Metabolic flux ratio analysis and multi-objective optimization revealed a globally conserved and coordinated metabolic response of E. coli to paraquat-induced oxidative stress
Tie
Shen
a,
Bin
Rui
b,
Hong
Zhou
c,
Ximing
Zhang
a,
Yin
Yi
a,
Han
Wen
b,
Haoran
Zheng
c,
Jihui
Wu
c and
Yunyu
Shi
*c
aSchool of Life Science and Key Laboratory of Plant Physiology and Development Regulation, Guizhou Province, Guizhou Normal University, 550001, Guiyang, China
bSchool of Life Science, Anhui Agricultural University, 230026, Hefei, China
cSchool of Life Science and Hefei National Laboratory for Physical Science at Microscale, University of Science and Technology of China, 230026, Hefei, China. E-mail: yyshi@ustc.edu.cn; Fax: +86-551-3601443; Tel: +86-551-3606364
First published on 11th October 2012
Abstract
The ability of a microorganism to adapt to changes in the environment, such as in nutrient or oxygen availability, is essential for its competitive fitness and survival. The cellular objective and the strategy of the metabolic response to an extreme environment are therefore of tremendous interest and, thus, have been increasingly explored. However, the cellular objective of the complex regulatory structure of the metabolic changes has not yet been fully elucidated and more details regarding the quantitative behaviour of the metabolic flux redistribution are required to understand the systems-wide biological significance of this response. In this study, the intracellular metabolic flux ratios involved in the central carbon metabolism were determined by fractional 13C-labeling and metabolic flux ratio analysis (MetaFoR) of the wild-type E. coli strain JM101 at an oxidative environment in a chemostat. We observed a significant increase in the flux through phosphoenolpyruvate carboxykinase (PEPCK), phosphoenolpyruvate carboxylase (PEPC), malic enzyme (MEZ) and serine hydroxymethyltransferase (SHMT). We applied an ε-constraint based multi-objective optimization to investigate the trade-off relationships between the biomass yield and the generation of reductive power using the in silico iJR904 genome-scale model of E. coli K-12. The theoretical metabolic redistribution supports that the trans-hydrogenase pathway should not play a direct role in the defence mounted by E. coli against oxidative stress. The agreement between the measured ratio and the theoretical redistribution established the significance of NADPH synthesis as the goal of the metabolic reprogramming that occurs in response to oxidative stress. Our work presents a framework that combines metabolic flux ratio analysis and multi-objective optimization to investigate the metabolic trade-offs that occur under varied environmental conditions. Our results led to the proposal that the metabolic response of E. coli to paraquat-induced oxidative stress is globally conserved and coordinated.
Introduction
The ability of a microorganism to rapidly adapt to changes in the environment, such as in nutrient or oxygen availability, is essential for its competitive fitness and survival.1,2 This adaptation requires dynamic reprogramming and changes in the intracellular physiological state of the microorganism.3 An extensive physiological representation involved the flux distribution through the central carbon metabolism, which also provides the energy, cofactors, and building blocks for the maintenance and synthesis of biomass.4 Accordingly, the redistribution of metabolic flux in the adaptation to a varied environment has been of tremendous interest and, thus, has been increasingly explored.5–9Reactive oxygen species (ROS), such as superoxide anion radicals (O2−), could be derived from the endogenous cellular aerobic metabolism or the exogenous agents, such as redox active drugs, near-UV radiation, and heavy metals.10 Since ROS destroy proteins, nucleic acids and lipids, cells possess numerous mechanisms to repair these types of impairments and to adapt to oxidative stress.11 Several such adaptive mechanisms have been revealed in E. coli.12,13 Among these mechanisms, changes in the activities of certain (and most likely other) transcription factors, can alter the global gene expression patterns in cells, which may lead to global changes in protein activities and metabolic fluxes.13–15
Therefore, the influence of oxidative stress on the central carbon metabolism has emerged as an amazing system for the investigation of both the regulatory response of metabolic systems to oxidative stress and the cellular objectives and strategies used by cells in their adaptation to a changing environment. A number of studies focussing on individual protein activity levels have been carried out on key enzymes, such as glucose-6-phosphate dehydrogenase, aconitase and α-ketoglutarate dehydrogenase (AKGDH).16–18 Global responses have previously been investigated using proteomics and gene microarrays.13,14 A genome-wide transcriptional profiling analysis of E. coli probed the activation of the sets of coordinately regulated genes that are required for cellular homeostasis in response to superoxide stress.19 More generally, a time-series microarray experiment was conducted to reveal the transcriptional networks involved in the response to superoxide stress.13
Recently, we used 13C-labelling metabolic flux analysis (13C MFA) to follow the adjustment of the intracellular fluxes of the central metabolism in the defence against superoxide stress.7 The activities of enzymes that are important in the regulation of the flux changes were assayed. However, the quantitative behaviour of the metabolic flux in response to oxidative stress should be analyzed in more detail to determine its systems biology significance taking into account the following issues. (1) In the previous study, because of the large confidence intervals of some of the estimated flux values, the adjustment in certain branch pathways was not precisely processed and discussed. (2) The cellular objective of the complex regulatory structure of the metabolic response has not yet been fully elucidated. Understanding the cellular objective and the strategy of the metabolic response to an extreme environment is therefore of interest because it can offer clues to the molecular mechanisms that regulate the reorganization of metabolic networks and the molecular apparatuses that enable cells to adapt to new environments.20,21
The intracellular fluxes in the central carbon metabolism can be predicted in silico under the assumption that cellular systems function in an optimized manner with respect to a certain biological objective through Flux Balance Analysis (FBA).22 FBA is a commonly used linear optimization method in which a specific objective function is optimized and mass balance and enzyme capacity constraints are used to limit the allowable functional states.23,24 The use of FBA requires a stoichiometric model of the metabolism network and the appropriate definition of the cellular objectives.25–27 However, because neither nature nor cell has a single goal, diverse cellular objectives may exist in the metabolic response to an environmental stimulus.28 A more desirable and realistic theoretical treatment is to simultaneously consider two or more objectives, which may be conflicting. As a consequence, this strategy will result in a set of solutions that represent the optimal trade-offs among the different objectives.29 The search for trade-offs between these objectives is regarded as a multi-objective optimization (MOO) problem.30,31 The results of MOO problems can be represented as the point/curve in the space known as the Pareto-optimal set or the Pareto front.30
Based on these requirements, we combined metabolic flux ratio analysis and multi-objective optimization to obtain more detailed and macroscopic insight into the metabolic adaptation to oxidative stress. The intracellular metabolic flux ratios in the central carbon metabolism were determined by fractional 13C-labelling and metabolic flux ratio (MetaFoR) analysis of a wild-type strain (JM101) exposed to 0, 70 μM paraquat in a chemostat. Paraquat (N,N′-dimethyl-4,4′-bipyridinium dichloride) is readily reduced by E. coli to form a radical, which reacts very rapidly with dioxygen to yield O2−. As such, it is frequently adopted to induce superoxidative stress in previous work. Due to the precision of the flux ratio analysis, we were able to quantify a range of changes in the carbon metabolic flux as a response to PQ stress that had not been previously confirmed in E. coli. In addition to glucose-6-phosphate dehydrogenase (GDH), we observed significant elevation in the fluxes through phosphoenolpyruvate carboxykinase (PEPCK), phosphoenolpyruvate carboxylase (PEPC), malic enzyme (MEZ) and serine hydroxymethyltransferase (SHMT). We applied ε-constraint-based flux balance analysis as an implementation of multi-objective optimization to investigate the trade-off relationship between the biomass yield and the generation of reductive power using an in silico genome-scale model of E. coli K-12, iJR904.32 The results of analysis of the theoretical metabolic flux redistribution implied that the trans-hydrogenase pathway should not play a direct role in the defence against oxidative stress. The agreement between the measured ratio and the theoretical redistribution established the significance of NADPH synthesis as the primary goal of the metabolic reprogramming that occurs in response to oxidative stress. Our work presents a framework that combines metabolic flux ratio analysis and multi-objective optimization for investigation of the metabolic trade-offs that occur under varied environmental conditions.
Materials and methods
Strains and growth conditions
The E. coli K-12 strain JM101 [F−traD36 lacIq Δ (lacZ) M15 proA+B+supE thi Δ (lac-proAB)] was employed in this study. This strain has all wildtype restriction systems of E. coli with no antibiotics resistance and has been extensively used in prior research on metabolic pathways.7,33 Paraquat (PQ), which mediates the transfer of electrons from NADPH or NADH to O2, thereby generating a flux of O2−, was utilized to produce intracellular superoxide.13Batch cultivations were performed overnight in 20 ml tubes with 5 ml of LB medium at 200 rpm and 37 °C. The total batch medium was harvested and used for reactor inoculation.
Continuous cultivation was carried out in a M9 medium, containing 3 g of glucose, 48 mM Na2HPO4, 22 mM KH2PO4, 10 mM NaCl, and 30 mM (NH4)2SO4, as a control experiment. The following ingredients were sterilized and then added separately to the culture (1 ml per litre of the final medium): 1 M MgSO4, 0.1 mM CaCl2, and 1 mg ml−1 vitamin B1 (filter sterilized). In addition, 1 ml per litre of polypropylene glycol 2000 was added to prevent foaming. Cultivation was performed at 37 °C in a 1.3 litre aerobic chemostat in a 1.5 litre fermentor (B. Braun Biotech, Germany) equipped with pH, dissolved oxygen, and temperature probes. The pH was maintained at 7.00 using 1 M NaOH, and the fermentation volume was kept constant with a volume-controlled pump. The agitation was set to 400 rpm, with an airflow of 2 l min−1. The dilution rate (D) was 0.17 h−1. A steady state nature of the continuous cultures can be inferred (i) after approximately 4 to 5 volume changes after new conditions are introduced and (ii) from a stable optical density at 600 nm (OD600) and stable dissolved oxygen for at least two volume changes. Labelling experiments were then initiated and performed as described by Sauer et al.8 The feeding medium containing unlabelled glucose was replaced by an identical medium containing 2.7 g of unlabelled glucose and 0.3 g of [U-13C6] glucose (13C, >99%; CIL) per litre. The biomass samples for MetaFoR were processed after two volume changes, such that 86% of the biomass was labelled. For continuous cultivation of the cells under superoxide stress conditions, 2 mM paraquat was added through a substrate pump with a constant dilution rate. Cultivations with paraquat were maintained with 70 μM paraquat concentrations. 70 μM of paraquat is half of the critical value at which E. coli maintains a growth rate equal to or greater than the dilution rate (0.17 h−1). The other manipulations and parameters used in these cultivations were the same as those in the control experiment.
NMR sample preparation, spectroscopy measurements and data analysis
Preparation of the samples for NMR spectral analysis was performed as described by Szyperski et al.8,34–36 For MetaFoR by NMR, 200 ml of culture was withdrawn and centrifuged at 7000 × g for 10 min at 4 °C. The pellet was washed with 20 ml of a 20 mM Tris–HCl (pH 7.6) solution and centrifuged again. The pellets were then resuspended in 20 ml of the same buffer and sonicated for 5 min. The cell debris was removed by centrifugation at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 5 min. The sonication and centrifugation steps were repeated until cell lysis was complete. Small debris was removed by an additional centrifugation at 14000 × g for 30 min. Pure HCl was added to the supernatant to obtain a final concentration of 6 M. Next, aliquots were hydrolyzed in sealed Pyrex glass tubes for 24 h at 110 °C. The samples were then filtered through a 0.2 mm pore-size filter and lyophilized, and the dried material was dissolved in 600 μl of 20 mM deuterium chloride (DCl) in D2O and incubated for 2 h at room temperature.
000 × g for 5 min. The sonication and centrifugation steps were repeated until cell lysis was complete. Small debris was removed by an additional centrifugation at 14000 × g for 30 min. Pure HCl was added to the supernatant to obtain a final concentration of 6 M. Next, aliquots were hydrolyzed in sealed Pyrex glass tubes for 24 h at 110 °C. The samples were then filtered through a 0.2 mm pore-size filter and lyophilized, and the dried material was dissolved in 600 μl of 20 mM deuterium chloride (DCl) in D2O and incubated for 2 h at room temperature.
Two-dimensional proton-detected heteronuclear single-quantum 13C–1H correlation NMR spectroscopy ([13C, 1H]-HSQC) was performed as described by Szyperski.34 The NMR spectra were documented at 21 °C with a 13C resonance frequency of 150.8 MHz using a Bruker AV600 spectrometer. The integration volume generated by the regression algorithm in SPARKY was adopted as the intensity of each peak here. The NMR intensity was corrected according to the first-order washout kinetics, as previously described.37 The resulting intensities of the individual multiplet components in the 13C–13C scalar coupling fine structures were used as the relative abundances of the specific groups of isotopomers for different amino acids.
Bioreaction network
A model of the central carbon metabolism network was previously developed and is shown in Fig. 1.7,38 This metabolic pathway includes the Embden–Meyerhof (EM), pentose phosphate (PP), and Entner–Doudoroff (ED) pathways, the tricarboxylic acid (TCA) cycle, the anaplerotic reaction, and the glyoxylate shunt. The pathways relevant to the metabolic flux ratio that we measured are listed below.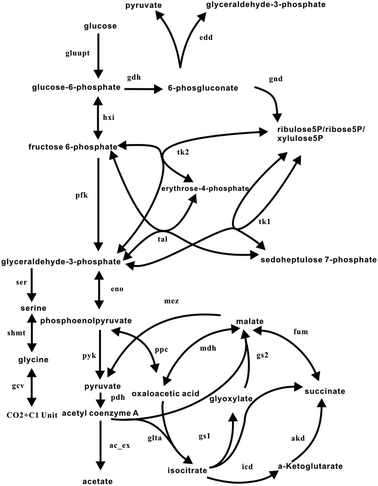 | ||
| Fig. 1 Reactions of the central carbon metabolism network in E. coli. This information was compiled from the EcoCyc database40 and previous work.38,41 The sets of reactions involved in glycolysis, the PP pathway, the glyoxylate shunt, the TCA cycle, the ED pathway and the C1 metabolism are included. The direction of the reaction is denoted by the arrow. Double arrows represent reversible reactions. The abbreviations for the reactions are presented in the appendix. | ||
The EM pathway is generalized into four successive basic steps: hxi (the step from glucose-6-phosphate to fructose-6-phosphate), pfk (the step from fructose-6-phosphate to 2-glyceraldehyde-3-phosphate), eno (the step from glyceraldehyde-3-phosphate to phosphoenolpyruvate), and pyk (the step from phosphoenolpyruvate to pyruvate).
The PP pathway consists of five steps: gdh (the step from glucose-6-phosphate to 6-phosphogluconate (6PG)), gnd (the step from 6PG to CO2 and ribulose 5-phosphate), tk1 (the step from xylulose 5-phosphate and ribose 5-phosphate to sedoheptulose 7-phosphate and glyceraldehyde-3-phosphate (GAP)), tk2 (the step from xylulose 5-phosphate and erythrose 4-phosphate to GAP and fructose 6-phosphate), and tal (the step from GAP and sedoheptulose 7-phosphate to erythrose 4-phosphate and fructose 6-phosphate).
The TCA cycle includes six steps: glta (the step from acetyl-CoA and oxaloacetic acid to citrate and then to isocitrate), icd (the step from isocitrate to α-ketoglutarate and CO2), akd (the step from alpha-ketoglutarate to succinate and CO2), fum (the step from succinate to malic acid), and mdh (the step from malate to oxaloacetate) and ac_ex (the step from acetyl-CoA to acetate).
The anaplerotic reaction includes two steps: ppc (the step from phosphoenolpyruvate to oxaloacetate) and mez (the step from malic acid to pyruvate).
The pathway of serine and glycine synthesis includes 3 steps: ser (the step from glyceraldehyde-3-phosphate to serine), glyA (the step from serine to glycine, which forms 5-10-methylenetetrahydrofolate) and gcv (the cleavage of glycine into CO2 and 5-10-methylenetetrahydrofolate).
Metabolic Flux Ratio (MetaFoR) analysis
The overall degree of 13C labelling in the sample (Pl) and the fraction of the labelled substrate (Pf) were determined as described by Szyperski et al. and confirmed by analysis of the scalar coupling fine structure of leucine C.36,39Table 1 presents the definition of the experimentally determined flux ratios. We calculated the relative abundances of intact carbon fragments originating from a single molecule of glucose as previously described.34 The flux ratios of key pathways were adopted as described previously.8,34,42,43 The experimental error (error bars) was estimated from the signal-to-noise ratio of the 13C–1H COSY spectra according to the Gaussian law of error propagation.
| Symbol | Description | Definition |
|---|---|---|
| a V represents the absolute value of the forward or backward velocity of a reaction. b The subscript denotes the corresponding metabolic step in Fig. 1. No sign symbol represented a irreversible reaction. + Refers to the forward velocity of a reaction. − Refers to the backward velocity of the reaction. | ||
| XPPC | Oxaloacetate from PEP | V ppc +, , /(Vppc+ + Vmdh+) |
| XOAA | PEP from oxaloacetate | V ppc −/(Vppc− + Veno+) |
| Xmalate | Pyruvate from malate | V mez/(Vmez + Vpyk) |
| XPP | G3P from PP pathway | (Vtk1+ + Vtk2+ + Vtal+)/(2 × Vhxi+ + Vtk1+ + Vtk2+ + Vtal+ + Vedd) |
| XSER | Glycine from serine | V ser/(Vser + VglyA−) |
| XGLY | Serine from glycine | V glyA +/(VglyA+ + Vgcv−) |
| Xexchange | OAA exchanged to FUM | (Vfum−)/(Vfum− + Vakd) (Vfum+)/(Vmdh− + Vfum+ + Vgs2) (Vmdh+)/(Vmdh+ + Vppc+) |
Multi-objective optimization
Assuming that the metabolic network is operating at a steady state, the flux balance model can be set up as follows:| S·v = b | (1) |
An objective function for FBA can be denoted as
| Z = c·v | (2) |
| Z = (Z1, Z2, …, Zp) | (3) |
Using eqn (1) and (3), the multi-objective optimization can be stated as follows:
| Max/Min Z = (Z1, Z2, …,Zp) | (4) |
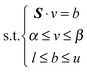 | (5) |
The objectives may be in conflict with each other. Consequently, multi-objective optimization, unlike traditional single-objective optimization, does not result in a unique solution that simultaneously optimizes all of the objectives. The key concept is the Pareto-optimal set. A point, v*, is included in the Pareto-optimal solution if there does not exist any other feasible point, v, such that Zi(v) ≤ Zi(v*) for all i = 1, …, p and Zj(v) < Zj (v*) for at least one j. Namely, v* is optimal if an objective can only be reached by sacrificing one or more of the others. Thus, the solution to a multi-objective optimization is a set of potentially infinite points, none of which behaves better than another. This set is known as the Pareto-optimal set or Pareto front.
Due to the linear objectives and the convex solution space, we can apply the well-known ε-constraint method to obtain the Pareto front.29,44 The multi-objective optimization is then transformed into a single-objective problem by maximizing one of the objectives, while the other is incorporated as the inequality constraint ε. The stepwise change in the parameter ε allows identification of the Pareto curve. The formulation was stated as follows:
| max(Z1 or Z2) | (6) |
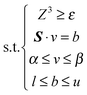 | (7) |
The optimization was performed in COBRA TOOLBOX 2.0, a MatLab Package for implementing the COBRA method. The method was applied using iJR904, which is a widely used genome-scale metabolic model of E. coli.32,45 The term Z3 represented the biomass yield. The ε step for Z3 was 0.006, and the Z3 values ranged from 0.84 to 1.18, which encompasses the biomass yield for both normal and stressed cells. Two separate optimizations were conducted: one for Z1 and the other for Z2. The term Z1 represents the weighted sum of all of the NADPH-producing reactions in the model,26 whereas Z2 was the weighted sum of all NADPH-producing reactions with the exception of the trans-hydrogenase reaction, which was kept at a constant value. Flux balance analysis was performed using the optimized CbModel in the COBRA TOOLBOX.23,46 When the flux balance analysis could not return a single solution, a flux variability analysis was employed, and the resulting flux range was averaged to obtain the metabolic flux. This computation was performed using the flux variability component in the COBRA TOOLBOX.
Results
The changes in some general growth parameters and redistribution of metabolic flux ratios under PQ stress
Certain physiological parameters of E. coli under PQ stress were measured in chemostat cultivations. Table 2 shows cell proliferation rate and dry weight increases determined for wild type E. coli growing in normal and PQ-containing minimum media. Under PQ exposure, the rate of biomass accumulation was reduced. In addition, the rate of acetic acid production, which is represented by the efflux AC_EX in the metabolic model, increased dramatically.| Strain | JM101 | JM101 |
|---|---|---|
| a Yield of biomass on glucose. b Specific acetic acid production rate. | ||
| Dilution rate (h−1) | 0.17 | 0.17 |
| Y biomass/glc (g g−1) | 0.374 ± 0.04 | 0.323 ± 0.05 |
| Q ace (mmol g−1 h−1) | — | 6.43 ± 1.2 |
| Paraquat (mmol l−1) | 0 | 70 |
The metabolic flux ratios were calculated for wild-type JM101 exposed to 0, 70 μM paraquat stress. At the PQ levels tested, the intracellular environment should be sufficiently disturbed, while the cells maintain a growth rate equal to or greater than the dilution rate of 0.17 h−1.
The metabolic flux ratios of the wild type E. coli in normal and PQ-containing media are summarized in Fig. 2. The indication of the ratios in the normal and PQ-stressed cells exhibited observable shifts among a range of pathways in the central carbon metabolic network. These include the following:
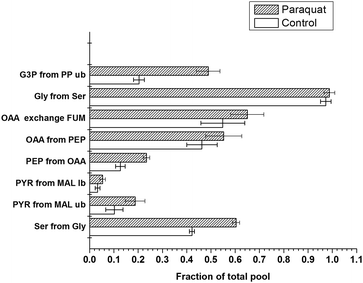 | ||
| Fig. 2 Flux ratios of E. coli JM101 associated with different concentrations of paraquat. The E. coli strains were grown in glucose-limited chemostats with 0, 70 μM paraquat. The white box represents the flux ratio under no-paraquat conditions. The black twill represents the flux ratio in cells exposed to 70 μM paraquat. The experimental error (error bars) was estimated from the signal-to-noise ratio of the 13C–1H COSY spectra according to the Gaussian law of error propagation. The fraction of the total pool for a particular metabolite represents the ratio of this metabolite derived from a specified substrate to the sum of all other substrates that contribute to the pool of this metabolite, as presented in Table 1. Abbreviations: G3P, 3-phosphoglycerate; PEP, phosphoenolpyruvate; PYR, pyruvate; SER, serine; GLY, glycine; OAA, oxaloacetate; MAL, malate; FUM, fumarate. The terms lb and ub refer to the lower and upper limits of the flux ratio, respectively. | ||
(1) The fraction of G3P derived from the PP pathway increased significantly to 0.49 after exposure to 70 μM PQ stress (from 0.20 under normal conditions).
(2) The fraction of oxaloacetic acid from PEP rose from a baseline of 0.46 to fractions of 0.55 in response to 70 μM PQ. Similarly, the fraction of PEP from oxaloacetic acid significantly increased from 0.13 to 0.23.
(3) An increase in the fraction of pyruvate from malate was also detected after exposure to both PQ levels. The lower and upper limits for this fraction in normal medium were 0.03 and 0.05, respectively, whereas these values were 0.05 and 0.18, respectively, after exposure to 70 μM PQ stress.
(4) An increase in the fraction of serine derived from glycine from 0.41 to 0.60 was observed after exposure to 70 μM PQ.
(5) The fraction of pyruvate from the edd pathway was not investigated since the ED pathway does not play a significant role in oxidative stress based upon our previous work. The indication of activation glyoxylate shunt was not clear, as such the fraction of oxaloacetic acid glyoxylate shunt was not shown.34
Metabolic flux shift predicted by ε-constraint flux balance analysis
The response of metabolic enzymes to oxidative stress has been exhaustively mined in recent years.41,47–50 Some researchers believe that a number of enzymes related to NADPH generation are induced to increase NADPH yields. However, whether the metabolic redistribution is the simple combination of a number of individual events or represents the globally concerted emergence of systems-wide behaviour is unknown. What is the trade-off relationship between the multiple conflicting objectives under oxidative stress? To address these questions, multiple common cellular functions are simultaneously optimized through flux balance analysis to predict the intracellular flux response without imposing additional constraints. Maximization of biomass was incorporated as Z3, which is one of two conflicting objectives. The other objective was selected from the following two objectives: Z1, which represents maximization of the sum of all NADPH-producing reactions, and Z2, representing maximization of the sum of all NADPH-producing reactions with the exception of the trans-hydrogenase reaction, which is kept constant.The ε-constraint method in the 2D direction provides a good approximation of the overall shape of the Pareto optimal solution set for the two objectives. The resulting projection of Pareto surfaces (interpolated) on biomass yield and NADPH yield is shown in Fig. 3. The first pareto curve corresponded to the optimization process with maximization of Z1. The second pareto curve corresponded to the optimization process with maximization of Z2. From these figures, it can be observed that there is a conflict between the production of NADPH and the biomass yield. The maximum NADPH yields were associated with reduced biomass yields.
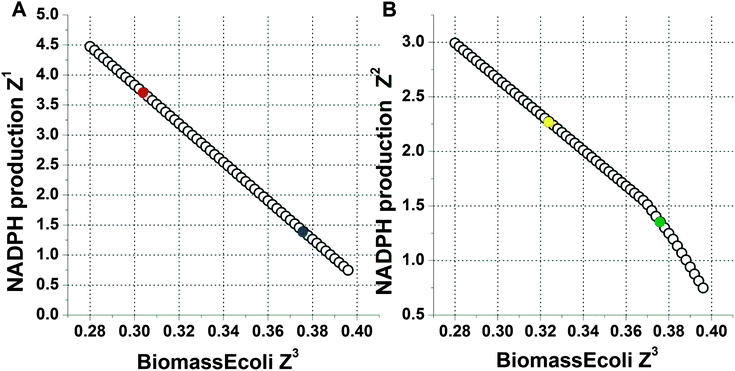 | ||
| Fig. 3 The Pareto curve obtained via the ε-constraint method for the NADPH production rate vs. the biomass production rate. The NADPH-producing rate was normalized with respect to the rate of glucose uptake. The x-axis indicates the biomass yield on glucose, whose unit is g g−1 as denoted in Table 1. (A) The y-axis indicates the value of Z1, which is the weighted sum of all of the NADPH-producing reactions in the model. In this curve, the circle filled with red colour represented the cell in normal media while the circle filled with navy blue colour represented the cell in PQ-containing media. (B) The y-axis indicates the value of Z2, which is the weighted sum of all of the NADPH-producing reactions with a constant value for the trans-hydrogenase reaction. In this curve, the circle filled with yellow colour represented the cell in normal media. The circle filled with green colour represented the cell in PQ-containing media. | ||
The trade-off between the intracellular flux and the NADPH yield (denoted by biomass yield) under the experimental conditions is also depicted, as shown in Fig. 4. The black circles (○) corresponded to the optimization process with maximization of Z1. In this curve, the circle filled with red colour represented the cell in normal media while the circle filled with navy blue colour represented the cell in PQ-containing media. The metabolic flux shift predicted with maximization of Z1 was based on the data extracted from the two points. The violet circles (○) corresponded to the optimization process with maximization of Z2. In this curve, the circle filled with yellow colour represented the cell in normal media. The circle filled with green colour represented the cell in PQ-containing media. The metabolic flux shift predicted with maximization of Z2 was based on the data extracted from the two points. When the red and yellow circles overlapped, they were represented by the circle filled with violet colour. When navy blue and green circles overlapped, they were represented by the circle filled with black colour. The trade-off relationship between the corresponding flux and NADPH production (denoted by biomass yield) could be drawn from these curves. Additionally, in the maximization of Z1, a decrease was predicted for the flux through GDH, MEZ, SHMT and PPC and an increase for AC_EX, whereas maximization of Z2 predicted an increase in the flux through GDH, MEZ, PPC and AC_EX and a decrease in the flux through SHMT.
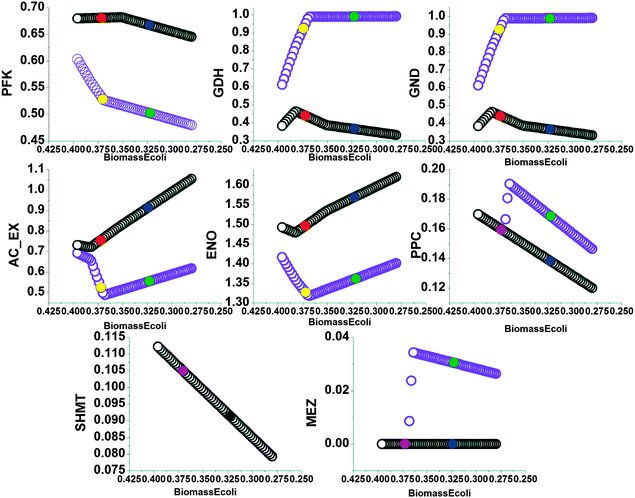 | ||
Fig. 4 The trade-off in the flux relationships with respect to the changes in the Pareto optimal solution. The x-axis is the biomass yield on glucose, whose unit is g g−1 as presented in Table 1. The tick labels on the x-axis decrease from 0.47 to 0.21. The flux values are relative to the specific glucose uptake rate (16.7 mmol h−1). The abbreviations for the reactions are presented in the appendix. The black circles (○) represent the result of maximizing the weighted sum of all NADPH-producing reactions. In this curve, the circle filled with red colour represented the cell in normal media while the circle filled with navy blue colour represented the cell in PQ-containing media. The violet circles ( ) represent the result of maximizing the weighted sum of all NADPH-producing reactions with a constant value for the trans-hydrogenase reaction. In this curve, the circle filled with yellow colour represented the cell in normal media. The circle filled with green colour represented the cell in PQ-containing media. When the red and yellow circles overlapped, they were represented by the circle filled with violet colour. When navy blue and green circles overlapped, they were represented by the circle filled with black colour. ) represent the result of maximizing the weighted sum of all NADPH-producing reactions with a constant value for the trans-hydrogenase reaction. In this curve, the circle filled with yellow colour represented the cell in normal media. The circle filled with green colour represented the cell in PQ-containing media. When the red and yellow circles overlapped, they were represented by the circle filled with violet colour. When navy blue and green circles overlapped, they were represented by the circle filled with black colour. | ||
Discussion
Possible regulatory mechanism involved in changes in local pathways
The addition of paraquat to the cellular environment triggered changes in the overall central carbon metabolism, which were manifested in the redistribution of the flux around several key branch points. The flux through the PP pathway (including GDH and GND) has been confirmed to increase in response to superoxidative stress and acts as a major source of NADPH, an antioxidant that opposes PQ stress.7,14,16,47 Our results are in agreement with this previous research.The fractions of serine from glycine and glycine from serine both increased in cells exposed to paraquat. Because these fractions depend on the forward and reverse fluxes for the conversion of serine into glycine, this shift reflects an enhancement of the in vivo serine hydroxymethyltransferase (SHMT) activity and its operating net flux. Serine hydroxymethyltransferase is a ubiquitous enzyme that is found in all prokaryotes and eukaryotes. Elevated SHMT activity is associated with an increased demand for DNA synthesis, particularly in tumour cells.51 SHMT has thus arisen as an attractive target for cancer chemotherapy due to its central role in nucleotide biosynthesis.51,52 Vijaya Lakshmi et al. showed that the activity of a mutant form of SHMT conferred protection against oxidative stress induced by coronary artery disease in human cells.53 Jin Endo reported that oxidative stress was stimulated by mitochondrial aldehyde in murine cardiovascular cells, in which the gene encoding SHMT was shown to be significantly upregulated.54 Jeon et al. found that upregulation of SHMT was induced in murine kidneys treated with TiO2 nanoparticles, which trigger oxidative stress.55 These findings were all obtained in mammalian cells. To the best of our knowledge, our result is the first direct observation of elevated SHMT activity induced by oxidative stress in vivo in prokaryotic organisms.
SHMT catalyses the reversible retro-aldol cleavage of serine to produce glycine, which is a precursor of glutathione, and results in the hydroxymethyl group being transferred to tetrahydrofolate to form 5,10-methylene-tetrahydrofolate. Eukaryotic cSHMT has been demonstrated to sequester 5-methyltetrahydrofolate and inhibit SAM synthesis, which competes with glutathione synthesis.54 Therefore, we suggest that the upregulation of SHMT plays a role in the response to oxidative stress. Increased oxidative stress induced enhanced expression of SHMT and, hence, an upregulation of the flux through the reaction catalyzed by SHMT. This increased flux resulted in an increased glycine level and inhibited SAM synthesis. The inhibition of SAM synthesis in turn led to an increase in the availability of cysteine, which plays a pivotal role in glutathione production. The increased glycine and cysteine levels therefore result in an elevated glutathione level. Thus, we suggest that the SHMT flux increases in E. coli to meet the high GSH demand. The fractions of oxaloacetic acid from PEP and PEP from oxaloacetic acid were enhanced under paraquat stress. Because the fraction of PEP derived from oxaloacetic acid is due exclusively to the flux through phosphoenolpyruvate carboxykinase (PEPCK) in E. coli, the increase in this fraction demonstrates an upregulation of the activity of this enzyme. The phosphoenolpyruvate carboxykinase enzyme catalyses the reversible dissociation of oxaloacetate to yield phosphoenolpyruvate and CO2. This enzyme plays a key role in several metabolic processes and has been widely studied in mammalian cells.56,57 Our results are concordant with the enhanced PEPCK expression observed in H4IIE cells under oxidative stress.58 The upregulation of this enzyme activity mainly results from elevated gene expression because PEPCK has been shown to be almost entirely regulated at the transcriptional level.57 The metabolic involvement of PEPCK in oxidative stress is poorly understood and has yet to be fully delineated.
PEPCK usually operated exclusively forwardly in E. coli grown on glucose and accounts for a small portion of the carbon flux.38 Hence, the elevation in the fraction of oxaloacetic acid from PEP is mainly the result of increased phosphoenolpyruvate carboxylase (PEPC) activity. PEPC activity was found to be induced in rice under oxidative stress.59 However, this is the first report that shows that oxidative stress increases PEPC activity in E. coli. PEPC is an allosteric enzyme because it possesses many types of effectors and has distinct binding sites for each of them. Cysteinyl, histidyl, lysyl and arginyl residues are essential for its catalytic activity.60 Therefore, the regulation of this enzyme in response to oxidative stress is most likely complicated and, thus, needs to be explored further. Phosphoenolpyruvate carboxylase catalyzes the irreversible carboxylation of phosphoenolpyruvate (PEP) to form oxaloacetate (OAA). Recent studies have reported that oxidative stress decreases the cellular levels of pyruvate dehydrogenase activity.61,62 The increased PEPC flux therefore offers a different fate for phosphoenolpyruvate as an alternative to the impairment of the pyruvate dehydrogenase pathway in response to oxidative stress. An elevation of the fraction of pyruvate from malate was detected, which indicates an enhancement in the flux toward pyruvate through malic enzyme (MEZ). Malic enzyme is responsible for the reversible oxidative decarboxylation of L-malate, which produces NAD(P)H. Two malic enzymes have been found in E. coli, one of which is NADP-dependent, while the other is NAD-dependent. Enhancement of NADP-dependent malic enzyme activity in response to oxidative stress has been observed in Pseudomonas fluorescens.50 Expression of the NAD(P)-malic enzyme gene was shown to increase in response to oxidative stress in plants.63 NADPH is regarded as the most relevant molecule in protection against oxidative stress. The reaction catalyzed by malic enzyme combined with the reaction catalyzed by PEPC and the reverse function of malate dehydrogenase produce NADPH and decouple the formation of NADPH from the TCA cycle.64 Hence, the coordinated increase in the fluxes through malic enzyme and PEPC might serve as another important source of NADPH production.
Global conservation of metabolic flux redistribution in response to super-oxidative stress
Oxidative stress has been shown to be strongly associated with diverse human diseases, such as PD and the development of cancer. The role of metabolic reprogramming in cancer development and diseased cells has been the subject of increasing numbers of investigations in recent years. Therefore, the metabolic reprogramming that occurs in response to oxidative stress has drawn heightened interest.7,47,50,65 The adjustment of the pentose pathway that occurs in response to oxidative stress has been shown to be conserved among a number of organisms.47,49,50 In this study, we showed that the changes in the flux through SHMT, PEPC, PEPCK and MEZ in response to oxidative stress are also conserved among both prokaryotic and eukaryotic organisms. This implies that the metabolic reprogramming that occurs in response to oxidative stress is widely conserved throughout evolution at the level of the global central carbon metabolism, instead of at a local pathway level.The trade-off relationship between biomass yield and the recycling of reductive power
Maximization of the weighted sum of all NADPH-producing reactions resulted in a theoretical metabolic flux redistribution that exhibited limited overlap with the experimental results. In fact, the predicted response of the GDH, SHMT, MEZ and PPC steps was the opposite of the measured values with the exception of the AC_EX. The metabolic capacity shift in this situation is therefore not a good approximation of the physiological reality within the cell. However, theoretical maximization of the weighted sum of all NADPH-producing reactions, with the exception of the trans-hydrogenase (THD) reaction, resulted in a metabolic flux redistribution that coincided well with the experimental results for all reactions, except the flux through SHMT.The incorporation of THD resulted in nearly completely opposite flux responses. Discrepancy between the Z1 and Z2 predictions was only generated by removal of the THD step from the objective function. The significant decrease in the amount of correctly predicted fluxes using the Z1 objective function led to the exclusion of THD as an objective for the NADPH maximization. The THD step consists of the trans-hydrogenase reaction, which transfers electrons directly from NADH to NADP+ and vice versa. The membrane-bound trans-hydrogenase PntAB catalyses the energy-dependent transfer of reducing power from NADH to NADP+, whereas the soluble trans-hydrogenase UdhA catalyses the reverse, energy-independent reaction.66 Reduced pntA transcription was observed concomitant with excess NADPH formation; similarly, downregulation of udhA transcription occurs when NADPH formation is reduced. The role of PntAB and UdhA was found to be involved in the restoration of the balance between NADH and NADPH, as shown by Canonaco and Sauer et al.67 Therefore, the THD step is incompatible with the goal of maximizing NADPH production.68 Additionally, the expression of udhA and pntA was found to be simultaneously slightly induced by paraquat stress in previous study.41 These data support that the THD reaction should not play a direct role in the defence against oxidative stress.
With the exception of the flux through SHMT, the results obtained from the maximization of Z2 were consistent with the measured experimental changes, as shown in Fig. 5. NADPH primarily provides the reductive power required for biosynthesis and serves as the reductive source for the recycling of antioxidants. The metabolic responses coordinated to fulfil the requirement for NADPH coincide well with the experimental results. Our results therefore establish the significance of NADPH production as the purpose of the metabolic reprogramming that occurs in response to oxidative stress. Previous research did not ascertain whether this metabolic reprogramming is a simple combination of a number of individual events or represents the globally concerted emergence of systems-wide behaviour. Based on the data obtained from our multi-objective optimization, we tend to support the hypothesis that under oxidative stress conditions, metabolic systems undergo a globally concerted reorganization in favour of maximizing the production of NADPH.
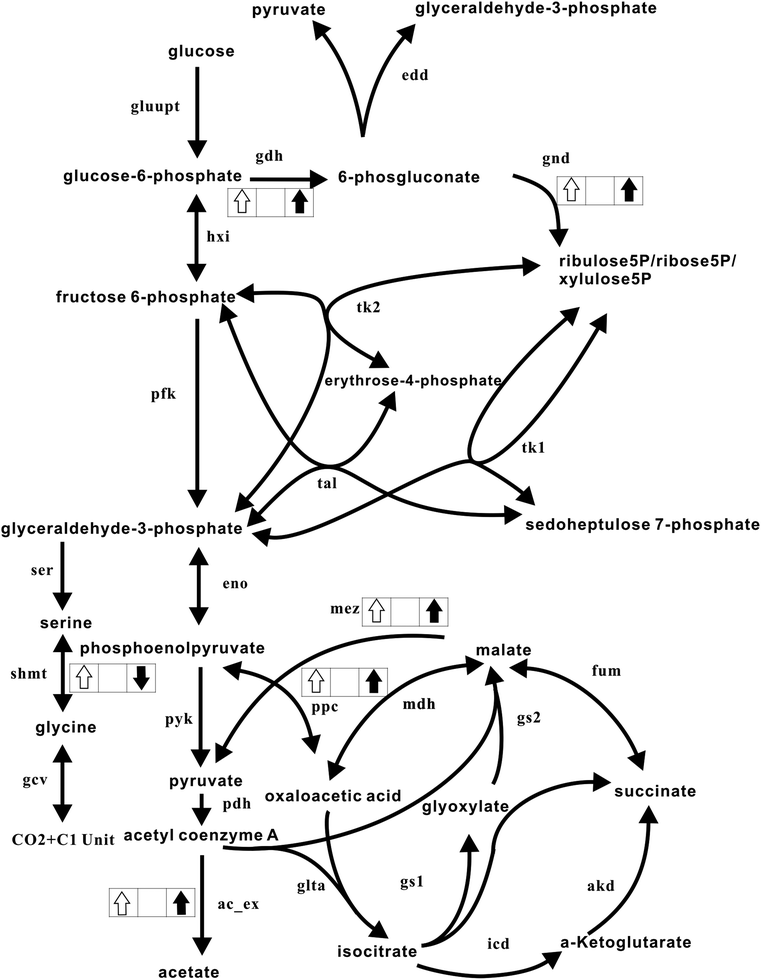 | ||
| Fig. 5 Comparison of the measured and theoretically predicted metabolic responses. The metabolic network is the same as in Fig. 1. An upward white arrow in the box illustrates that the flux was shown to be enhanced experimentally. A downward white arrow in the box shows that the flux was experimentally reduced. Similarly, an upward black arrow depicts that the flux was theoretically predicted to increase, whereas a downward black arrow illustrates that the flux was theoretically reduced. | ||
Conclusions
It has been reported that E. coli undergoes reprogramming of its metabolic systems in defence against and adaptation to the oxidative stress induced by paraquat. Although a number of studies have been performed addressing the quantitative behaviour and cellular objective of metabolic responses, further research is required to understand their functional significance.We carried out quantitative measurements on the changes in the metabolic flux ratios that occur due to exposure of E. coli to PQ. In addition to the established increase in the flux catalyzed by GDH and efflux of acetate, we observed significant elevation of the flux through PEPCK, PEPC, MEZ and SHMT. These adjustments, whether occurring individually, such as for SHMT, or coordinately, such as for PPC and MEZ, contribute to the increased requirement for reductive power that occurs in response to oxidative stress. More generally, our results and other studies performed on eukaryotic cells support the conclusion that the metabolic reprogramming that occurs in response to oxidative stress exhibits universality among prokaryotic and eukaryotic organisms.
Environmental perturbations cause fluctuations in the survival stress on microorganisms. Thus, the metabolic systems of microorganisms might assume conflicting cellular functions depending on environmental conditions. Performing ε constraint-based flux balance analysis enabled us to obtain the Pareto curve and perform a trade-off analysis of the flux distributions between these different objectives. We applied this method to investigate the trade-off between the biomass yield and the generation of reductive power using the iJR904 in silico genome-scale model of E. coli K-12. The obtained theoretical metabolic responses supported that the trans-hydrogenase pathway should not play a direct role in the defence against oxidative stress. The agreement between the measured ratio and the theoretical redistribution established the significance of NADPH production as the purpose of the metabolic reprogramming in response to oxidative stress. Consequently, multi-objective optimization demonstrated that metabolic systems underwent a globally concerted reorganization, as opposed to individual pathways being regulated. In addition, our results present a framework that combines metabolic flux ratio analysis and multi-objective optimization for examination of the metabolic trade-offs that occur under varied habitats. This combined approach can provide a comprehensive and informative picture of the response of microorganisms to oxidative stress and other severe environmental challenges.69
Abbreviations
1. Metabolic steps in Fig. 1 and in the text
(1) Embden–Meyerhof–Parnas (EM) pathway:| hxi | glucose6P ⇒ fructose6P |
| pfk | fructose6P ⇒ 2glyceraldehyde3P |
| eno | glyceraldehyde3P ⇒ phosphoenolpyruvate |
| pyk | phosphoenolpyruvate ⇒ pyruvate |
| (2) | Serine and glycine biosynthesis: |
| ser | glyceraldehyde-3-phosphate ⇒ serine |
| glyA | serine ⇒ glycine + 5-10-methylenetetrahydrofolate |
| gcv | glycine ⇒ CO2 + 5-10-methylenetetrahydrofolate |
| (3) | Tricarboxylic acid cycle: |
| glta | acetyl-CoA + oxaloacetate + H2O ⇒ isocitrate + CoA |
| icd | isocitrate + NADP ⇒ α-ketoglutarate + NADPH + CO2 |
| akd | α-ketoglutarate + CoA + NAD ⇒ succinyl-CoA + CO2 + NADH |
| fum | succinyl-CoA ⇒ malate |
| mdh | malate + NAD ⇒ oxaloacetate + NADH |
| ac_ex | acetyl-CoA ⇒ acetate |
| (4) | Pentose phosphate (PP) pathway: |
| gdh | glucose6P + H2O + NADP ⇒ 6-phosphogluconic acid + NADPH |
| gnd | 6-phosphogluconic acid + NADP ⇒ ribulose5P + CO2 + NADPH |
| rpe | ribulose5P ⇒ ribose5P |
| rpi | ribulose5P ⇒ xylulose5P |
| tk1 | xylulose5P + ribose5P ⇒ sedoheptulose7P + glyceraldehyde3P |
| tk2 | xylulose5P + erythrose4P ⇒ fructose6P + glyceraldehyde3P |
| tal | sedoheptulose7P + glyceraldehyde3P ⇒ fructose6P + erythrose4P |
| (5) | Anaplerotic reactions: |
| ppc | phosphoenolpyruvate + CO2 + ATP + H2O ⇒ oxaloacetate + ADP |
| mez | malic acid ⇒ CO2 + pyruvate |
| (6) | Glyoxylate shunt: |
| gs1 | isocitrate ⇒ glyox + succinic acid |
| gs2 | glyOx + acetyl-CoA ⇒ malic acid |
| (7) | Entner–Dourodouf pathway: |
| edd | 6-phoshogluconic acid ⇒ glyceraldehyde3P + pyruvate |
2. Metabolites and cofactors
| ACA | acetyl coenzyme A |
| AKG | α-ketoglutarate |
| E4P | erythrose-4-phosphate |
| Fru6P | fructose 6-phosphate |
| GLC | glucose |
| Glc6P | glucose-6-phosphate |
| G3P | glyceraldehyde-3-phosphate |
| OAA | oxaloacetic acid |
| SUC | succinate |
| GLY | glycine |
| Glyoxy | glyoxylate |
| ICIT | isocitrate |
| MAL | malate |
| PYR | pyruvate |
| PEP | phosphoenolpyruvate |
| Rib5P | ribose 5-phosphate |
| Rul5 | ribulose 5-phosphate |
| Sed7P | sedoheptulose 7-phosphate |
| Xul5P | xylulose 5-phosphate |
| Ery4P | erythrose 4-phosphate |
| SER | serine |
Acknowledgements
This work was supported by the Chinese National Natural Science Foundation (grant 31200626), Guizhou Lianhe Foundation, LKS(2012)22, the National Basic Research Program of China (973 Program) (grants 2011CB966302 and 2011CB911104) and the Docterate stuff Foundation at the Guizhou Normal University.Notes and references
- A. Mitchell and Y. Pilpel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7271–7276 CrossRef CAS.
- A. Mitchell, G. H. Romano, B. Groisman, A. Yona, E. Dekel, M. Kupiec, O. Dahan and Y. Pilpel, Nature, 2009, 460, U220–U280 CrossRef.
- J. M. Buescher, W. Liebermeister, M. Jules, M. Uhr, J. Muntel, E. Botella, B. Hessling, R. J. Kleijn, L. Le Chat, F. Lecointe, U. Mader, P. Nicolas, S. Piersma, F. Rugheimer, D. Becher, P. Bessieres, E. Bidnenko, E. L. Denham, E. Dervyn, K. M. Devine, G. Doherty, S. Drulhe, L. Felicori, M. J. Fogg, A. Goelzer, A. Hansen, C. R. Harwood, M. Hecker, S. Hubner, C. Hultschig, H. Jarmer, E. Klipp, A. Leduc, P. Lewis, F. Molina, P. Noirot, S. Peres, N. Pigeonneau, S. Pohl, S. Rasmussen, B. Rinn, M. Schaffer, J. Schnidder, B. Schwikowski, J. M. Van Dijl, P. Veiga, S. Walsh, A. J. Wilkinson, J. Stelling, S. Aymerich and U. Sauer, Science, 2012, 335, 1099–1103 CrossRef CAS.
- U. Sauer, Mol. Syst. Biol., 2006, 2, 62 CrossRef.
- Y. J. Tang, J. S. Hwang, D. E. Wemmer and J. D. Keasling, Appl. Environ. Microbiol., 2007, 73, 718–729 CrossRef CAS.
- L. M. Blank and U. Sauer, Microbiology, 2004, 150, 1085–1093 CrossRef CAS.
- B. Rui, T. Shen, H. Zhou, J. P. Liu, J. S. Chen, X. S. Pan, H. Y. Liu, J. H. Wu, H. R. Zheng and Y. Y. Shi, BMC Syst. Biol., 2010, 4, 122 CrossRef.
- U. Sauer, D. R. Lasko, J. Fiaux, M. Hochuli, R. Glaser, T. Szyperski, K. Wuthrich and J. E. Bailey, J. Bacteriol., 1999, 181, 6679–6688 CAS.
- Y. Toya, K. Nakahigashi, M. Tomita and K. Shimizu, Mol. BioSyst., 2012, 8, 2593–2604 RSC.
- R. G. Allen and M. Tresini, Free Radical Biol. Med., 2000, 28, 463–499 CrossRef CAS.
- J. A. Imlay, Annu. Rev. Biochem., 2008, 77, 755–776 CrossRef CAS.
- G. Storz and J. A. Imlay, Curr. Opin. Microbiol., 1999, 2, 188–194 CrossRef CAS.
- J. L. Blanchard, W. Y. Wholey, E. M. Conlon and P. J. Pomposiello, PLoS One, 2007, 2, e1186 Search PubMed.
- P. J. Pomposiello, M. H. Bennik and B. Demple, J. Bacteriol., 2001, 183, 3890–3902 CrossRef CAS.
- J. T. Greenberg and B. Demple, J. Bacteriol., 1989, 171, 3933–3939 CAS.
- D. L. Rowley and R. E. Wolf, Jr., J. Bacteriol., 1991, 173, 968–977 CAS.
- P. R. Gardner and I. Fridovich, J. Biol. Chem., 1991, 266, 19328–19333 CAS.
- L. Tretter and V. Adam-Vizi, Philos. Trans. R. Soc., B, 2005, 360, 2335–2345 CrossRef CAS.
- J. T. Greenberg, P. Monach, J. H. Chou, P. D. Josephy and B. Demple, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 6181–6185 CrossRef CAS.
- M. J. Herrgard, B. S. Lee, V. Portnoy and B. O. Palsson, Genome Res., 2006, 16, 627–635 CrossRef CAS.
- J. Sainz, F. Pizarro, J. R. Perez-Correa and E. Agosin, Biotechnol. Bioeng., 2003, 81, 818–828 CrossRef CAS.
- M. W. Covert, E. M. Knight, J. L. Reed, M. J. Herrgard and B. O. Palsson, Nature, 2004, 429, 92–96 CrossRef CAS.
- S. A. Becker, A. M. Feist, M. L. Mo, G. Hannum, B. O. Palsson and M. J. Herrgard, Nat. Protoc., 2007, 2, 727–738 CrossRef CAS.
- A. M. Feist, C. S. Henry, J. L. Reed, M. Krummenacker, A. R. Joyce, P. D. Karp, L. J. Broadbelt, V. Hatzimanikatis and B. O. Palsson, Mol. Syst. Biol., 2007, 3, 121 CrossRef.
- R. Schuetz, L. Kuepfer and U. Sauer, Mol. Syst. Biol., 2007, 3, 119 CrossRef.
- A. L. Knorr, R. Jain and R. Srivastava, Bioinformatics, 2007, 23, 351–357 CrossRef CAS.
- D. Y. Lee, H. Yun, S. Park and S. Y. Lee, Bioinformatics, 2003, 19, 2144–2146 CrossRef CAS.
- R. Mahadevan and C. H. Schilling, Metab. Eng., 2003, 5, 264–276 CrossRef CAS.
- Y. G. Oh, D. Y. Lee, S. Y. Lee and S. Park, Biotechnol. Prog., 2009, 25, 999–1008 CrossRef CAS.
- D. Nagrath, M. Avila-Elchiver, F. Berthiaume, A. W. Tilles, A. Messac and M. L. Yarmush, Metab. Eng., 2010, 12, 429–445 CrossRef CAS.
- D. Nagrath, M. Avila-Elchiver, F. Berthiaume, A. W. Tilles, A. Messac and M. L. Yarmush, Ann. Biomed. Eng., 2007, 35, 863–885 CrossRef.
- J. L. Reed, T. D. Vo, C. H. Schilling and B. O. Palsson, Genome Biol., 2003, 4, R54 CrossRef.
- M. Emmerling, M. Dauner, A. Ponti, J. Fiaux, M. Hochuli, T. Szyperski, K. Wuthrich, J. E. Bailey and U. Sauer, J. Bacteriol., 2002, 184, 152–164 CrossRef CAS.
- T. Szyperski, Eur. J. Biochem., 1995, 232, 433–448 CrossRef CAS.
- C. Yang, Q. Hua, T. Baba, H. Mori and K. Shimizu, Biotechnol. Bioeng., 2003, 84, 129–144 CrossRef.
- P. Jouhten, E. Pitkanen, T. Pakula, M. Saloheimo, M. Penttila and H. Maaheimo, BMC Syst. Biol., 2009, 3, 104 CrossRef.
- W. van Winden, D. Schipper, P. Verheijen and J. Heijnen, Metab. Eng., 2001, 3, 322–343 CrossRef CAS.
- L. Peng, M. J. Arauzo-Bravo and K. Shimizu, FEMS Microbiol. Lett., 2004, 235, 17–23 CrossRef CAS.
- A. Sola, P. Jouhten, H. Maaheimo, F. Sanchez-Ferrando, T. Szyperski and P. Ferrer, Microbiology, 2007, 153, 281–290 CrossRef CAS.
- R. Caspi, H. Foerster, C. A. Fulcher, P. Kaipa, M. Krummenacker, M. Latendresse, S. Paley, S. Y. Rhee, A. G. Shearer, C. Tissier, T. C. Walk, P. Zhang and P. D. Karp, Nucleic Acids Res., 2008, 36, D623–D631 CrossRef CAS.
- B. Rui, T. Shen, H. Zhou, J. Liu, J. Chen, X. Pan, H. Liu, J. Wu, H. Zheng and Y. Shi, BMC Syst. Biol., 2010, 4, 122 CrossRef.
- Q. Hua, C. Yang, T. Baba, H. Mori and K. Shimizu, J. Bacteriol., 2003, 185, 7053–7067 CrossRef CAS.
- P. Jouhten, E. Rintala, A. Huuskonen, A. Tamminen, M. Toivari, M. Wiebe, L. Ruohonen, M. Penttila and H. Maaheimo, BMC Syst. Biol., 2008, 2, 60 CrossRef.
- N. S. Sharma, M. G. Ierapetritou and M. L. Yarmush, Biotechnol. Bioeng., 2005, 92, 321–335 CrossRef CAS.
- X. Chen, A. P. Alonso, D. K. Allen, J. L. Reed and Y. Shachar-Hill, Metab. Eng., 2011, 13, 38–48 CrossRef CAS.
- J. Schellenberger, R. Que, R. M. Fleming, I. Thiele, J. D. Orth, A. M. Feist, D. C. Zielinski, A. Bordbar, N. E. Lewis, S. Rahmanian, J. Kang, D. R. Hyduke and B. O. Palsson, Nat. Protoc., 2011, 6, 1290–1307 CrossRef CAS.
- J. M. Sandoval, F. A. Arenas and C. C. Vasquez, PLoS One, 2011, 6, e25573 CAS.
- S. Wang, K. Deng, S. Zaremba, X. Deng, C. Lin, Q. Wang, M. L. Tortorello and W. Zhang, Appl. Environ. Microbiol., 2009, 75, 6110–6123 CrossRef CAS.
- C. J. Baxter, H. Redestig, N. Schauer, D. Repsilber, K. R. Patil, J. Nielsen, J. Selbig, J. Liu, A. R. Fernie and L. J. Sweetlove, J. Plant Physiol., 2007, 143, 312–325 CAS.
- R. Singh, R. J. Mailloux, S. Puiseux-Dao and V. D. Appanna, J. Bacteriol., 2007, 189, 6665–6675 CrossRef CAS.
- S. B. Renwick, K. Snell and U. Baumann, Structure, 1998, 6, 1105–1116 CrossRef CAS.
- A. Aneiros-Guerrero, A. M. Lendinez, A. R. Palomares, B. Perez-Nevot, L. Aguado, A. Mayor-Olea, M. Ruiz-Galdon and A. Reyes-Engel, BMC Med. Genet., 2011, 12, 75 CrossRef CAS.
- S. V. Vijaya Lakshmi, S. M. Naushad, D. Seshagiri Rao and V. K. Kutala, Cell Biochem. Biophys., 2011, 13, 1–8 Search PubMed.
- K. Herbig, E. P. Chiang, L. R. Lee, J. Hills, B. Shane and P. J. Stover, J. Biol. Chem., 2002, 277, 38381–38389 CrossRef CAS.
- Y.-M. Jeon, S.-K. Park, S.-K. Rhee and M.-Y. Lee, Mol. Cell. Toxicol., 2010, 6, 419–425 CrossRef.
- J. Yang, S. C. Kalhan and R. W. Hanson, J. Biol. Chem., 2009, 284, 27025–27029 CrossRef CAS.
- J. Cheong, J. E. Coligan and J. D. Shuman, J. Biol. Chem., 1998, 273, 22714–22718 CrossRef CAS.
- Y. Ito, S. Oumi, T. Nagasawa and N. Nishizawa, Biosci., Biotechnol., Biochem., 2006, 70, 2191–2198 CrossRef CAS.
- X. L. D. M. Jiao and B. H. Ji, Biomed. Life Sci., 2005, 43, 501–508 Search PubMed.
- Y. Kai, H. Matsumura, T. Inoue, K. Terada, Y. Nagara, T. Yoshinaga, A. Kihara, K. Tsumura and K. Izui, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 823–828 CrossRef CAS.
- L. J. Sweetlove, J. L. Heazlewood, V. Herald, R. Holtzapffel, D. A. Day, C. J. Leaver and A. H. Millar, Plant J., 2002, 32, 891–904 CrossRef CAS.
- R. K. Dagda, S. J. Cherra, 3rd, S. M. Kulich, A. Tandon, D. Park and C. T. Chu, J. Biol. Chem., 2009, 284, 13843–13855 CrossRef CAS.
- I. Ślesak, Z. Miszalski, B. Karpinska, E. Niewiadomska, R. Ratajczak and S. Karpinski, Plant Physiol. Biochem., 2002, 40, 669–677 CrossRef.
- U. Sauer and B. J. Eikmanns, FEMS Microbiol. Rev., 2005, 29, 765–794 CrossRef CAS.
- M. Valdivia-Gonzalez, J. M. Perez-Donoso and C. C. Vasquez, Biometals, 2012, 25, 451–458 CrossRef CAS.
- F. Canonaco, T. A. Hess, S. Heri, T. Wang, T. Szyperski and U. Sauer, FEMS Microbiol. Lett., 2001, 204, 247–252 CrossRef CAS.
- U. Sauer, F. Canonaco, S. Heri, A. Perrenoud and E. Fischer, J. Biol. Chem., 2004, 279, 6613–6619 CrossRef CAS.
- M. Rühl, D. L. Coq, S. Aymerich and U. Sauer, J. Biol. Chem., 2012, 287, 27959–27970 CrossRef.
- R. Schuetz, N. Zamboni, M. Zampieri, M. Heinemann and U. Sauer, Science, 2012, 336, 601–604 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |