Optimisation of aqueous solubility in a series of G protein coupled receptor 119 (GPR119) agonists†‡
James S.
Scott
*a,
Alan M.
Birch
a,
Katy J.
Brocklehurst
a,
Hayley S.
Brown
a,
Kristin
Goldberg
a,
Sam D.
Groombridge
a,
Julian A.
Hudson
a,
Andrew G.
Leach
a,
Philip A.
MacFaul
a,
Darren
McKerrecher
a,
Ruth
Poultney
a,
Paul
Schofield
a and
Per H.
Svensson
b
aCardiovascular & Gastrointestinal Innovative Medicines Unit, AstraZeneca, Mereside, Alderley Park, Macclesfield, Cheshire, SK10 4TG, UK. E-mail: jamie.scott@astrazeneca.com; Fax: +44 (0)1625 516667; Tel: +44 (0)1625 232567
bDepartment of Applied Physical Chemistry, Royal Institute of Technology, SE-100 44 Stockholm, Sweden
First published on 21st June 2012
Abstract
Improving aqueous solubility is a challenge frequently faced within drug discovery programs. Herein we describe increases in solubility in two sub-series of GPR119 agonists through reduction of lipophilicity together with hydrogen bond acceptor modulation. Small molecule X-ray crystallography was utilised to investigate effects on solid state interactions.
Introduction
G Protein coupled receptor 119 (GPR119) is a class A type receptor that is expressed predominantly in pancreatic islets and intestinal enteroendocrine cells.1 It has been demonstrated that agonism of the GPR119 receptor increases incretin secretion from the gut2 and stimulates insulin release from β-cells in the pancreas.3 Consequently, GPR119 agonists have generated considerable interest in their potential to provide a therapeutic intervention in the treatment of diabetes.4 A number of research groups have recently disclosed details of their efforts, including Arena,5 GSK,6 Pfizer,7 Merck,8 and Astellas.9 Additionally, initial clinical data has been published on APD-597 (JNJ-38431055).10We have described our work in this area11 leading to the optimisation of an initial lead sulfone 1via a key cyano-pyridyl 2 to the development candidate 3 (Scheme 1).12 Oxadiazole 3 had many attractive features however, the aqueous solubility was sub-optimal (6 μM) and improvement of this parameter was seen as a key focus for a subsequent optimisation campaign. Our strategy to achieve higher solubility was to target lower lipophilicity space and to disrupt intermolecular interactions in the solid state.
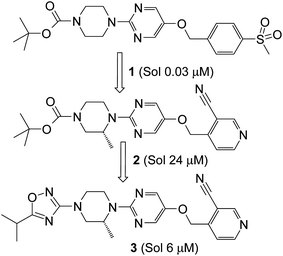 | ||
| Scheme 1 Evolution of CN-pyridyl start points 2 and 3 from sulphone 1. | ||
Results and discussion
Our initial efforts focussed on identifying alternatives to the tert-butyl group of the carbamate present in 2 (Table 1). We identified that cyclic ethers 4–7 resulted in significant reductions in lipophilicity leading to improvements in solubility and stability, albeit at the expense of potency13 against the receptor. An examination of the Ligand-Lipophilicity Efficiency (LLE)14 (pEC50−log D) indicated that the 4- and 5-ring ethers had lost less than a unit of LLE and this encouraged us to further optimise in this region. Substitution of a methyl group onto the oxetane 8 gave an increase in potency. However, further expansion to the ethyl 9 or isopropyl 10 analogues resulted in no increase in potency but an increase in metabolic instability as measured in human liver microsomes. Metabolite-identification studies showed that the metabolism was occurring predominantly on the alkyl oxetane and, in an effort to block this, the trifluoromethyl-oxetane 11 was synthesised. This compound was potent against both the human and mouse forms of the enzyme and retained the favourable solubility (110 μM) associated with the ethers. Compound 11 was also lower in lipophilicity (log D 2.5) than the initial start point 2 leading to an increase in LLE (5.2). Compound 11 was also more stable in human liver microsomes and metabolite-identification studies indicated that, in contrast to 9 and 10, no metabolism of the oxetane was observed. The ring expanded homologue 12 was also examined but found to give no significant benefit in terms of greater potency. The higher lipophilicity (log D 3.1) resulted in lower solubility, higher clearance in human liver microsomes together with an erosion of LLE.|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cpd | R | Human pEC50 | Human IA (%) | Mouse pEC50 | Mouse IA (%) | Log D7.4a | Solubilityb (μM) | LLE pEC50−log D | Human micsc (μl min−1 mg−1) | Melting point (°C) |
| a Distribution coefficient between 1-octanol and aqueous phosphate buffer at pH 7.4. b Solubility of compounds in aqueous phosphate buffer at pH 7.4 after 24 hours at 25 °C (μM). c Human microsome metabolism intrinsic clearance (μl min−1 mg−1). | ||||||||||
| 2 |
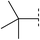
|
8.3 | 171 | 7.4 | 97 | 3.3 | 24 | 5.0 | 43 | 91–93 |
| 4 |
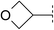
|
5.8 | 142 | <4.5 | 31 | 1.6 | >2200 | 4.2 | 5 | 117–118 |
| 5 |
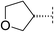
|
5.9 | 95 | <5.7 | 13 | 1.9 | 690 | 4.1 | 27 | 91–92 |
| 6 |
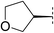
|
6.3 | 133 | 5.5 | 40 | 1.9 | 1100 | 4.4 | 17 | 76–77 |
| 7 |
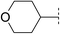
|
6.2 | 102 | <5.1 | 52 | 2.2 | 110 | 4.0 | 37 | 107–108 |
| 8 |
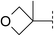
|
6.7 | 183 | 6.2 | 67 | — | >860 | — | 23 | 98–99 |
| 9 |
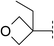
|
6.7 | 150 | 6.0 | 71 | — | — | — | 103 | — |
| 10 |
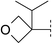
|
6.7 | 150 | 6.3 | 47 | — | — | — | 308 | — |
| 11 |
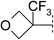
|
7.7 | 171 | 6.8 | 76 | 2.5 | 110 | 5.2 | 44 | 53–55 |
| 12 |
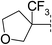
|
7.8 | 142 | 6.8 | 72 | 3.1 | 34 | 4.7 | 107 | 75–76 |
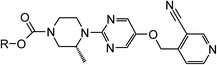
The synthesis of the carbamate of 11 initially proved problematic with synthetic routes utilising N-succinimidocarbonate or phenyl carbonate coupling methodology proving unsuitable for this hindered tertiary alcohol. To overcome this issue, pentafluoro-carbonate chemistry was developed and this allowed multi-gram quantities of 11 to be synthesised (Scheme 2).
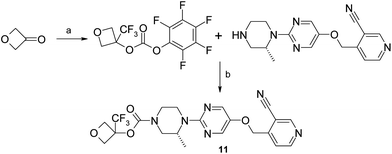 | ||
| Scheme 2 Synthesis of trifluoro-oxetane 11. (a) (i) (nBu)4NF, Si(CH3)3CF3, THF, 20 °C, 2 h; (ii) bis(perfluorophenyl) carbonate, NEt3, CH3CN, 20 °C, 18 h, 83%; (b) NEt3, CHCl3, 110 °C, 30 min, 39%. | ||
In a previous publication, we had highlighted the importance of crystal packing on the aqueous solubility in this series.12 In order to understand the effects of the ether carbamates on the solid state structure, we grew crystals of compounds in an analogous des-methyl piperizine series (these were found to give crystals more suitable for X-ray diffraction). Compound 13 is the racemic, des-methyl analogue of compound 5.
The crystal structure of 13 (solubility 180 μM; mp 129 °C; log D 1.5) (Fig. 1a) shows that the molecules are flat as has been observed for other molecules in this series12 with two enantiomers (with carbons coloured light blue and purple respectively) lying side by side in a plane. The molecules stack (Fig. 1b) in a slightly offset fashion that has been observed previously for molecules bearing a methyl substituent on the piperazine ring.12 That the molecules do not stack directly on top of one another suggests that π–π type interactions are counteracted which may be caused by the protrusion out of the plane of the molecule of the cyclic ether. The final view of the molecule in Fig. 1c shows that within the plane of the molecule there are only relatively weak interactions holding the plane together involving the CH bonds of the pyrimidine interacting with carbonyl oxygen or pyrimidine nitrogens of adjacent molecules. The cyano group interacts with an aromatic CH of an adjacent pyridine ring. The pyridyl nitrogen and the cyclic ether oxygen are not able to interact with any particularly polarized CH bonds (the pyridyl's closest approach is to a CH bond in an adjacent cyclic ether and the ether oxygen with a CH bond in a piperazine in the layer above). Both of these groups will be well solvated in water and so will contribute to the solubility enhancement observed for the ethers.
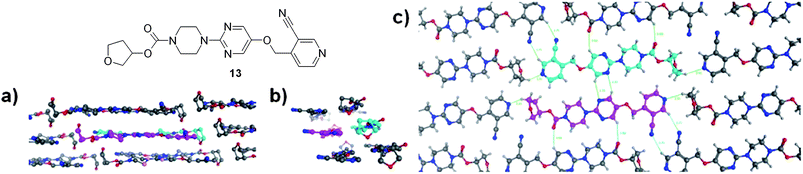 | ||
| Fig. 1 Crystal structure of cyano-pyridyl 13. | ||
As an alternative strategy to improve solubility, we also investigated the introduction of ether functionality into the alkyl substituent of oxadiazole 3 (Table 2). The methyl oxetane analogue 14 resulted in a reduction in potency and LLE (4.2) but pleasingly a ten-fold gain in solubility. Acyclic analogues 15 and 16 resulted in comparable potencies and LLE (4.9 and 5.0) and were also significantly more soluble (>10-fold) than oxadiazole 3. Introduction of a second ether with dioxane 17 led to a less lipophilic compound (log D 2.3) but with lower potencies and LLE than the acyclic ethers. A switch to tertiary alcohol 18 resulted in a reduction in potency and LLE (3.8) indicating this was poorly tolerated. Despite its low log D (2.4), this compound was poorly soluble. The melting point of this compound (235–236 °C) was significantly higher than others in the series. This may be attributable to the addition of a hydrogen bond donor that could potentially form interactions in the solid state with a number of acceptors present in the molecule.
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cpd | R | Human pEC50 | Human IA (%) | Mouse pEC50 | Mouse IA (%) | Log D7.4a | Solubilityb (μM) | LLE pEC50−log D | Human micsc (μl min−1 mg−1) | Melting point (°C) |
| a Distribution coefficient between 1-octanol and aqueous phosphate buffer at pH 7.4. b Solubility of compounds in aqueous phosphate buffer at pH 7.4 after 24 hours at 25 °C (μM). c Human microsome metabolism intrinsic clearance (μl min−1 mg−1). | ||||||||||
| 3 |
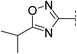
|
8.1 | 269 | 7.3 | 92 | 3.3 | 6 | 4.8 | 23 | 117–118 |
| 14 |
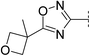
|
7.2 | 188 | 7.0 | 38 | 3.0 | 59 | 4.2 | 43 | 128–129 |
| 15 |
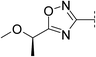
|
7.8 | 239 | 6.8 | 86 | 2.9 | 61 | 4.9 | 41 | 79–80 |
| 16 |
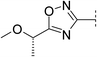
|
7.8 | 216 | 6.8 | 83 | 2.8 | 72 | 5.0 | 35 | 77–78 |
| 17 |
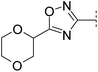
|
7.0 | 279 | 6.2 | 45 | 2.3 | 30 | 4.7 | 26 | 162–163 |
| 18 |
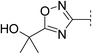
|
6.2 | 171 | 6.2 | 31 | 2.4 | 8 | 3.8 | 14 | 235–236 |
| 19 |
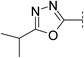
|
6.8 | 212 | <5.4 | 96 | 2.6 | 150 | 4.2 | 22 | 67–68 |
| 20 |
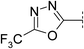
|
7.6 | 220 | 6.4 | 94 | 2.7 | 120 | 4.9 | 12 | 92 |
It is known that 1,3,4-oxadiazoles are less lipophilic than their 1,2,4-regioisomeric counterparts.15 We have reported that this is in part due to the increase in acceptor strength as determined by the log Kβ values.16 As expected, 1,3,4-oxadiazole 19 was found to be considerably less lipophilic (Δlog D 0.7) than the starting point 3 but had greater solubility (150 μM) than compounds from the same series with similar lipophilicity. The LLE was lower (4.2) however, replacement of the iPr group with a CF3 in compound 20 restored the LLE to a level comparable with 3 (4.9). This change maintained the high solubility (120 μM), in addition to increasing stability in human liver microsomes.
Fig. 2 shows the solubilities of matched molecular pairs17 differing only in the carbamate (Fig. 2A) and oxadiazole (Fig. 2B) portion of the molecule plotted against measured log D7.4. The ether containing compounds and the 1,3,4-oxadiazoles occupy the upper region of these plots indicating that these are more soluble at comparable lipophilicities than other members of this series that do not contain these structural features.
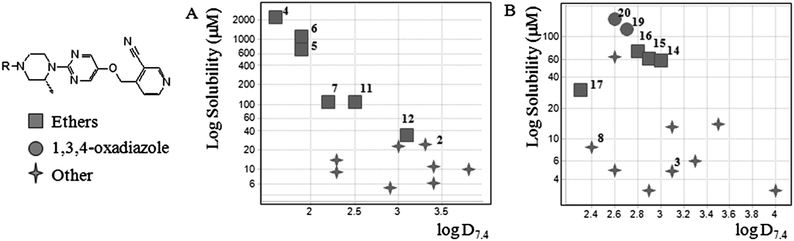 | ||
| Fig. 2 Graph of log aqueous solubility against log D7.4 for a series of matched pairs of cyanopyridyl containing compounds. Ethers are labelled (■), 1,3,4-oxadiazoles (●) and other members of the series with a cross. Individual compounds are labelled according to their numbering in the manuscript. (A) Carbamates (Table 1) and (B) oxadiazoles (Table 2). | ||
The structural modifications together with the reduction in lipophilicity resulted in large increases in solubility for 11 and 20 relative to 3. LLE has remained relatively high and any lowering of absolute potency values has been offset by increases in free levels as a consequence of the lowered lipophilicity (Table 3). Permeability as measured in an in vitro CACO-2 assay remains high with no evidence of efflux. No liabilities in terms of five major isoforms of cytochrome P450 enzymes (CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4) were introduced during these structural modifications. A small benefit in terms of reduction of hERG inhibition was observed with compound 20.
| Cpd | Log D7.4a | Solubilityb (μM) | LLE pEC50−log D | Mouse PPBc (% free) | Rat PPBc (% free) | CACO permeabilityd Papp (×10−6 cm s−1) | CYPSe (μM) | hERGf (μM) |
|---|---|---|---|---|---|---|---|---|
| a Distribution coefficient between 1-octanol and aqueous phosphate buffer at pH 7.4. b Solubility of compounds in aqueous phosphate buffer at pH 7.4 after 24 hours at 25 °C (μM). c %free compound measured when dialysed with appropriate plasma proteins. d Compound permeability measured using CACO-2 cells (10−6 cm s−1). e Inhibition of cytochrome P450 enzymes (μM). f Inhibition of hERG channel (μM). | ||||||||
| 3 | 3.3 | 6 | 4.8 | 1.9 | 1.8 | 15 (A2B); 6 (B2A) | All >25 | 10 |
| 11 | 2.5 | 110 | 5.2 | 8.1 | 4.7 | 16 (A2B); 9 (B2A) | All >25 | 12 |
| 20 | 2.7 | 120 | 4.9 | 12 | 9.7 | 18 (A2B); 21 (B2A) | All >25 | 18 |
The stabilities of the carbonate 11 and oxadiazole 20 were compared to the initial Boc containing starting compound 1 by incubating samples at 65 °C for 18 hours at pH 1, 4, 6, 8 and 10, using a typical starting concentration of 10 μM, and employing 5% 2-methoxyethanol as co-solvent (Table 4).18 As expected, and in contrast to 1, both 11 and 20 were completely stable across the pH range 4–8.
| Cpd | pH 1 | pH 4 | pH 6 | pH 8 | pH 10 |
|---|---|---|---|---|---|
| t 1/2 (d) | t 1/2 (d) | t 1/2 (d) | t 1/2 (d) | t 1/2 (d) | |
| 1 | <0.075 | 0.06 | 0.12 | 0.11 | <0.006 |
| 11 | 0.41 | >10 | >10 | >10 | 0.04 |
| 20 | 0.45 | >10 | >10 | >10 | 0.56 |
On the basis of their improved aqueous solubilities, compounds 11 and 20 were selected for further evaluation, the results of which will be reported in due course.
Conclusions
In summary, we have described improvements in solubility in a series of GPR119 agonists through a strategy of reduction of lipophilicity together with either judicious inclusion of ether functionality or modulation of heterocyclic hydrogen bond acceptor strength. A small molecule X-ray crystal structure allowed an understanding of the effects on the solid state interactions. A comparison of matched molecular pairs with other members of the series highlighted the importance of these structural features in allowing the synthesis of potent and soluble agonists of GPR119.Acknowledgements
Rolf Walker and Adrian Pickup are thanked for their synthetic contributions.Notes and references
- R. Fredriksson, P. J. Höglund, D. E. I. Gloriam, M. C. Lagerström and H. B. Schiöth, FEBS Lett., 2003, 554, 381–388 CrossRef CAS.
- Z. Chu, C. Carroll, J. Alfonso, V. Gutierrez, H. He, A. Lucman, M. Pedraza, H. Mondala, H. Gao, D. Bagnol, R. Chen, R. M. Jones, D. P. Behan and J. Leonard, Endocrinology, 2008, 149, 2038–2047 CrossRef CAS.
- Z. Chu, R. M. Jones, H. He, C. Carroll, V. Gutierrez, A. Lucman, M. Moloney, H. Gao, H. Mondala, D. Bagnol, D. Unett, Y. Liang, K. Demarest, G. Semple, D. P. Behan and J. Leonard, Endocrinology, 2007, 148, 2601–2609 CrossRef CAS.
- (a) M. C. T. Fyfe, J. G. McCormack, H. A. Overton, M. J. Procter and C. Reynet, Expert Opin. Drug Discovery, 2008, 3, 403–413 CrossRef CAS; (b) U. Shah, Curr. Opin. Drug Discovery Dev., 2009, 12, 519–532 CAS; (c) R. M. Jones and J. N. Leonard, in Annual Reports in Medicinal Chemistry, ed. John E. Macor, Academic Press, 2009, pp. 149–170 Search PubMed.
- (a) G. Semple, B. Fioravanti, G. Pereira, I. Calderon, J. Uy, K. Choi, Y. Xiong, A. Ren, M. Morgan, V. Dave, W. Thomsen, D. J. Unett, C. Xing, S. Bossie, C. Carroll, Z. Chu, A. J. Grottick, E. K. Hauser, J. Leonard and R. M. Jones, J. Med. Chem., 2008, 51, 5172–5175 CrossRef CAS; (b) G. Semple, A. Ren, B. Fioravanti, G. Pereira, I. Calderon, K. Choi, Y. Xiong, Y. Shin, T. Gharbaoui, C. R. Sage, M. Morgan, C. Xing, Z. Chu, J. N. Leonard, A. J. Grottick, H. Al-Shamma, Y. Liang, K. T. Demarest and R. M. Jones, Bioorg. Med. Chem. Lett., 2011, 21, 3134–3141 CrossRef CAS; (c) G. Semple, J. Lehmann, A. Wong, A. Ren, M. Bruce, Y. Shin, C. R. Sage, M. Morgan, W. Chen, K. Sebring, Z. Chu, J. N. Leonard, H. Al-Shamma, A. J. Grottick, F. Du, Y. Liang, K. Demarest and R. M. Jones, Bioorg. Med. Chem. Lett., 2012, 22, 1750–1755 CrossRef CAS.
- Y. Wu, J. D. Kuntz, A. J. Carpenter, J. Fang, H. R. Sauls, D. J. Gomez, C. Ammala, Y. Xu, S. Hart and S. Tadepalli, Bioorg. Med. Chem. Lett., 2010, 20, 2577–2581 CrossRef CAS.
- (a) K. F. McClure, E. Darout, C. R. W. Guimarães, M. P. DeNinno, V. Mascitti, M. J. Munchhof, R. P. Robinson, J. Kohrt, A. R. Harris, D. E. Moore, B. Li, L. Samp, B. A. Lefker, K. Futatsugi, D. Kung, P. D. Bonin, P. Cornelius, R. Wang, E. Salter, S. Hornby, A. S. Kalgutkar and Y. Chen, J. Med. Chem., 2011, 54, 1948–1952 CrossRef CAS; (b) V. Mascitti, B. D. Stevens, C. Choi, K. F. McClure, C. R. W. Guimarães, K. A. Farley, M. J. Munchhof, R. P. Robinson, K. Futatsugi, S. Y. Lavergne, B. A. Lefker, P. Cornelius, P. D. Bonin, A. S. Kalgutkar, R. Sharma and Y. Chen, Bioorg. Med. Chem. Lett., 2011, 21, 1306–1309 CrossRef CAS; (c) A. S. Kalgutkar, V. Mascitti, R. Sharma, G. W. Walker, T. Ryder, T. S. McDonald, Y. Chen, C. Preville, A. Basak, K. F. McClure, J. T. Kohrt, R. P. Robinson, M. J. Munchhof and P. Cornelius, Chem. Res. Toxicol., 2011, 24, 269–278 CrossRef CAS; (d) R. Sharma, H. Eng, G. S. Walker, G. Barreiro, A. F. Stepan, K. F. McClure, A. Wolford, P. D. Bonin, P. Cornelius and A. S. Kalgutkar, Chem. Res. Toxicol., 2011, 24, 2207–2216 CrossRef CAS.
- (a) Y. Xia, S. Chackalamannil, W. J. Greenlee, C. Jayne, B. Neustadt, A. Stamford, H. Vaccaro, X. L. Xu, H. Baker, K. O'Neill, M. Woods, B. Hawes and T. Kowalski, Bioorg. Med. Chem. Lett., 2011, 21, 3290–3296 CrossRef CAS; (b) J. W. Szewczyk, J. Acton, A. D. Adams, G. Chicchi, S. Freeman, A. D. Howard, Y. Huang, C. Li, P. T. Meinke, R. Mosely, E. Murphy, R. Samuel, C. Santini, M. Yang, Y. Zhang, K. Zhao and H. B. Wood, Bioorg. Med. Chem. Lett., 2011, 21, 2665–2669 CrossRef CAS.
- (a) S. Yoshida, T. Ohishi, T. Matsui, H. Tanaka, H. Oshima, Y. Yonetoku and M. Shibasaki, Diabetes, Obes. Metab., 2011, 13, 34–41 CrossRef CAS; (b) S. Yoshida, T. Ohishi, T. Matsui and M. Shibasaki, Biochem. Biophys. Res. Commun., 2010, 400, 437–441 CrossRef CAS; (c) S. Yoshida, T. Ohishi, T. Matsui, H. Tanaka, H. Oshima, Y. Yonetoku and M. Shibasaki, Biochem. Biophys. Res. Commun., 2010, 402, 280–285 CrossRef CAS; (d) S. Yoshida, H. Tanaka, H. Oshima, T. Yamazaki, Y. Yonetoku, T. Ohishi, T. Matsui and M. Shibasaki, Biochem. Biophys. Res. Commun., 2010, 400, 745–751 CrossRef CAS; (e) K. Negoro, Y. Yonetoku, T. Maruyama, S. Yoshida, M. Takeuchi and M. Ohta, Bioorg. Med. Chem., 2012, 20, 2369–2375 CrossRef CAS.
- (a) L. B. Katz, J. J. Gambale, P. L. Rothenberg, S. R. Vanapalli, N. Vaccaro, L. Xi, D. C. Polidori, E. Vets, T. C. Sarich and P. P. Stein, Clin. Pharmacol. Ther., 2011, 90, 685–692 CrossRef CAS; (b) L. B. Katz, J. J. Gambale, P. L. Rothenberg, S. R. Vanapalli, N. Vaccaro, L. Xi, T. C. Sarich and P. P. Stein, Diabetes, Obes. Metab., 2012 DOI:10.1111/j.1463-1326.2012.01587.x.
- K. J. Brocklehurst, A. Broo, R. J. Butlin, H. S. Brown, D. S. Clarke, Ö. Davidsson, K. Goldberg, S. D. Groombridge, E. E. Kelly, A. Leach, D. McKerrecher, C. O'Donnell, S. Poucher, P. Schofield, J. S. Scott, J. Teague, L. Westgate and M. J. M. Wood, Bioorg. Med. Chem. Lett., 2011, 21, 7310–7316 CrossRef CAS.
- J. S. Scott, A. M. Birch, K. J. Brocklehurst, A. Broo, H. S. Brown, R. J. Butlin, D. S. Clarke, Ö. Davidsson, A. Ertan, K. Goldberg, S. D. Groombridge, J. A. Hudson, D. Laber, A. G. Leach, P. A. MacFaul, D. McKerrecher, A. Pickup, P. Schofield, P. H. Svensson, P. Sörme and J. Teague, J. Med. Chem., 2012 DOI:10.1021/jm300592r.
- GPR119 agonists were tested on HEK293S cells over-expressing human GPR119. Changes in cAMP concentrations were assessed using the cAMP dynamic 2 HTRF kit (Cisbio) and pEC50 values calculated. The intrinsic activity (IA) was expressed as the percent effect compared to that of the control, 50 μM oleoylethanolamide, defined as 100%. For full details see ESI‡.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS.
- J. Boström, A. Hogner, A. Llinàs, E. Wellner and A. T. Plowright, J. Med. Chem., 2012, 55, 1817–1830 CrossRef.
- K. Goldberg, S. Groombridge, J. Hudson, A. G. Leach, P. A. MacFaul, A. Pickup, R. Poultney, J. S. Scott, P. H. Svensson and J. Sweeney, Med. Chem. Commun., 2012, 3, 600–604 RSC.
- E. Griffen, A. G. Leach, G. R. Robb and D. J. Warner, J. Med. Chem., 2011, 54, 7739–7750 CrossRef CAS.
- P. A. MacFaul, L. Ruston and J. M. Wood, Med. Chem. Commun., 2011, 2, 140–142 RSC.
Footnotes |
| † This article is part of a MedChemComm ‘New Talents’ issue highlighting the work of outstanding rising scientists in medicinal chemistry research. |
| ‡ Electronic supplementary information (ESI) available: Synthetic details for the synthesis of 11 and crystallographic data for 13. See DOI: 10.1039/c2md20130e |
| This journal is © The Royal Society of Chemistry 2013 |