Carr, P. M. Foley, E. Kimoto, M. T. Leininger, A. Lowe, M. K. Klenotic, J. I. MacDonald, R. J. Maguire, V. M. Masterson, T. S. Maurer, Z. Miao, J. D. Patel, C. Préville, M. R. Reese, L. She, C. M. Steppan, B. A. Thuma and T. Zhu, C-Aryl glycoside inhibitors of SGLT2: exploration of sugar modifications including C-5 spirocyclization, Bioorg. Med. Chem. Lett., 2010, 20, 1569–1572 CrossRef CAS.
- For another independent report see: G. R. Bebernitz, M. G. Bock, D. Bhuniya, L. Datrange, S. E. Kurhade, P. V. Palle and D. S. Reddy, Preparation of glycoside derivative as antidiabetic agents, PCT Int. Appl. WO2011048148, 2011, Chem. Abstr., 2011, 154, 486619 Search PubMed.
- V. Mascitti, R. P. Robinson, C. Préville, B. A. Thuma, C. L. Carr, M. R. Reese, R. J. Maguire, M. T. Leininger, A. Lowe and C. M. Steppan, Syntheses of C-5-spirocyclic C-glycoside SGLT2 inhibitors, Tetrahedron Lett., 2010, 51, 1880–1883 CrossRef CAS.
- For aldol-Cannizzaro reaction see: R. Schaffer, Branched-chain higher sugars. III. A 4-C-(hydroxymethy1)-pentose, J. Am. Chem. Soc., 1959, 81, 5452–5454 Search PubMed.
- For oxetane synthesis see: P. Picard, D. Leclercq, J.-P. Bats and J. Moulines, An efficient one-pot synthesis of oxetanes from 1,3-diols, Synthesis, 1981, 550–551 Search PubMed.
- T. S. Maurer, A. Ghosh, N. Haddish-Berhane, A. Sawant-Basak, C. M. Boustany-Kari, L. She, M. T. Leininger, T. Zhu, M. Tugnait, X. Yang, E. Kimoto, V. Mascitti and R. P. Robinson, Pharmacodynamic model of sodium-glucose transporter 2 (SGLT2) inhibition: implications for quantitative translational pharmacology, AAPS J., 2011, 13, 576–585 Search PubMed.
- V. Mascitti and B. M. Collman, Preparation of dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives as antidiabetic agents, PCT Int. Appl. WO2010023594, 2010, Chem. Abstr., 2010, 152, 311862 Search PubMed.
- The addition of anisole avoids the formation of side products arising from Friedel–Craft-type reactions between the PMB cation and the aryl ring of the substrate.
- A. S. Kalgutkar, M. Tugnait, T. Zhu, E. Kimoto, Z. Miao, V. Mascitti, X. Yang, B. Tan, R. L. Walsky, J. Chupka, B. Feng and R. P. Robinson, Preclinical species and human disposition of PF-04971729, a selective inhibitor of the sodium-dependent glucose cotransporter 2 and clinical candidate for the treatment of type 2 diabetes mellitus, Drug Metab. Dispos., 2011, 39, 1609–1619 CrossRef CAS.
- S. Hanessian, The Total Synthesis of Natural Products: The Chiron Approach, Pergamon Press, Oxford, 1983 Search PubMed.
- E. J. Corey, and X. M. Cheng, The Logic of Chemical Synthesis, Wiley, New York, 1989 Search PubMed.
- V. Mascitti and C. Préville, Stereoselective synthesis of a dioxa-bicyclo[3.2.1]octane SGLT2 inhibitor, Org. Lett., 2010, 12, 2940–2943 CrossRef CAS.
- G. Nucci, Presented at the 47th EASD Meeting, Lisbon, September 2011, (abstract A-11-1092-EASD) Search PubMed.
- N. B. Amin, Presented at the 71st ADA Meeting, San Diego, CA, June 2011 (abstract 0048-LB) Search PubMed.
- Further clinical data will be reported in due course.
Footnotes |
| † This article is dedicated to professor Stephen Hanessian for his contribution to organic synthesis. |
| ‡ This article is part of a MedChemComm ‘New Talents’ issue highlighting the work of outstanding rising scientists in medicinal chemistry research. |
| § Electronic supplementary information (ESI) available: X-ray crystallographic data for compound 33. CCDC 888909. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2md20163a |
| ¶ Abbreviations: SGLT, sodium-dependent glucose cotransporter; UGE, urinary glucose excretion; PK, pharmacokinetics; PD, pharmacodynamics; CLplasma, total plasma clearance; Vss, steady state volume of distribution; CLint app, in vitro apparent intrinsic clearance; HLM, human liver microsomes; HHEP, human hepatocytes; F, oral bioavailability; IC50, half maximal inhibitory concentration; ED50, half maximal effective dose. |
| This journal is © The Royal Society of Chemistry 2013 |