Design of membrane targeting tobramycin-based cationic amphiphiles with reduced hemolytic activity†‡
Ido M.
Herzog
a,
Mark
Feldman
a,
Anat
Eldar-Boock
b,
Ronit
Satchi-Fainaro
b and
Micha
Fridman
*a
aSchool of Chemistry, Tel Aviv University, Tel Aviv 69978, Israel. E-mail: mfridman@post.tau.ca.il; Fax: +972 3-6409293; Tel: +972 3-6408687
bDepartment of Physiology and Pharmacology, Sackler School of Medicine, Tel Aviv University, Tel Aviv 69978, Israel
First published on 13th August 2012
Abstract
Tobramycin-based cationic amphiphiles differing in the chemical bond linking their hydrophobic and hydrophilic parts were synthesized and biologically evaluated. Several compounds demonstrated potent antimicrobial activities compared to the parent drug. One analogue exhibited a significant reduction in red blood cells hemolysis, demonstrating that it is possible to maintain the antimicrobial potency of these molecules while reducing their undesired hemolytic effect through chemical modifications.
Membranes and cell walls are essential constituents required for the viability of bacterial cells, and therefore serve as attractive targets for the development of antibiotics. Amongst cell-wall-targeting antibiotics are several families of peptidoglycan biosynthesis inhibitors, including β-lactams that irreversibly inhibit the activity of the peptidoglycan trans-peptidation biosynthetic step,1,2 glyco-peptide antibiotics such as vancomycin that competitively inhibit the trans-peptidation step, and the glyco-lipid antibiotic agent moenomycin A that inhibits the peptidoglycan trans-glycosylation step.3–6 To date, disruption of the bacterial membrane bi-layer has been poorly exploited as a strategy for the development of antibiotics. Bacterial membrane-disrupting antibiotics offer several advantages over antimicrobial agents that target intracellular bacterial targets: first, membrane disruption is not dependent on the bacterial cell cycle state and is therefore a promising strategy for the eradication of dormant bacteria and treatment of persistent infections.7 Second, antimicrobial agents that act in the extracellular bacterial environment evade intracellular resistance mechanisms and are expected to maintain prolonged clinical efficacy. Finally, cell permeability consideration, which is often a significant challenge for drug designers, is not necessary for the design of membrane-targeting antibiotics. Although peptidoglycan exists solely in bacteria, membranes composed of lipid bi-layers are common to all cells; therefore, avoiding cytotoxicity to eukaryotic cells through non-selective membrane disruption is a major challenge. In contrast to most eukaryotic cell membranes, both Gram-positive and Gram-negative bacterial membranes are highly negatively charged due to high content of anionic lipids such as cardiolipin and phosphatidylglycerol.8,9 Gram-negative bacterial membranes also have the negatively charged core of lipopolysaccharide (LPS), while negatively charged techoic acids are major constituents of Gram-positive bacterial cell walls.10,11
Hence, both Gram-positive and Gram-negative bacterial membranes attract positively charged organic compounds through ionic interactions. LPS that constitutes the Gram-negative outer membrane leaflet is unique to bacteria and serves as a target for the antimicrobial agent polymyxin B1 (Fig. 1). Polymyxin B1 composed of a cyclic cationic decapeptide with an N-terminal hydrophobic residue is a potent and clinically used antibiotic that binds to the negatively charged LPS core and disrupts the outer membrane of Gram-negative bacteria.12,13 The potency and broad-spectrum activity of polymyxin B1 against Gram-negative bacteria demonstrate the potential that lies in the development of membrane-targeting antibiotics. In recent years, several studies have demonstrated the potential of positively charged aminoglycosides (AGs) as scaffolds for the development of membrane-targeting cationic amphiphilic antimicrobial agents by the attachment of hydrophobic residues to one or more positions on the AG.14–17
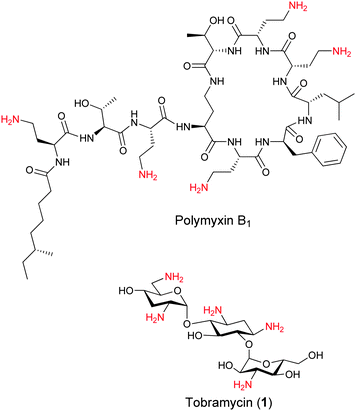 | ||
| Fig. 1 Structures of the Gram-negative bacteria targeting polymyxin B1 and the bacterial ribosome targeting aminoglycoside tobramycin. | ||
We have been particularly interested in tobramycin (1) based cationic amphiphiles since similar to polymyxin B1, this AG also contains five primary amines which are positively charged under physiological conditions (Fig. 1). We recently demonstrated that the attachment of aliphatic chains to the 6′′-position of tobramycin resulted in potent antimicrobial agents and provided evidence for their membrane-disruption activity.18
The most potent and broad-spectrum antimicrobial activity was observed for thioether analogues containing C12-, C14-, and C16-linear alkyl chains (Scheme 1A, 2a–c). The aliphatic chain length affected not only the antimicrobial activity but also the level of undesired red blood cell (RBC) hemolysis; the C12 analogue had the least hemolytic activity. We hypothesized that the aliphatic alkyl chains and the AG scaffold are required for optimal antimicrobial activity but that altering the link between these two segments should not have a dramatic effect on the antimicrobial performance, yet may affect the specificity of these compounds towards different membranes. To test this hypothesis, we chose to evaluate several types of chemical bonds between the aliphatic chain and tobramycin (1). We compared the thioether-linked analogues (Scheme 1A, 2a–c) to sulfone-linked analogues (3a–c), triazole ring-linked analogues (Scheme 1B, 4a–c), and amide bond-linked analogues (Scheme 1C, 5a–c). The thioethers 2a–c were prepared from the penta-NH-Boc-6′′-O-trisyl tobramycin (Scheme 1A, 1a) as previously reported.18,19 Oxidation of the protected thioether analogues (1b–d) using mCPBA followed by the removal of the NH-Boc protecting groups in neat TFA yielded the sulfone analogues (3a–c). The 6′′-O-trisyl group of 1a was replaced by an azide to yield compound 1e,20 which served as a precursor for the preparation of the triazole analogues (Scheme 1B, 4a–c). Microwave-heated click reaction using 1e and terminal alkynyl aliphatic chains, followed by the removal of the NH-Boc groups, yielded the desired triazole analogues 4a–c. Reduction of the 6′′-azido group of 1e under the Staudinger reaction conditions resulted in superior yields (80%) of the 6′′-amino tobramycin analogue 1f (Scheme 1C) compared to the reduction of the azide under catalytic hydrogenation conditions (H2, Pd/C, MeOH). Compound 1f served as the precursor for the preparation of the amide analogues (Scheme 1C); 1f was coupled to linear aliphatic carboxylic acids using HBTU (71–86% yield), and the NH-Boc groups were removed to yield the amide-linked analogues (Scheme 1C, 5a–c).
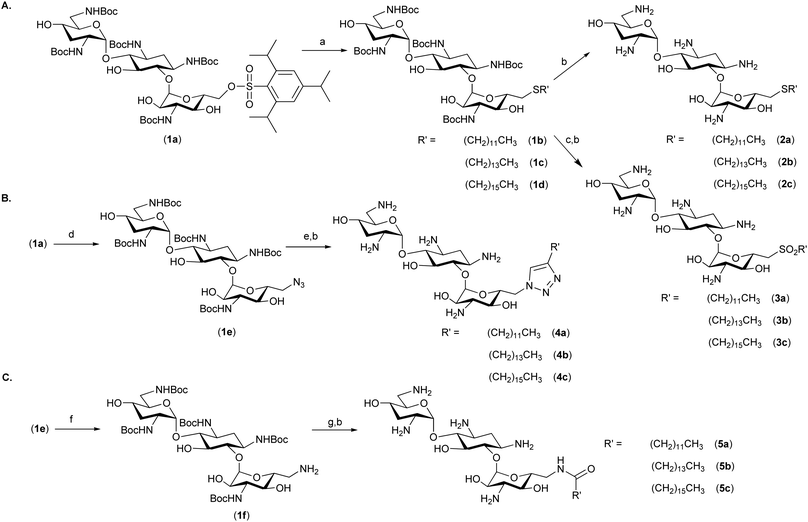 | ||
| Scheme 1 Synthesis of amphiphilic tobramycin analogues: Reagents and conditions: (a) R′SH, Cs2CO3, DMF, 25–60 °C, 63–92%; (b) neat TFA, rt; (c) mCPBA (3 equiv.), CHCl3, rt; (d) NaN3, DMF, 60 °C, 12 h, 91%; (e) R′CCH, CuSO4·5H2O (0.1 equiv.), sodium ascorbate (0.2 equiv.), DMF, microwave irradiation, 87–94%; (f) PMe3 (1 M in THF, 1.1 equiv.), 0.01 M aqueous NaOH/THF: 1/20, rt, 80%; and (g) R′COOH, HBTU, DIEA, DMF, 71–86%. | ||
The minimum inhibitory concentrations (MICs) of the semi-synthetic tobramycin amphiphiles were determined for 11 Gram-positive and Gram-negative strains (Table 1). Amongst the Gram-positive bacteria were pathogenic strains such as Streptococcus pyogenes M12 (strain A), a hospital isolate of methicillin-resistant Staphylococcus aureus (MRSA; strain B), and vancomycin-resistant Enterococcus (VRE; strain D) with high levels of resistance to tobramycin (MIC = 64 μg mL−1 for strain A and >128 μg mL−1 for strains B and D). Amongst the Gram-negative isolates were the pathogenic and highly tobramycin resistant (MIC > 128 μg mL−1) Pseudomonas aeruginosa (ATCC33347; strain I) and Shigella sonnei, which is responsible for the severe foodborne disease shigellosis. Two types of S. sonnei were tested: O-antigen positive (strain J), and O-antigen negative (strain K).21 In general, analogues with a C14 linear aliphatic chain (2b, 3b, 4b, and 5b) exhibited the most potent antimicrobial activity, which was in most cases one to two double dilutions more potent than the activity of the corresponding C12 and C16 linear aliphatic chain analogues. The chemical links between the AG and the aliphatic chain did not have significant effects on MIC values against the tested strains with the exception of the sulfone linked analogues 3a–c. These analogues were less potent than the corresponding un-oxidized thioether analogues 2a–c. Some of the amphiphilic tobramycin analogues demonstrated high potency against strains that were highly tobramycin resistant: the MIC of tobramycin against S. pyogenes M12 (strain A) was 64 μg mL−1; the thioether 2b, triazole 4b, and the amide analogue 5b were 16 to 32 times more potent against this strain (2 μg mL−1 for 2b, and 4 μg mL−1 for 4b and 5b). A significant improvement in antimicrobial activity of the semi-synthetic analogues compared to that of tobramycin was also observed in the case of S. mutans UA159 and S. epidermidis ATCC35984 (strains C and G, respectively). Although most of the synthetic analogues were not active against the tested P. aeruginosa (strain I), the C12 chain triazole analogue 4a and amide analogue 5a demonstrated improved antimicrobial activity against this strain relative to tobramycin (MICs = 64 and 32 μg mL−1, respectively, and MIC > 128 μg mL−1 for tobramycin).
| AGa | MICs (μg mL−1) for tested bacterial strainsb | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | G | H | I | J | K | |
| a AG = aminoglycoside. b MIC values were determined against Gram-positive bacterial strains: A, S. pyogenes serotype M12 (strain MGAS9429); B, MRSA; C, S. mutans UA159; D, VRE; E, E. faecalis ATCC29212; F, S. aureus ATCC9144; G, S. epidermidis ATCC35984; H, S. epidermis ATCC12228 and Gram-negative bacterial strains: I, P. aeruginosa ATCC33347; J, S. sonnei clinical isolate 6831 (O-antigen positive); and K, S. sonnei clinical isolate 6831 (O-antigen negative). All strains were tested by using the double-dilution method (from a starting concentration of 128 μg mL−1). All experiments were performed in triplicate, and results were obtained from two different sets of experiments. | |||||||||||
| 1 | 64 | >128 | 128 | >128 | >128 | 16 | 128 | <1 | >128 | 16 | 32 |
| 2a | 8 | 64 | 8 | 64 | 128 | 16 | 8 | 8 | 128 | 32 | 64 |
| 2b | 2 | 32 | 2 | 16 | 64 | 8 | 4 | 4 | 128 | 16 | 32 |
| 2c | 4 | 64 | 4 | 64 | 128 | 16 | 8 | 4 | >128 | 32 | 64 |
| 3a | 32 | >128 | 16 | 64 | >128 | 64 | 64 | 128 | >128 | >128 | >128 |
| 3b | 4 | 64 | 4 | 32 | 64 | 16 | 16 | 16 | 128 | 32 | 64 |
| 3c | 8 | 32 | 8 | 32 | 64 | 8 | 16 | 16 | >128 | 32 | 128 |
| 4a | 8 | 128 | 4 | 32 | 128 | 16 | 8 | 16 | 64 | 64 | 64 |
| 4b | 4 | 64 | 4 | 32 | 128 | 8 | 8 | 4 | 128 | 32 | 32 |
| 4c | 16 | 32 | 8 | 16 | 128 | 16 | 8 | 4 | >128 | 128 | 64 |
| 5a | 16 | 128 | 4 | 64 | 128 | 16 | 8 | 8 | 32 | 128 | 128 |
| 5b | 4 | 32 | 4 | 32 | 64 | 8 | 4 | 4 | 128 | 16 | 32 |
| 5c | 8 | 32 | 4 | 32 | 128 | 8 | 4 | 4 | 128 | 32 | 32 |
The antibacterial activity of tobramycin and six out of the 12 synthetic analogues was better against O-antigen positive S. sonnei (strain J) than against the corresponding O-antigen negative (strain K). This difference may be explained by the higher overall negative charge of the membrane of the O-antigen positive S. sonnei, which contains the negatively charged 2-acetamido-2-deoxy-L-altruronic acid.22
It was previously demonstrated that low micromolar concentrations of saturated fatty acids inhibit the formation of biofilms formed by S. aureus and Listeria monocytogenes strains.23 The most potent biofilm growth inhibitors were C12–C14 aliphatic chain carboxylic acids. We therefore determined the minimal biofilm inhibition concentration (MBIC) values for each of the C12 and C14 chain tobramycin analogues (Table 2). MBIC tests were performed using S. mutans UA159 and S. epidermidis ATCC35984 grown under biofilm-forming conditions. Compared to tobramycin (MBIC range of 64–128 μg mL−1), the tested analogues demonstrated improved biofilm growth inhibition properties (MBIC range of 4–32 μg mL−1) against the tested strains (Table 2). However, the MBIC values of the tested compounds were identical or no more than one double dilution lower than their MIC values against strains C and G. We therefore conclude that these compounds have no specific biofilm growth inhibition properties against the tested strains, and that their MBIC values result from their antibacterial activity.
| AGa | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Strainb | 1 | 2a | 2b | 3a | 3b | 4a | 4b | 5a | 5b |
a AG = aminoglycoside.
b
S. mutans UA159, C; S. epidermidis ATCC35984, G. All strains were tested by using the double-dilution method (starting from 128 μg mL−1). S. mutans biofilm was grown in BHI + sucrose 2%, at final dilution 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100. S. epidermidis biofilm was grown in TSB + glucose 1%, at final dilution 1:100. Biofilms were stained using crystal violet. All experiments were performed in triplicate and results were obtained from two different sets of experiments. :100. S. epidermidis biofilm was grown in TSB + glucose 1%, at final dilution 1:100. Biofilms were stained using crystal violet. All experiments were performed in triplicate and results were obtained from two different sets of experiments.
|
|||||||||
| C | 64 | 4 | 4 | 16 | 4 | 4 | 4 | 8 | 4 |
| G | 128 | 16 | 8 | 32 | 8 | 16 | 16 | 8 | 8 |
Finally, the hemolytic activity was determined using a hemolysis assay using laboratory rat RBCs (Fig. 2A–C).18 The MIC and MBIC values were significantly lower than the concentrations required for 100% hemolysis for some of the analogues (Fig. 2). In most cases, the MIC range of analogues with the C14 aliphatic chain was 2–32 μg mL−1; these analogues caused significant hemolysis (∼23 to 43%) at 32 μg mL−1. All of the C14 aliphatic chain analogues caused extensive hemolysis (74.4 ± 5.5% to 100%) at a concentration of 64 μg mL−1. The C16 aliphatic chain analogues also caused high levels of hemolysis at 64 μg mL−1 (37.9 ± 5.1% to 81.8 ± 2.3%).
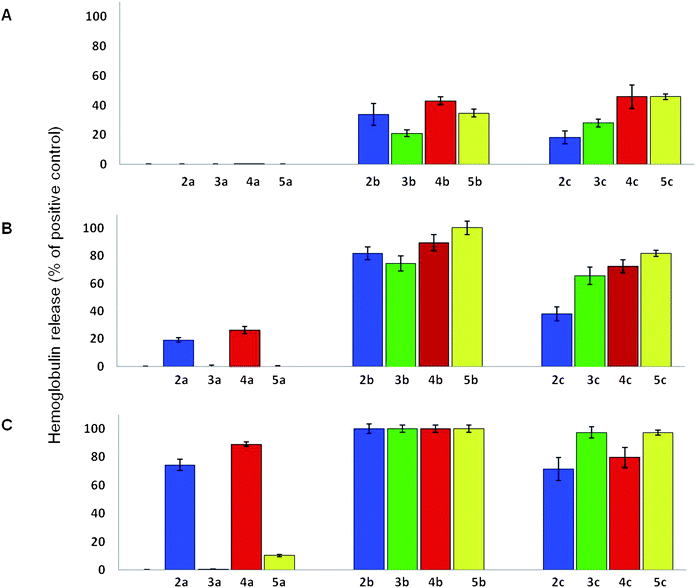 | ||
| Fig. 2 Laboratory rat RBC hemolysis assay. Amphiphilic tobramycin analogues were incubated with RBCs isolated from a laboratory rat at concentrations of (A) 32 μg mL−1, (B) 64 μg mL−1, and (C) 128 μg mL−1 for 1 hour at 37 °C. All experiments were performed in triplicate, and results are the average from two different sets of experiments using blood samples from two laboratory rats. | ||
No direct correlation between the antibacterial potency and the hemolytic activity was detected for the thioether, triazole, or amide analogues. As initially hypothesized, the hemolytic activity of the different tobramycin analogues was affected by the type of bond between the aliphatic chain and the AG scaffold. The most dramatic effect was observed for the C12 aliphatic chain analogues. At 64 μg mL−1, the triazole C12 aliphatic chain analogue 4a demonstrated the highest hemolytic effect (26.3 ± 2.7%) of the C12 aliphatic chain tobramycin analogues. The C12 aliphatic chain amide analogue 5a caused almost no hemolysis at the same concentration (0.0 ± 0.4%). At 128 μg mL−1, the triazole analogue 4a caused extensive hemolysis (89.1 ± 1.6%), the thioether 2a caused 71.6 ± 8.3% hemolysis, yet the amide analogue 5a caused significantly less hemolysis (10.2 ± 0.8%). The lowest hemolytic activity at all of the tested concentrations was observed for the C12 sulfone analogue 3a, however, this compound had poor antimicrobial activity against the tested strains. In contrast, while the C12 amide analogue 5a was potent against several of the tested bacterial strains, and was the most potent analogue against the tested P. aeruginosa (strain I), it caused the lowest levels of hemolysis at a concentration which was 16–32 times higher than the MIC values of this compound against several of the tested strains.
Conclusions
In conclusion, 12 6′′-aliphatic chain tobramycin analogues differing in the chemical linkage between the AG and the hydrophobic chain (thioether, sulfone, triazole, and amide bonds) and in the length of their hydrophobic linear aliphatic chain (C12, C14, and C16 chains) were synthesized and evaluated for their antimicrobial activity against 11 bacterial strains. Of the three chain lengths tested, the C14 aliphatic chain analogues were the most potent antimicrobial agents, and were in most cases one or two double dilutions more potent than the corresponding C12 and C16 chain analogues. In some cases, the antimicrobial activity was at least 32-fold more potent than that of the parent AG tobramycin (1). Finally, RBC hemolysis tests revealed that there was no linear correlation between the antimicrobial potency and the hemolytic activity of the amphiphilic tobramycin analogues. Both the aliphatic chain length and the type of chemical linkage between the hydrophilic and hydrophobic parts of the molecule affect the specificity towards bacterial membranes. The C12 linear aliphatic chain 6′′-amide analogue 5a is of particular interest. This analogue was significantly more potent than tobramycin and caused little measurable hemolysis of laboratory rat RBCs at concentrations up to 32 times higher than the MIC values of this compound against some of the tested bacterial strains. The results of this study demonstrate that the choice of the hydrophobic segment and the chemical group that links the hydrophobic region to the AG is an important factor in the design of such membrane targeting antibiotics. Hence, further improvement in the selectivity of these compounds towards bacterial membranes through chemical modifications is worth pursuing.Acknowledgements
This work was supported by the FP7-PEOPLE-2009-RG Marie Curie Action: Reintegration Grants (grant 246673). We thank Profs Itzhak Ofek, Dani Cohen (Tel Aviv University) and Doron Steinberg (The Hebrew University of Jerusalem) for the gift of bacterial strains.Notes and references
- H. S. Chung, Z. Yao, N. W. Goehring, R. Kishony, J. Beckwith and D. Kahne, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21872–21877 CrossRef CAS.
- J. F. Fisher, S. O. Meroueh and S. Mobashery, Chem. Rev., 2005, 105, 395–424 CrossRef CAS.
- V. L. Healy, I. A. D. Lessard, D. I. Roper, J. R. Knox and C. T. Walsh, Chem. Biol., 2000, 7, R109–R119 CrossRef CAS.
- M. Ge, Z. Chen, H. R. Onishi, J. Kohler, L. L. Silver, R. Kerns, S. Fukuzawa, C. Thompson and D. Kahne, Science, 1999, 284, 507–511 CrossRef CAS.
- Y. Yuan, S. Fuse, B. Ostash, P. Sliz, D. Kahne and S. Walker, ACS Chem. Biol., 2008, 3, 429–436 CrossRef CAS.
- S. Fuse, H. Tsukamoto, Y. Yuan, T.-S. A. Wang, Y. Zhang, M. Bolla, S. Walker, P. Sliz and D. Kahne, ACS Chem. Biol., 2010, 5, 701–711 CrossRef CAS.
- J. G. Hurdle, A. J. O'Neill, I. Chopra and R. E. Lee, Nat. Rev. Microbiol., 2011, 9, 62–75 CrossRef CAS.
- J. Weghuber, M. C. Aichinger, M. Brameshuber, S. Wieser, V. Ruprecht, B. Plochberger, J. Madl, A. Horner, S. Reipert, K. Lohner, T. Henics and G. J. Schutz, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2581–2590 CrossRef CAS.
- R. F. Epand, P. B. Savage and R. M. Epand, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 2500–2509 CrossRef CAS.
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, 000414 CrossRef.
- J. G. Swoboda, J. Campbell, T. C. Meredith and S. Walker, ChemBioChem, 2010, 11, 35–45 CrossRef CAS.
- H. Tsubery, I. Ofek, S. Cohen, M. Eisenstein and M. Fridkin, Mol. Pharmacol., 2002, 62, 1036–1042 CrossRef CAS.
- H. Tsubery, I. Ofek, S. Cohen and M. Fridkin, J. Med. Chem., 2000, 43, 3085–3092 CrossRef CAS.
- M. Ouberai, F. El Garch, A. Bussiere, M. Riou, D. Alsteens, L. Lins, I. Baussanne, Y. F. Dufrene, R. Brasseur, J.-L. Decout and M.-P. Mingeot-Leclercq, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 1716–1727 CrossRef CAS.
- S. Bera, G. G. Zhanel and F. Schweizer, J. Med. Chem., 2010, 53, 3626–3631 CrossRef CAS.
- I. Baussanne, A. Bussiere, S. Halder, C. Ganem-Elbaz, M. Ouberai, M. Riou, J.-M. Paris, E. Ennifar, M. P. Mingeot-Leclercq and J.-L. Decout, J. Med. Chem., 2010, 53, 119–127 CrossRef CAS.
- S. Hanessian, K. Pachamuthu, J. Szychowski, A. Giguere, E. E. Swayze, M. T. Migawa, B. Francois, J. Kondo and E. Westhof, Bioorg. Med. Chem. Lett., 2010, 20, 7097–7101 CrossRef CAS.
- I. M. Herzog, K. D. Green, Y. Berkov-Zrihen, M. Feldman, R. R. Vidavski, A. Eldar-Boock, R. Satchi-Fainaro, A. Eldar, S. Garneau-Tsodikova and M. Fridman, Angew. Chem., Int. Ed., 2012, 51, 5652–5656 CrossRef CAS.
- K. Michael, H. Wang and Y. Tor, Bioorg. Med. Chem., 1999, 7, 1361–1371 CrossRef CAS.
- M. D. Disney and O. J. Barrett, Biochemistry, 2007, 46, 11223–11230 CrossRef CAS.
- J. G. Shepherd, L. Wang and P. R. Reeves, Infect. Immun., 2000, 68, 6056–6061 CrossRef CAS.
- B. Liu, Y. A. Knirel, L. Feng, A. V. Perepelov, S. N. Senchenkova, Q. Wang, P. R. Reeves and L. Wang, FEMS Microbiol. Rev., 2010, 34, 606 CAS.
- U. T. Nguyen, I. B. Wenderska, M. A. Chong, K. Koteva, G. D. Wright and L. L. Burrows, Appl. Environ. Microbiol., 2012, 78, 1454–1465 CrossRef CAS.
Footnotes |
| † This article is part of a MedChemComm ‘New Talents’ issue highlighting the work of outstanding rising scientists in medicinal chemistry research. |
| ‡ Electronic supplementary information (ESI) available: Synthetic protocols, 1H, 13C NMR assignments and spectra, HRMS, MIC, MBIC, hemolysis assay procedures are provided. See DOI: 10.1039/c2md20162c |
| This journal is © The Royal Society of Chemistry 2013 |