DOI:
10.1039/C2MB25389E
(Paper)
Mol. BioSyst., 2013,
9, 113-120
Identification of polyoxometalates as inhibitors of basic fibroblast growth factor†
Received
22nd May 2012
, Accepted 12th October 2012
First published on 19th October 2012
Abstract
Angiogenesis is the process of new blood vessel formation from pre-existing ones. Angiogenic factors contribute to neovascularization that takes place in angiogenesis-dependent diseases, including cancer. Inhibiting the activity of the angiogenic factors to block the angiogenesis pathways is the current strategy of cancer therapy. Basic fibroblast growth factor (bFGF) is regarded as one of the most important angiogenic factors. Herein, we selected polyoxometalates (POMs) with different structures to study the interactions between bFGF and POMs. The results show that POMs could bind to the protein with high affinity, causing detectable changes in conformation and biophysical properties of protein. In addition, POMs could effectively inhibit the cell proliferation induced by bFGF. Significantly, we found that the structure, size and composition of POMs play a key role in the interactions between bFGF and POMs. This study will be meaningful for future screening and design of polyoxometalate-based anticancer drugs.
1. Introduction
Angiogenesis, the development of new blood vessels from the pre-existing vasculature, plays a key role in various physiological and pathological conditions.1,2 Tumor angiogenesis is thought to occur mostly via sprouting angiogenesis. This is a process through which a single endothelial cell, called the tip cell, is selected from the vasculature, overcoming its quiescent environment, and forming a new vessel.3,4 Inhibition of angiogenesis has been shown to significantly suppress certain disease processes including tumor growth.5 Basic fibroblast growth factor (basic FGF, FGF-2, bFGF) is one of the most important growth factors that induce angiogenesis.6–8 It belongs to a large FGF family.9–11 The three-dimensional structure of the human bFGF has been elucidated by means of X-ray crystallographic spectroscopy.12–15 bFGF can be divided into three submotifs each consisting of four antiparallel β-strands.16 Heparan sulfate proteoglycans (HSPGs) modulate bFGF activity by low affinity interaction with specific bFGF and fibroblast growth factor receptors (FGFRs) binding sites, facilitating bFGF binding to FGFR.17,18 Ligands that recognize and bind to bFGF may disrupt its interaction with HSPGs and FGFRs, and would represent a new class of potential therapeutic agents.19–23 Heparin fragments,24 suramin,25,26 pentosan polysulfate (PPS),27 sulfonated derivatives of distamycin A,19,20 nonsulfated polyanionic aromatic compounds,28 and porphyrins23 have been tested for their anti-FGF and anti-angiogenic potential. However, these inhibitors are always difficult to synthesize, which makes them very expensive and thus may limit their widespread application. Besides, very large doses of these compounds are required to show activity, and their efficacy is limited by anticoagulant side effects.29 Therefore, it is necessary to look for novel agents with more potent antiangiogenic activity and lower toxicity.
Polyoxometalates (POMs) are early transition metal oxygen anion clusters. They have been reported for their promising antiviral, antitumoral and antibacterial activities for years.30 Their properties of versatility, simple synthesis, easy modification, and cheapness make them attractive in pharmaceuticals markets.31 In recent years, POMs were investigated based on cell-culture assay, enzymatic-activities in vitro, and molecular modeling studies. As demonstrated by Hill, POMs are minimally toxic, and selectively inhibit purified HIV-1 protease.32 Prudent et al. reported that POMs are powerful protein kinase CK2 inhibitors.33 Studies of interactions between POMs and human serum albumin (HSA) are one of the necessary steps toward an understanding of POMs' biological activity.34–36 In addition, we conducted primary and exploratory studies and reported that some POMs bind to bFGF and amyloid β-peptides.37,38 Among those compounds used, the Keggin, Keggin sandwich, Wells–Dawson and Wells–Dawson sandwich type POMs seem to have a higher biological activity and a lower toxicity, holding considerable promise as chemotherapeutic agents.39 While the inhibition of POMs has been well documented, the primary mechanism at a molecular level for the antiviral and antitumoral actions has remained elusive. More detailed experiments are required to better correlate the inhibitory effect of POMs to structural or compositional features. In this paper, we selected POMs with different structures for the further study. Our aim is to evaluate whether POMs could interact with angiogenic factor bFGF in a specific manner and affect the biophysical properties and cell proliferation activity of bFGF. For that, the interaction in vitro of selected POMs with bFGF was studied and began with an attempt to prove that their activity was directly related to their bFGF-binding properties.
2. Results and discussion
2.1 Characterization of polyoxometalates
POMs with different structures were selected for the present study, including Keggin, Wells–Dawson, and Anderson structure, whose basic anion backbones are illustrated in Fig. 1. Keggin type are a kind of classical polyoxoanions that have formula [XM12O40]n−, where X is a heteroatom and M represents Mo or W in most cases. The lacunary Keggin structure [XM11O39]n− and trivacant Keggin structure [XM9O34]n− are derived from [XM12O40]n− by removal of one or three of the twelve M atoms. The five Keggin structure POMs studied herein included La2K[PTi2W10O40], K7[PTi2W10O40], La2K4[GeTi3W9O40], K8[β-SiW11O39], and α-Na9H[SiW9O34]. A Wells–Dawson structure (NH4)6[P2W18O62] was used for comparison. Fourier-Transformed Infrared (FTIR) spectra show the characteristic peaks of POMs used (Fig. S1, ESI†). For example, the peaks of K7[PTi2W10O40] at 1084, 1064 and 1045 cm−1 are assigned to νas(P–Oa), and the peaks at 953, 875 and 788 cm−1 are assigned to νas(W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Od), νas(W–Ob–W) and νas(W–Oc–W), respectively, (where Oa is the oxygen of the central tetrahedral, Ob is the bridged oxygen of two octahedra sharing a corner, Oc is the bridged oxygen sharing an edge, and Od is the terminal oxygen). The stability of POMs in dilute aqueous solution is one issue that should be addressed. All POMs investigated showed typical bands between 200 and 300 nm. After degradation of the cluster structure these bands disappear. Thus, these peaks were chosen to study the hydrolytic stability of POMs using UV/Vis spectroscopy.34,35,38,40 As shown in Fig. S2 (ESI†), the POMs tested remained intact under the conditions used in our experiments. Therefore, they are suitably used as potential chemotherapeutic agents.
Od), νas(W–Ob–W) and νas(W–Oc–W), respectively, (where Oa is the oxygen of the central tetrahedral, Ob is the bridged oxygen of two octahedra sharing a corner, Oc is the bridged oxygen sharing an edge, and Od is the terminal oxygen). The stability of POMs in dilute aqueous solution is one issue that should be addressed. All POMs investigated showed typical bands between 200 and 300 nm. After degradation of the cluster structure these bands disappear. Thus, these peaks were chosen to study the hydrolytic stability of POMs using UV/Vis spectroscopy.34,35,38,40 As shown in Fig. S2 (ESI†), the POMs tested remained intact under the conditions used in our experiments. Therefore, they are suitably used as potential chemotherapeutic agents.
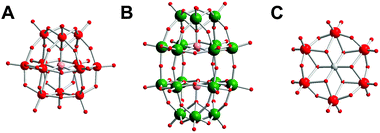 |
| | Fig. 1 Chemical structures of POMs. (A) Keggin structure; (B) Wells–Dawson structure; (C) Anderson structure. | |
2.2 Effects of POMs on bFGF
Understanding of interactions between ligands and biomolecules requires a complete characterization of thermodynamic data with the structural properties of the interacting molecules. The interaction between bFGF and POMs was first demonstrated by fluorescence quenching measurement. The fluorescence of native bFGF is dominated by tyrosines, since that of tryptophan is quenched by proximate groups. So bFGF was excited at 280 nm and emission was monitored at 305 nm. As shown in Fig. S3A and B (ESI†), the fluorescence intensity of bFGF was strongly quenched with an increase in the amount of POMs. The titration reached an end point at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for all POMs used. The apparent dissociation constants (Kd) for bFGF and POMs were yielded by using nonlinear least-squares fit of the data,19 as summarized in Table 1. Such Kd values were similar to that for PNU145156E, within the same order of magnitude.19 By comparison to other published bFGF inhibitors, we concluded that POM was one of the high-affinity bFGF binders. Also, we found the quenching efficiency in fluorescence titration decreased upon increase of ionic strength by NaCl addition (Fig. S3C, ESI†). It was assumed that the interaction between bFGF and POMs depended on the electrostatic effect. Besides, we found the structure and size of POMs play a more important role. The Keggin structure is 11.5 Å in diameter, and the Wells–Dawson structure is 11.5 × 15 Å.37 The Wells–Dawson structure showed higher affinity, although it contains the least number of charges compared to the Keggin compounds. The interaction of POMs with bFGF was also evident from the resonance light scattering (RLS) experiment. RLS is a simple and sensitive technique applied to studies of assembly phenomena and the identification of macromolecules.41 The RLS signals of bFGF and POMs were weak. Addition of POMs to bFGF enhanced the RLS spectra significantly (inset of Fig. S3, ESI†), which obviously resulted from the interaction between bFGF and POMs. At a molar ratio of 1:1 for POMs, the titration reached the end point, permitting a direct measurement of the interaction stoichiometry. The value was consistent with fluorescence quenching measurement.
:1 for all POMs used. The apparent dissociation constants (Kd) for bFGF and POMs were yielded by using nonlinear least-squares fit of the data,19 as summarized in Table 1. Such Kd values were similar to that for PNU145156E, within the same order of magnitude.19 By comparison to other published bFGF inhibitors, we concluded that POM was one of the high-affinity bFGF binders. Also, we found the quenching efficiency in fluorescence titration decreased upon increase of ionic strength by NaCl addition (Fig. S3C, ESI†). It was assumed that the interaction between bFGF and POMs depended on the electrostatic effect. Besides, we found the structure and size of POMs play a more important role. The Keggin structure is 11.5 Å in diameter, and the Wells–Dawson structure is 11.5 × 15 Å.37 The Wells–Dawson structure showed higher affinity, although it contains the least number of charges compared to the Keggin compounds. The interaction of POMs with bFGF was also evident from the resonance light scattering (RLS) experiment. RLS is a simple and sensitive technique applied to studies of assembly phenomena and the identification of macromolecules.41 The RLS signals of bFGF and POMs were weak. Addition of POMs to bFGF enhanced the RLS spectra significantly (inset of Fig. S3, ESI†), which obviously resulted from the interaction between bFGF and POMs. At a molar ratio of 1:1 for POMs, the titration reached the end point, permitting a direct measurement of the interaction stoichiometry. The value was consistent with fluorescence quenching measurement.
Table 1 Equilibrium dissociation constants Kd of bFGF and POMs
|
|
K
d
(M) |
Er (M) |
Charges |
Structure |
|
K
d
values were calculated from eqn (1).
|
| α-Na9H[SiW9O34] |
3.57 × 10−7 |
4.5 × 10−8 |
10 |
Trivacant Keggin |
| K8[β-SiW11O39] |
6.17 × 10−7 |
8.1 × 10−8 |
8 |
Lacunary Keggin |
| La2K[PTi2W10O40] |
1.73 × 10−7 |
3.4 × 10−8 |
7 |
Keggin |
| K7[PTi2W10O40] |
1.08 × 10−7 |
1.9 × 10−8 |
7 |
Keggin |
| La2K4[GeTi3W9O40] |
3.52 × 10−8 |
1.9 × 10−8 |
10 |
Keggin |
| (NH4)6[P2W18O62] |
9.35 × 10−8 |
2.2 × 10−8 |
6 |
Wells–Dawson |
| K8[P2CoW17O61] |
1.02 × 10−7 |
3.08 × 10−8 |
8 |
Wells–Dawson |
| K8[P2NiW17O61] |
8.88 × 10−8 |
1.20 × 10−8 |
8 |
Wells–Dawson |
We have determined the binding affinity and stoichiometry of the complexes using fluorescence spectroscopy. Subsequently, the thermodynamics and equilibrium of interaction were sought. Fig. 2 depicts the thermal denaturation of bFGF in the presence of POMs by employing a UV-melting method. Under the ionic-strength conditions described herein, bFGF alone showed a cooperative thermal unfolding with a mid-point denaturation temperature (Tm) at 60 °C. The increase of Tm in the presence of POMs was observed, suggesting that thermodynamic stability of bFGF was enhanced upon binding to POMs relative to the free protein. Moreover, the enhancement of thermal stability was dose-dependent. All of these POMs stabilized bFGF by approximately 10 °C when excess POMs were added. Among them, the degree of Wells–Dawson structure-based stabilization is higher than that of the Keggin structure. Temperature-dependent CD was also used to investigate the influence of POMs on thermal stability of bFGF secondary structure, and the similar results were obtained (Fig. S4, ESI†). The thermal denaturation experiments supported the binding studies discussed above and demonstrated the effects of the different POM structures.
![Thermal denaturation experiments. (A) UV-melting curves of bFGF (2 μM) with K8[β-SiW11O39]. [POM] = 0, 2.0, 4.0, 8.0, 12.0, 16.0 μM. (B) Effects of POMs with various structures on the thermal stability of bFGF.](/image/article/2013/MB/c2mb25389e/c2mb25389e-f2.gif) |
| | Fig. 2 Thermal denaturation experiments. (A) UV-melting curves of bFGF (2 μM) with K8[β-SiW11O39]. [POM] = 0, 2.0, 4.0, 8.0, 12.0, 16.0 μM. (B) Effects of POMs with various structures on the thermal stability of bFGF. | |
The results that binding of POMs stabilized bFGF are similar to the increase in stability of bFGF upon binding heparin. Binding of heparin induces conformational changes of bFGF. In order to gain an insight into the conformational changes occurring in bFGF upon binding POMs, far-UV CD spectroscopy was performed. bFGF is a β-sheet-rich protein,42 whose typical CD spectrum exhibits a weak positive band at 227–230 nm, and an intense negative band at 210–205 nm. Upon addition of POMs, the band intensities at 204 and 229 nm decreased, indicating a change in bFGF secondary structure (Fig. 3). Moreover, the Wells–Dawson-type POM exerted a more significant effect than Keggin-type POMs. The results further supported the fluorescence and thermal unfolding experiments, indicating that the size effect is significant for such an interaction.
![CD spectra of bFGF in the presence of POMs: (A) α-Na9H[SiW9O34], and (B) (NH4)6[P2W18O62]. [bFGF] = 5 μM, [POM] = 0, 2.5, 5, 10, 20 μM.](/image/article/2013/MB/c2mb25389e/c2mb25389e-f3.gif) |
| | Fig. 3 CD spectra of bFGF in the presence of POMs: (A) α-Na9H[SiW9O34], and (B) (NH4)6[P2W18O62]. [bFGF] = 5 μM, [POM] = 0, 2.5, 5, 10, 20 μM. | |
Although the thermal unfolding of bFGF had been examined, the thermodynamic parameters of the unfolding process were difficult to obtain from thermal denaturation experiments due to irreversible aggregation occurred at high temperatures after partial unfolding. Urea-induced unfolding was therefore employed to monitor the reversible unfolding of bFGF. The fluorescence of the single Trp in native bFGF is quenched, and the spectrum is dominated by multiple Tyr emission at 305 nm. As bFGF unfolds, the change of microenvironment causes an increase in fluorescence at 350 nm (Fig. S5, ESI†). Thus, the ratio of the 350 nm to the 305 nm emission could be reliably used to monitor the conformational transitions during the unfolding of bFGF. Fig. 4A shows the equilibrium unfolding curves of bFGF, indicating that the urea-induced unfolding of bFGF was a two-state process. Unliganded bFGF unfolded at a Cm of 1.7 M (the concentration of urea at the midpoint of the denaturant-induced transition from native to unfolded protein). Upon addition of POMs at a molar ratio of 1:1, the unfolding transition of bFGF shifted to notably higher urea concentrations. The results suggested that POMs protected bFGF from urea-induced unfolding. The stabilization of bFGF occurs presumably by shifting the equilibrium between folded and unfolded protein by the preferential binding of POMs to the native protein. It was noteworthy that the Cm was observed at 5.73 M in the presence of Wells–Dawson structure, which is much bigger than that in the presence of Keggin structures (Table 2). The larger shift in the transition midpoint suggested that the size of POMs is an important parameter in the urea-induced unfolding process.
![Urea-induced denaturation experiments. (A) Fraction of unfolded species at various concentrations of urea, as monitored by the changes in the ratio of 350/305 nm fluorescence. [bFGF] = 1 μM, [POMs] = 1 μM. (B) The curve of free energy of unfolding. bFGF(■), bFGF in the presence of α-Na9H[SiW9O34](●), K8[β-SiW11O39](▲), La2K[PTi2W10O40](▼), K7[PTi2W10O40](◆) and (NH4)6[P2W18O62](★).](/image/article/2013/MB/c2mb25389e/c2mb25389e-f4.gif) |
| | Fig. 4 Urea-induced denaturation experiments. (A) Fraction of unfolded species at various concentrations of urea, as monitored by the changes in the ratio of 350/305 nm fluorescence. [bFGF] = 1 μM, [POMs] = 1 μM. (B) The curve of free energy of unfolding. bFGF(■), bFGF in the presence of α-Na9H[SiW9O34](●), K8[β-SiW11O39](▲), La2K[PTi2W10O40](▼), K7[PTi2W10O40](◆) and (NH4)6[P2W18O62](★). | |
Table 2 Thermodynamic parameters for bFGF determined by urea-induced denaturationa
|
|
C
m
(M) |
ΔG0unf (kJ mol−1) |
m (kJ mol−1 M−1) |
|
ΔG0unf and m values were calculated from eqn (2)–(4).
|
| bFGF |
1.71 ± 0.02 |
10.9 ± 1.1 |
7.2 ± 0.7 |
| α-Na9H[SiW9O34] |
3.08 ± 0.02 |
16.4 ± 0.7 |
5.4 ± 0.2 |
| K8[β-SiW11O39] |
3.39 ± 0.05 |
16.5 ± 1.5 |
4.4 ± 0.4 |
| La2K[PTi2W10O40] |
2.73 ± 0.10 |
13.7 ± 1.5 |
2.0 ± 0.2 |
| K7[PTi2W10O40] |
2.91 ± 0.07 |
11.5 ± 2.7 |
3.2 ± 0.6 |
| (NH4)6[P2W18O62] |
5.73 ± 0.21 |
13.8 ± 0.6 |
2.5 ± 0.1 |
| K8[P2CoW17O61] |
5.16 ± 0.14 |
14.6 ± 0.7 |
2.7 ± 0.2 |
| K8[P2NiW17O61] |
5.30 ± 0.15 |
14.2 ± 0.7 |
2.6 ± 0.1 |
By assuming simple two-state behavior, the equilibrium denaturation profiles were analyzed by linear extrapolation method to obtain the thermodynamic parameters (Fig. 4B, Table 2). This analysis found the apparent free energy of unfolding ΔG0unf of 10.9 ± 1.1 kJ mol−1 for the unliganded protein. The increases of free energy ΔG0unf implied stabilization of the native bFGF upon binding to POMs. The m value reflects the amount of newly exposed surface area upon denaturation of proteins. Hence, the m value is a useful parameter to assess the cooperativity of a protein folding/unfolding reaction.43,44 The m values estimated for the urea-induced unfolding of bFGF was 7.2 ± 0.7 kJ mol−1 M−1. In the presence of POMs, the m values decreased significantly, indicating the unfolding of bFGF in the absence of POMs was more cooperative. It represented the less sensitivity of the protein in the presence of POMs to the chemically induced denaturation. Another approach to characterize the interaction was to examine the rate and extent of denaturant-induced unfolding of bFGF by fluorescence spectroscopy. As shown in Fig. S6 (ESI†), bFGF alone unfolded rapidly in the presence of 4 M urea; however, addition of POMs to bFGF decreased the rate and extent of unfolding. Moreover, the decrease in the presence of Wells–Dawson structure was smaller than that in the presence of Keggin structure. These results corroborated the other data obtained in this study.
To gain a better understanding of the interaction, we used limited protease-digestion analysis to probe the possible structural changes of bFGF induced by POMs. Trypsin generally cleaves proteins at the C-terminal ends of basic amino acids such as lysine and arginine residues. As bFGF is rich in lysine and arginine residues, trypsin is an apt choice. Time-dependent trypsin digestion of bFGF was monitored by SDS-PAGE analysis (Fig. S7A and B, ESI†). The undigested bFGF yielded a band on the gel that corresponds to a molecular mass of 18 kDa. The intensity of this band was used as an indicator of the degree of susceptibility of bFGF to trypsin action. It could be observed that the intensity of the band decreased with the increase of incubation time with trypsin. However, in the presence of POMs, more than 50% of the band remained undigested even after 2 hours. Dose-dependent digestion experiments revealed that, with the increase of POMs, protection of the protein from trypsin cleavage was enhanced (Fig. 5). MALDI-TOF mass spectra also confirmed a very different susceptibility to degradation (Fig. S7C, ESI†). The trypsin hydrolyzed bFGF extensively in the absence of POM, and many cleavage products were observed at a mass range from 1500 to 5500 Da. Upon the addition of POM, the fragments were altered significantly, and some of the fragments were missing, suggesting a part of the lysine and arginine residues were protected and could not be cleaved by trypsin (Table S1, ESI†). The enhanced resistance to trypsin cleavage could be due to masking of some of the potential enzyme cleavage sites in bFGF by POMs. The hampering of interaction between bFGF and trypsin was hypothesized to be related to a possible interaction of positively charged lysine and arginine residues with negatively charged POMs. Indeed, more than 20 basic residues are exposed on the surface of bFGF,13 including Lys 34, 127, 133, 137, 143, and Arg 52, 128 (Fig. S8, ESI,† full-length bFGF of 154 amino acids).12 This basic cluster, is a preferential site susceptible to trypsin cleavage,45 and corresponds very likely to the POMs-binding site. In the absence of POMs, cleavage of this site caused the protein to unfold, and the whole bFGF molecule was rapidly degraded. Upon binding to POMs, the primary site was not accessible to the enzyme and the trypsin digestion of the whole bFGF molecule was prevented. It may be argued that the difference observed in the digestion pattern could be due to the altered cleavage specificity of the enzyme in the presence of POMs. This contention could be discounted because control experiment using HSA in the presence of POMs revealed that POMs did not affect the activity of trypsin (Fig. S9, ESI†). In addition, POMs often induce protein aggregation through multivalent electrostatic interaction. Non-reducing PAGE experiments showed that increased resistance of the protein to proteolytic cleavage was not due to bFGF oligomerization induced upon POMs binding (Fig. S10, ESI†).
![Trypsin digestion of bFGF monitored by SDS-PAGE. (A) Lines 3–6: α-Na9H[SiW9O34]; lines 7–10: K8[β-SiW11O39]. (B) Lines 3–6: La2K[PTi2W10O40]; lines 7–10: K7[PTi2W10O40].](/image/article/2013/MB/c2mb25389e/c2mb25389e-f5.gif) |
| | Fig. 5 Trypsin digestion of bFGF monitored by SDS-PAGE. (A) Lines 3–6: α-Na9H[SiW9O34]; lines 7–10: K8[β-SiW11O39]. (B) Lines 3–6: La2K[PTi2W10O40]; lines 7–10: K7[PTi2W10O40]. | |
Detection of the cryptic structural propensities in the amino acid sequence is expected to provide useful insight into the problem of protein folding. Trifluorethanol (TFE) could disrupt the long-range interaction in the three-dimensional structure of the protein, leading to the formation of helix. Despite being an all-β protein, some segments of bFGF have a tendency to form α-helix in solution. As shown in Fig. S11 (ESI†), addition of TFE induced a change in the far-UV CD spectrum of the protein giving rise to a prominent α-helical shape, with two characteristic minima centered at 208 and 222 nm. The helical content of 4 μM bFGF in 40% (v/v) of TFE was 32.3% (an estimate of the α-helical content may be made from the mean residue ellipticity at 222 nm, fraction α-helix = −([θ] + 3000)/33000).46 Binding of POMs protected bFGF against TFE-induced denaturation. The helical content of same concentration of bFGF in the presence of POM K8[β-SiW11O39] (1:1) in TFE was 18.4%. POMs stabilized the structure of bFGF, so that a higher amount of TFE was needed to unfold the protein and induce the formation of α-helical. Binding of heparin also protected bFGF against TFE induced denaturation. The binding site of heparin is located in the turn/β11/turn region of bFGF,16 through a network of van der Waals, electrostatic, and hydrogen bands. TFE induces helix in specific regions of protein.47 Thus POMs may locate in the same site, and have the similar effect of heparin in the presence of TFE.48
2.3 Identification of the specific binding site
The identification of POMs binding site on bFGF is essential for the understanding of their biological function. Competition assays were performed to verify the putative binding site. Suramin, a prototype growth factor antagonist, could bind at or near the heparin binding site of bFGF and disrupt the interaction between bFGF and FGFR. It had weak fluorescence emission near 400 nm, as appeared in Fig. 6. Upon addition of bFGF, the fluorescence was significantly enhanced.49 However, the addition of POM to the bFGF–suramin complex resulted in a progressive decrease of the fluorescence intensity, suggesting POM displaced suramin from bFGF. Moreover, 50% of displacement occurred when the concentration of POM was approximately the same as that of suramin. This observation implied that both suramin and POMs competed for the same binding site, sharing at least one binding site. Another competition assay was to replace POMs from bFGF with heparin monitored by RLS (Fig. S12, ESI†). bFGF has a high affinity for heparin, a naturally occurring highly sulfated glycosaminoglycan. Heparin had little effect on RLS spectrum of bFGF, and POMs enhanced the RLS signal of bFGF significantly. Addition of heparin to the mixture of bFGF and POMs resulted in a visible decrease of their RLS spectrum. This result suggested that POMs bound to the heparin binding site, and the heparin kicked POMs out from bFGF due to its high affinity with bFGF. It is known that the binding site for anionic heparin is rich in positively charged lysine and arginine residues. As shown in Fig. S13 (ESI†), Asn35, Arg128, Lys133, Gln142, Lys34, Asn109 and Lys143 are potential contributors to heparin binding.17 POMs may bind to the same region of bFGF surface owing to their charged nature and bulky size. Besides, direct thiol titration studies using DTNB (5,5′-dithiobis(2-nitrobenzoic acid)) reagent were performed. Among the four cysteine residues in bFGF, only Cys78 and Cys96, which are located on opposing surfaces of bFGF, can be modified by thiolation or carboxy-methylation.32 The titration result revealed two free SH groups in the absence of POMs, and one free SH group in the presence of POMs. It indicated that POMs might bind near Cys96, which was located in the vicinity of the heparin binding site, and subsequently prevent bFGF from the thiol-mediated disulfide exchange reaction with the DTNB reagent.
![Effect of POM K8[β-SiW11O39] on the fluorescence of suramin–bFGF complex. [suramin] = 5 μM, [bFGF] = 3 μM. The addition of POMs produced the decrease in suramin–bFGF fluorescence. Inset shows the change of suramin–bFGF fluorescence intensity at 413 nm with the increase of POM's concentration.](/image/article/2013/MB/c2mb25389e/c2mb25389e-f6.gif) |
| | Fig. 6 Effect of POM K8[β-SiW11O39] on the fluorescence of suramin–bFGF complex. [suramin] = 5 μM, [bFGF] = 3 μM. The addition of POMs produced the decrease in suramin–bFGF fluorescence. Inset shows the change of suramin–bFGF fluorescence intensity at 413 nm with the increase of POM's concentration. | |
To the best of our knowledge, the crystal structure of POM–bFGF complex has not been published yet, therefore we lack a precise assignment of the binding hooks of drug on bFGF. Nevertheless, the data all together strongly suggested that the heparin site is probably the binding hook of POMs. The bFGF–heparin complex is essential for bFGF binding to its receptors, a prerequisite for the activation of the angiogenic process. POMs that recognize and bind to bFGF may disrupt the interactions between bFGF and its receptors, and block the angiogenic pathway, which renders POMs potentially efficient antiangiogenic agents.
2.4 Parameters for the interactions
Because of the distinct chemical structures, the affinities of heparin and POMs to bFGF were different. It was reported that decreasing the charges of heparin diminished its ability to stabilize aFGF (acidic fibroblast growth factor) against urea denaturation and decreasing the size did not appear to affect its ability to stabilize aFGF.50 Unlike the flexible organic polysulfonated polyanionic heparin-based compounds, POMs are fairly rigid, cage-like structures coated with oxygen atoms bearing partial negative charges. In contrast to the trend of heparin, our finding suggested that the structure of POMs plays more important role in the interaction with bFGF. The Wells–Dawson structure POM (NH4)6[P2W18O62] with less charges and bigger size exhibited stronger effect on bFGF demonstrated by fluorescence titration experiment, CD spectroscopy, thermal unfolding, and urea-induced unfolding studies. Besides (NH4)6[P2W18O62], two other Wells–Dawson structure POMs K8[P2CoW17O61] and K8[P2NiW17O61] were tested and similar results were obtained (Tables 1 and 2, and Fig. S14, ESI†). These results are consistent with our previous study,37 demonstrating that the conclusion is not drawn from a single example. The present data may be explained tentatively. Our results suggest that the heparin site is probably the binding site of POMs, whose width is measured to be more than 1 nm (Fig. S13, ESI†). The Wells–Dawson structure POM with bigger size would more fit to the site, thus protecting bFGF from thermal, urea, and protease more easily. The smaller Keggin structure POMs could not cover the site completely, and show weaker effects. Due to the complexity, in-depth studies of the detailed mechanism are currently underway in our laboratory.
It is reported previously that POMs containing elements W and Mo exhibited different antiviral activity and antitumor activity in cell culture, although the primary mechanisms have remained elusive.31 Therefore, we felt that the influence of metal atom in the POMs needs to be taken into consideration. The five Keggin structure POMs and a Wells–Dawson structure POM containing W atom affected the stability of bFGF. An Anderson structure POM Na5IMo6O24 and a Keggin structure POM H3PMo12O40 containing Mo atom had little effect on the stability of bFGF monitored by thermal denaturation and proteolytic digestion method. As shown in Fig. S15A (ESI†), the Tm value of bFGF in the presence of the two POMs containing Mo atom is similar to that of the protein alone. Meanwhile, Fig. S15B (ESI†) showed that trypsin could digest bFGF in the presence of the two POMs in 3 hours. Although these results we obtained revealed that the ability of POMs to stabilize bFGF might relate to POMs' composition, the correlation was not a strong one, with only few investigated cases and the small size of the Anderson structure. However, the study presented here contributed to a better understanding of the functioning of POMs, which will allow for the design of new drugs.
2.5 Inhibition of the cell proliferation induced by bFGF
The size, charge, and composition of POMs may all be interrelated in the inhibition of viral absorption/fusion and viral polymerase activity in a cell- or kinetics-based assay.51,52 Previous data indicated that POM structure mainly determines its inhibitory effect on protein kinase CK2.33 Small Keggin structures are inactive, larger Dawson compounds are moderately active, and the largest and most charged Pressler ions are the best subgroup of inhibitors. The biological activity of bFGF is mediated by interaction with high affinity receptors and HSPG on the cell surface. Many studies have suggested that the binding to HSPG facilitates bFGF receptor binding and activation.17,18 In order to demonstrate POMs could regulate the biological activity of bFGF, HUVEC, the most commonly used human endothelial cell for in vitro angiogenesis assay, were chosen as a model to further determine the effect of POMs on the mitogenic activity of bFGF using the MTT method. HUVEC were treated with bFGF that was first preincubated with POMs overnight at 4 °C. The assay results were normalized by using the results for cells untreated with bFGF as a control. As shown in Fig. 7A and Fig. S16, (ESI†) bFGF induced the proliferation of HUVEC by about 50% at least. However, POMs inhibited bFGF-induced proliferation of HUVEC in a dose-dependent manner. In addition, compared with the Keggin structure, the Wells–Dawson structure POM exhibited a higher inhibitory ability (Fig. S17, ESI†). The MTT assay suggested POMs could bind to the heparin binding site to block the binding of bFGF to cell surface receptors, interfere with its activation in signal transduction pathways, and then abrogate the bFGF-induced proliferation of HUVEC. It also consisted with the above result that the size of POMs played a key role in the interactions. Meanwhile, MTT assay was carried out to test the toxicity of POMs on the cells. No significant cytotoxicity for POMs was observed for 48 h co-incubation at the largest concentration used in inhibition assay (Fig. 7B). Such a low in vitro toxicity offers an essential premise for the desired high biocompatibility in vivo.
![MTT assay for inhibition of POMs on the HUVEC proliferation induced by bFGF and toxicity of POMs. (A) Effects of POMs (1) (NH4)6[P2W18O62] and (2) La2K[PTi2W10O40] on bFGF activity. Results are expressed as percentage of the control, and the data are expressed as the mean ± SD values (n = 5). Statistic results of ANOVA: #, p < 0.001 vs. the control group; *, p < 0.05, and ***, p < 0.001 vs. the bFGF stimulated group. (B) Toxicity of POMs (1) α-Na9H[SiW9O34], (2) K8[β-SiW11O39], (3) La2K[PTi2W10O40], (4) K7[PTi2W10O40], (5) La2K4[GeTi3W9O40], and (6) (NH4)6[P2W18O62] on the HUVEC.](/image/article/2013/MB/c2mb25389e/c2mb25389e-f7.gif) |
| | Fig. 7 MTT assay for inhibition of POMs on the HUVEC proliferation induced by bFGF and toxicity of POMs. (A) Effects of POMs (1) (NH4)6[P2W18O62] and (2) La2K[PTi2W10O40] on bFGF activity. Results are expressed as percentage of the control, and the data are expressed as the mean ± SD values (n = 5). Statistic results of ANOVA: #, p < 0.001 vs. the control group; *, p < 0.05, and ***, p < 0.001 vs. the bFGF stimulated group. (B) Toxicity of POMs (1) α-Na9H[SiW9O34], (2) K8[β-SiW11O39], (3) La2K[PTi2W10O40], (4) K7[PTi2W10O40], (5) La2K4[GeTi3W9O40], and (6) (NH4)6[P2W18O62] on the HUVEC. | |
DNA synthesis, an alternative measure of cell proliferation, was assayed. Analysis of DNA content was carried out by propidium iodide staining and assessing the amount of bound dye using flow cytometry. As shown in Fig. S18, (ESI†) mitogenic effects of bFGF on HUVEC could be blocked by POMs in a dose-dependent manner and the inhibition of cell proliferation in the presence of POMs was not due to toxicity. To further explore the biological effects of POMs on bFGF, a set of assays containing migration of cells, tube formation in vitro, and chicken chorioallantoic membrane assay (CAM) is necessary and is underway.
3. Conclusion
The design and screening of synthetic molecules that bind to angiogenic factors and block the angiogenesis pathways remain a major challenge in modern medicinal chemistry. Herein, the interactions of POMs with bFGF were investigated, showing that POMs could bind to the protein tightly at a molar ratio of 1:1, and cause detectable structure change in protein. The protein was dramatically stabilized against thermal- and denaturant-induced unfolding, and trypsin digestion. The structure, size and composition of POMs play a key role in the interactions. Besides, our results suggested that the heparin-binding site was probably the binding site of POMs. Importantly, the MTT assay indicated that POMs could inhibit bFGF-induced HUVEC proliferation and POMs themselves were nontoxic. The exact mechanism results most likely from the inhibition of the interactions between bFGF and its receptors by POMs, thus blocking the biological responses. A general goal at present is to obtain greater knowledge regarding the relationships between the physical and electronic structure of a POM and its behavior with bFGF. The study in depth for the interesting results is proceeding.
Despite numerous POMs were prepared and evaluated, the application of POMs in medicine is still in its infancy. Structure–activity information will facilitate the design and synthesis of POMs with more potent activity and lower toxicity by altering the properties. Moreover, encapsulation techniques have been developed for POMs to increase the efficacy of the delivery of the POMs to the target cell.53 Therefore, the POMs might be the highly attractive candidates for less expensive, easily administrable and more common angiogenesis inhibitors in the future.
4. Experimental section
4.1 Materials
Human bFGF was provided by Bio-Engineering Institute of Jinan University. bFGF samples were prepared in PBS buffer (20 mM sodium phosphate, 150 mM NaCl, pH 7.4). POMs were provided by Key Laboratory of Polyoxometalate Science of the Ministry of Education (Northeast Normal University). Heparin, suramin, and urea were purchased from BBI, Sigma, and Amersham Pharmacia Biotech AB, respectively. Solutions were all prepared in ultra pure water purified through a Milli-Q system. Human umbilical vascular endothelia cells (HUVEC) were obtained from Jilin University.
4.2 Steady-state fluorescence spectroscopy
Fluorescence spectra were obtained with a JASCO FP6500 spectrophotometer using an excitation wavelength of 280 nm. Fluorescence titration was used to determine the equilibrium dissociation constants (Kd) of the bFGF–POM complexes:| | | F0 = FI + ΔF{([P]t + Kd + [L]t) − [([P]t + Kd + [L]t)2 − 4[P]t[L]t]1/2}/(2[P]t) | (1) |
where F0 and FI are the observed and the initial fluorescence intensity, ΔF is the final change in fluorescence intensity, [P]t and [L]t are the total concentration of bFGF and the ligand, respectively.
4.3 Thermal denaturation experiments
Thermal curve of bFGF were determined using a JASCO V-550 spectrophotometer. bFGF and POMs were preincubated at room temperature for 1 h. Then, samples were heated at a gradient of 1 °C min−1 from 20 to 95 °C, while continuously monitoring the absorption at 280 nm.
4.4 Far-UV circular dichroism (CD)
The influence of POMs on protein secondary structure was recorded on a JASCO 810 spectrophotometer. The parameters were controlled as 0.05 nm intervals, and 8 seconds response. Samples of bFGF containing various ligands were placed into 0.1 cm path-length cells, in an average of 4 scans between 195 and 260 nm, with a 1.0 nm bandwidth, a scanning rate of 10 nm min−1.
4.5 Urea-induced denaturation experiments
The experiments were performed on a JASCO FP6500 spectrophotometer, using an excitation wavelength of 280 nm. Samples were incubated at 4 °C for 24 h prior to analysis. An equilibrium constant K was calculated for each urea concentration employed:where FN, FD, and F are ratios of the fluorescence intensities at 350 nm to 305 nm of native, unfolded, and intermediate states, respectively. Thermodynamic parameters were calculated by following equations:| | | ΔGunf = ΔG0unf – m[urea] | (4) |
where ΔGunf and ΔG0unf are Gibb's free energies of unfolding in the presence and absence of urea, respectively. [urea] is the concentration of urea, and m is a parameter reflecting the cooperativity of unfolding.
4.6 Proteolytic digestion assay
bFGF (10 μM, 5 μL) was incubated with trypsin (1 μg μL−1) at room temperature in the absence or presence of POMs. The digestion was stopped after a desired time interval by the addition of the gel loading dye. Then samples were heated at 90 °C for 5 min and subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) by using a 12% gel. After electrophoresis, protein was stained with Coomassie Brilliant Blue (0.1% Coomassie Brilliant Blue G-250, 4% H3PO4, 10% (NH4)2SO4, 20% ethanol).
4.7 Suramin competition assays
Suramin and bFGF were mixed at room temperature. Then titrations were conducted by adding a certain amount of POMs into bFGF–suramin complex. The fluorescence measurement was taken in a JASCO FP6500 spectrophotometer with excitation at 315 nm and emission from 350 to 550 nm.
4.8 Cell culture
HUVEC were maintained in RPMI 1640 medium with 10% fetal bovine serum (FBS) in a humidified 5% CO2 environment at 37 °C. We used the third generation of HUVEC to perform all the experiments.
4.9 MTT assay for inhibition of the cell proliferation induced by bFGF
HUVEC were seeded at a density of 3000 cells per well on 96-well plates for 24 h. Then the medium was replaced with RPMI 1640 medium containing 0.5% FBS. After 24 h, the effects of POMs on the HUVEC proliferation induced by bFGF were determined. Briefly, POM and bFGF with different molar ratios (0:1, 0.25:1, 0.5:1, 1:1, 2:1, and 4:1, [bFGF] = 1.0 nM) were incubated in PBS buffer at 4 °C overnight. Then the complexes were introduced into the 96-well plates with HUVEC. After incubation for 48 h at 37 °C, the cells were treated with 10 μL of MTT (5 mg mL−1 in PBS) for 4 h at 37 °C and were then lysed in DMSO for 10 min at room temperature in the dark. Absorbance values of formazan were determined at 570 nm with a Bio-Rad model-680 microplate reader.
4.10 Statistical analysis
The statistical significance of the differences among groups was determined by one-way analysis of variance (ANOVA). A p value of <0.05 was considered as statistically significant.
Acknowledgements
Financial support was provided by the National Basic Research Program of China (Grant 2012CB720602) and the National Natural Science Foundation of China (Grants 20831003, 90813001, 20833006, 90913007, 21072182).
Notes and references
- H. Malonne, I. Langer, R. Kiss and G. Atassi, Clin. Exp. Metastasis, 1999, 17, 1 CrossRef CAS.
- P. Carmeliet, Nature, 2005, 438, 932 CrossRef CAS.
- F. Hillen and A. W. Griffioen, Cancer Metastasis Rev., 2007, 26, 489 CrossRef.
- P. Correa de Sampaio, D. Auslaender, D. Krubasik, A. V. Failla, J. N. Skepper, G. Murphy and W. R. English, PLoS One, 2012, 7, e30753 CAS.
- M. A. Moses and R. Langer, Biotechnology, 1991, 9, 630 CrossRef CAS.
- J. Folkman and M. Klagsbrun, Science, 1987, 235, 442 CAS.
- M. Rusnati and M. Presta, Curr. Pharm. Des., 2007, 13, 2025 CrossRef CAS.
- R. E. Friesel and T. Maciag, FASEB J., 1995, 9, 919 CAS.
- M. Guzman-Casado, M. M. Garcia-Mira, J. M. Sanchez-Ruiz, G. Gimenez-Gallego and A. Parody-Morreale, Int. J. Biol. Macromol., 2002, 31, 45 CrossRef CAS.
- M. Guerrini, M. Hricovini and G. Torri, Curr. Pharm. Des., 2007, 13, 2045 CrossRef CAS.
- D. M. Ornitz and N. Itoh, Genome Biol., 2001, 2 CrossRef CAS , reviews3005.1.
- A. E. Eriksson, L. S. Cousens, L. H. Weaver and B. W. Matthews, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 3441 CrossRef CAS.
- J. D. Zhang, L. S. Cousens, P. J. Barr and S. R. Sprang, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 3446 CrossRef CAS.
- X. Zhu, H. Komiya, A. Chirino, S. Faham, G. M. Fox, T. Arakawa, B. T. Hsu and D. C. Rees, Science, 1991, 251, 90 CAS.
- H. Ago, Y. Kitagawa, A. Fujishima, Y. Matsuura and Y. Katsube, J. Biochem., 1991, 110, 360 CAS.
- A. E. Eriksson, L. S. Cousens and B. W. Matthews, Protein Sci., 1993, 2, 1274 CrossRef CAS.
- S. Faham, R. E. Hileman, J. R. Fromm, R. J. Linhardt and D. C. Rees, Science, 1996, 271, 1116 CAS.
- C. P. Kwan, G. Venkataraman, Z. Shriver, R. Raman, D. Liu, Y. Qi, L. Varticovski and R. Sasisekharan, J. Biol. Chem., 2001, 276, 23421 CrossRef CAS.
- M. Zamai, V. R. Caiolfa, D. Pines, E. Pines and A. H. Parola, Biophys. J., 1998, 75, 672 CrossRef CAS.
- M. Zamai, C. Hariharan, D. Pines, M. Safran, A. Yayon, V. R. Caiolfa, R. Cohen-Luria, E. Pines and A. H. Parola, Biophys. J., 2002, 82, 2652 CrossRef CAS.
- M. Zamai, C. Hariharan, D. Pines, M. Safran, A. Yayon, V. R. Caiolfa, M. Mariani, E. Pines, R. Cohen-Luria and A. H. Parola, J. Mol. Struct., 2006, 792–793, 23 CrossRef CAS.
- F. Manetti, F. Corelli and M. Botta, Curr. Pharm. Des., 2000, 6, 1897 CrossRef CAS.
- D. Aviezer, S. Cotton, M. David, A. Segev, N. Khaselev, N. Galili, Z. Gross and A. Yayon, Cancer Res., 2000, 60, 2973 CAS.
- J. Folkman and Y. Shing, Adv. Exp. Med. Biol., 1992, 313, 355 CAS.
- S. Takano, S. Gately, M. E. Neville, W. F. Herblin, J. L. Gross, H. Engelhard, M. Perricone, K. Eidsvoog and S. Brem, Cancer Res., 1994, 54, 2654 CAS.
- P. A. Raj, E. Marcus and R. Rein, Angiogenesis, 1998, 2, 183 CrossRef CAS.
- G. Zugmaier, M. E. Lippman and A. Wellstein, J. Natl. Cancer Inst., 1992, 84, 1716 CrossRef CAS.
- H. Q. Miao, D. M. Ornitz, E. Aingorn, S. A. Ben-Sasson and I. Vlodavsky, J. Clin. Invest., 1997, 99, 1565 CrossRef CAS.
- A. Wellstein and F. Czubayko, Breast Cancer Res. Treat., 1996, 38, 109 CrossRef CAS.
- T. Yamase, J. Mater. Chem., 2005, 15, 4773 RSC.
- J. T. Rhule, C. L. Hill, D. A. Judd and R. F. Schinazi, Chem. Rev., 1998, 98, 327 CrossRef CAS.
- D. A. Judd, J. H. Nettles, N. Nevins, J. P. Snyder, D. C. Liotta, J. Tang, J. Ermolieff, R. F. Schinazi and C. L. Hill, J. Am. Chem. Soc., 2001, 123, 886 CrossRef CAS.
- R. Prudent, V. Moucadel, B. Laudet, C. Barette, L. Lafanechere, B. Hasenknopf, J. Li, S. Bareyt, E. Lacote, S. Thorimbert, M. Malacria, P. Gouzerh and C. Cochet, Chem. Biol., 2008, 15, 683 CrossRef CAS.
- G. Zhang, B. Keita, C. T. Craescu, S. Miron, P. de Oliveira and L. Nadjo, J. Phys. Chem. B, 2007, 111, 11253 CrossRef CAS.
- G. J. Zhang, B. Keita, C. T. Craescu, S. Miron, P. de Oliveira and L. Nadjo, Biomacromolecules, 2008, 9, 812 CrossRef CAS.
- G. J. Zhang, B. Keita, J. C. Brochon, P. de Oliveira, L. Nadjo, C. T. Craescu and S. Miron, J. Phys. Chem. B, 2007, 111, 1809 CrossRef CAS.
- Q. Wu, J. Wang, L. Zhang, A. Hong and J. Ren, Angew. Chem., Int. Ed., 2005, 44, 4048 CrossRef CAS.
- J. Geng, M. Li, J. Ren, E. Wang and X. Qu, Angew. Chem., Int. Ed., 2011, 50, 4184 CrossRef CAS.
- Y. Inouye, Y. Fujimoto, M. Sugiyama, T. Yoshida and T. Yamase, Biol. Pharm. Bull., 1995, 18, 996 CAS.
- C. E. Muller, J. Iqbal, Y. Baqi, H. Zimmermann, A. Rollich and H. Stephan, Bioorg. Med. Chem. Lett., 2006, 16, 5943 CrossRef.
- C. Z. Huang and Y. F. Li, Anal. Chim. Acta, 2003, 500, 105 CrossRef CAS.
- C. S. C. Wu, S. A. Thompson and J. T. Yang, J. Protein Chem., 1991, 10, 427 CrossRef CAS.
- T. Srimathi, T. K. Kumar, K. M. Kathir, Y. H. Chi, S. Srisailam, W. Y. Lin, I. M. Chiu and C. Yu, Biophys. J., 2003, 85, 459 CrossRef CAS.
- I. V. Baskakov and D. W. Bolen, Biochemistry, 1998, 37, 18010 CrossRef CAS.
- D. Coltrini, M. Rusnati, G. Zoppetti, P. Oreste, A. Isacchi, P. Caccia, L. Bergonzoni and M. Presta, Eur. J. Biochem., 1993, 214, 51 CrossRef CAS.
- J. T. Pelton and L. R. McLean, Anal. Biochem., 2000, 277, 167 CrossRef CAS.
- T. Sivaraman, T. K. Kumar, K. W. Hung and C. Yu, J. Protein Chem., 1999, 18, 481 CrossRef CAS.
- J. M. Sanz, M. A. Jimenez and G. Gimenez-Gallego, Biochemistry, 2002, 41, 1923 CrossRef CAS.
- C. R. Middaugh, H. Mach, C. J. Burke, D. B. Volkin, J. M. Dabora, P. K. Tsai, M. W. Bruner, J. A. Ryan and K. E. Marfia, Biochemistry, 1992, 31, 9016 CrossRef CAS.
- C. J. Burke, D. B. Volkin, H. Mach and C. R. Middaugh, Biochemistry, 1993, 32, 6419 CrossRef CAS.
- S. Shigeta, S. Mori, J. Watanabe, M. Baba, A. M. Khenkin, C. L. Hill and R. F. Schinazi, Antiviral Chem. Chemother., 1995, 6, 114 CAS.
- S. Shigeta, S. Mori, J. Watanabe, T. Yamase and R. F. Schinazi, Antiviral Chem. Chemother., 1996, 7, 346 CAS.
- X. Wang, F. Li, S. Liu and M. T. Pope, J. Inorg. Biochem., 2005, 99, 452 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supporting figures. See DOI: 10.1039/c2mb25389e |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.