Acoustofluidics 21: ultrasound-enhanced immunoassays and particle sensors
Martin
Wiklund
*a,
Stefan
Radel
b and
Jeremy J.
Hawkes
c
aDept. of Applied Physics, Royal Institute of Technology, SE 106 91 Stockholm, Sweden. E-mail: martin@biox.kth.se; Fax: +46 8 5537 8466; Tel: +46 8 5537 8134
bInstitute of Chemical Technologies and Analytics & Institute of Applied Physics, Vienna University of Technology, Getreidemarkt 8/164 AC, A-1060 Vienna, Austria. E-mail: stefan.radel@tuwien.ac.at; Fax: +43 58801 15199; Tel: +43 58801 15142
cManchester Interdisciplinary Biocentre, The University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK. E-mail: J.Hawkes@manchester.ac.uk.; Tel: +44 161 3068884
First published on 9th November 2012
Abstract
In part 21 of the tutorial series “Acoustofluidics – exploiting ultrasonic standing wave forces and acoustic streaming in microfluidic systems for cell and particle manipulation”, we review applications of ultrasonic standing waves used for enhancing immunoassays and particle sensors. The paper covers ultrasonic enhancement of bead-based immuno-agglutination assays, bead-based immuno-fluorescence assays, vibrational spectroscopy sensors and cell deposition on a sensor surface.
 Martin Wiklund | Martin Wiklund is Associate Professor in applied physics at the Dept. of Applied Physics, Royal Institute of Technology (KTH), Stockholm, Sweden. He received his M.Sc. in engineering physics from Lund University in 1999 and his Ph.D. in physics from the Dept. of Physics, KTH, Stockholm, in 2004. In 2004–2005, he was a postdoctoral fellow at the Fraunhofer Institute for Biomedical Engineering (IBMT), Berlin, Germany. He returned to KTH in 2005, becoming an Assistant Professor in 2006, and Associate Professor in 2009. Currently, Wiklund's research is focused on acoustic and optical methods for micro-scaled handling and characterization of cells, and applications of the methods to biomedical research. He is also a lecturer in various courses in basic physics, optics, ultrasound physics and diagnostic ultrasound. He is author of >25 peer-reviewed journal papers, >50 conference contributions including >20 invited talks, two patents and a recent book chapter (“Ultrasonic Manipulation of Single Cells”, in: Single-Cell Analysis: Methods and Protocols, Methods in Molecular Biology, vol. 853, Springer, 2012). |
 Stefan Radel | Stefan Radel was born in Vienna, Austria, in 1967. He received the M.S. degree in physics from the Vienna University of Technology (VUT), Austria, in 1998. He conducted his Ph.D. research within a “Training and Mobility of Researchers” program of the European Commission at University College Dublin, Ireland, the University of Wales, Cardiff, U.K., and the VUT on the effects of ultrasonic waves on yeast cells in suspension. He received the Ph.D. degree in physics from the VUT in 2002. Since 2003, he has been with the Institute of Applied (former General) Physics, VUT, working on various applications of ultrasonic resonators including acoustic filters and bulk acoustic wave (BAW) sensors. Since 2007, he is with the Institute of Chemical Technologies and Analytics at VUT. He is member of the “Young Scientist Advisory Board” of the International Congress on Ultrasonics (ICU) and member of the committee of Ultrasonic Standing Wave Network (USWNet) as well as occasionally a scientific committee member to conferences in the regime of acoustics and ultrasound. His main research interest presently is the application of ultrasonic particle manipulation in vibrational spectroscopy. |
 Jeremy J. Hawkes | Dr Jeremy Hawkes has been part of the Manchester University Miniaturisation group since 2002, he holds a BSc in Biophysics and received a PhD (1988) from UCNW, Bangor, Electronics Department for a study of protein dielectric properties. He jointly established a Dielectrophoresis Cell Characterisation laboratory in the Biology Department at York University before a 9 month post doc in Southeast University, Nanjing (1993) where he first learnt how to manipulate particles with ultrasonic standing waves. This knowledge was greatly advanced working with Professor Coakley in Cardiff until 2002. Together they helped found the USWNet (the USW community forum), which he currently chairs. His research interests now are: particle manipulation in air and water using DEP and USW. Recently a return to scale-up has become a significant topic. |
ForewordIn the twenty-first paper of 23 in the Lab on a chip tutorial series of Acoustofluidics, Martin Wiklund, Stefan Radel and Jeremy J. Hawkes presents different techniques for analysing biological samples which all benefit from acoustic standing waves to enhance the performance. Topics covered are bead-based assays, such as agglutination and fluorescence assays, vibrational spectroscopy, such as Raman and FT-IR spectroscopy, and particle sensors where acoustics are used to move cells toward a surface.Andreas Lenshof – coordinator of the Acoustofluidics series |
I. Introduction
Many diagnostic methods are based on detecting the binding or immobilization of molecules, cells or other bio-particles onto a substrate. Examples include immunoassays where antigens bind to capture antibodies immobilized on beads in suspension or on the surface of a microplate, and biosensors based on immobilized antibodies against cell membrane-bound antigens for cell or bacteria detection. For clinical diagnostics, it is important to identify antigens or other ligands where the existence or concentration level of the specific antigen can be correlated with a certain disease or health state. Such antigens or ligands are often called biomarkers.1This tutorial review discusses the utilization of ultrasonic standing waves in order to enhance the performance of immunoassays and cell/particle sensors. In these applications, the function of the ultrasound is to induce particle–particle contact, to concentrate particles or cells, and/or to deposit particles or cells onto a sensor surface. The review includes ultrasound-enhanced bead-based immunoassays (Part II), ultrasound-enhanced vibrational-spectroscopy sensing (Part III) and ultrasound-enhanced cell/particle sensors (Part IV). The review focuses on the bioapplications of the technology, while the fundamental principles and device designs of ultrasonic standing wave particle manipulation are found in previous parts of the Acoustofluidics series, e.g. in ref. 2 and 3.
II. Ultrasound-enhanced bead-based immunoassays
Enhancement of immunoassays by the use of ultrasonic standing wave manipulation has been demonstrated for two different assay formats: agglutination assays, and fluorescence assays, see Fig. 1. Both these assay formats utilize antibody-functionalized polystyrene beads (Fig. 1b) as a solid substrate for capturing and concentrating the target analyte (Fig. 1a). The binding events are indirectly detected by either measuring the degree of clumping of beads (Fig. 1d), or by measuring the fluorescence light from secondary tracer antibodies (Fig. 1c and e). Both assays discussed here are of sandwich-type, and have been operated in the homogeneous (wash-free) format when enhanced by ultrasound, which makes them simpler and less labour-intensive.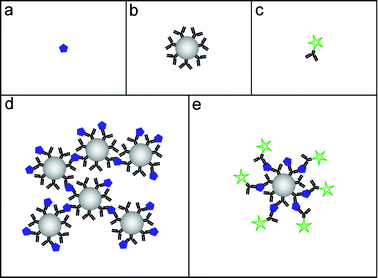 | ||
| Fig. 1 The building blocks of a bead-based immunoassay: The analyte (a), bead-immobilized capture antibodies (b) and fluorophore-labeled tracer antibodies (c). The two bead-based assay formats that have been enhanced by ultrasound: the agglutination assay (d) and the fluorescence assay (e). | ||
In this section we will discuss the basic principles of bead-based immunoassays and briefly review the field of ultrasound-enhancement of such assays. In this context, enhancement means to improve the speed and sensitivity of the assay. More detailed reviews of this particular topic are found in ref. 4 and 5.
A. Agglutination assays
The agglutination assay was invented in the 1950s by Singer and Plotz.6 They used 0.8 μm polystyrene beads (latex) coated with human gamma globulin (HGG), and noticed the beads were clumped when mixed with serum from patients having rheumatoid arthritis.7 Since then, a large number of latex agglutination tests (LATs) have been developed for a range of different diagnostic purposes.8In its simplest format, a sample containing the analyte (Fig. 1a) is mixed with a solution of capture beads functionalized with a suitable analyte-specific receptor molecule, e.g. an antibody (Fig. 1b). Initially, this analyte–bead mixture has a uniform color and texture. Following a gentle agitation of the mixture, the interaction between the analyte and the capture beads causes the formation of multi-bead aggregates (Fig. 1d). These aggregates can be detected qualitatively by the naked eye, by observing the change in texture from the uniform to a more turbid appearance of the mixture, see Fig. 2. Although this simple “yes/no”-format is the most common for commercially available LAT kits, it is also possible to perform quantitative LATs. Methods include turbidimetry,9 nephelometry,10 particle counting detectors11 and flow cytometry.12
 | ||
| Fig. 2 Latex agglutination test (LAT) on cerebrospinal fluid from a patient with culture-confirmed meningococcal group B infection. The slides show positive agglutination (A) and negative control (B). The figure is taken from Sobanski et al.16 | ||
The major limitation of LATs is the sensitivity. Typically, the detection limit is in the nanomolar range. The reason for this is the relatively small beads (<1 μm) and high bead concentrations needed for obtaining sufficiently high diffusion and bead collision rates, respectively. Furthermore, smaller beads have a higher degree of non-specific agglutination which increases the background noise.
Ultrasound can be used to enhance latex agglutination tests.5 The main function of the ultrasound is to increase the probability of bead collisions by pushing and concentrating the beads into the pressure nodes of the standing wave. The most obvious enhancement caused by the increased collision rate is improved speed of the assay (about one to two orders of magnitude).13 However, if the concentration of capture beads is decreased an increase in sensitivity is also achieved (between one to three orders of magnitude).14 Without ultrasound, decreased concentration of capture beads would directly lead to increased incubation times. Thus, there is a mutual benefit of the ultrasound for both the assay speed and the sensitivity.
The device used in ref. 13 and 14 was based on a 2 mm inner-diameter glass capillary inserted into a cylindrical ultrasonic resonator operated at 1.97 MHz or 4.59 MHz. This device, described in more detail in ref. 5, was later commercialized under the name “Immunosonic”, Electro-Medical Supplies, Wantage, UK, see Fig. 3. However, despite the benefits of ultrasound-enhanced LATs the commercial device was outcompeted by standard laboratory culture and PCR-based methods,15 and is no longer available for sale. A possible reason for this was the choice of target disease (which was meningococcal meningitis) when launching the Immunosonic instrument.16 On the other hand, ultrasound-enhanced agglutination assays still have great potential and would also be very suitable to implement in the lab-on-a-chip format. Thus, micro-scaled ultrasound-enhanced LATs are yet to be demonstrated.
 | ||
| Fig. 3 The Immunosonic instrument from Electro-Medical Supplies, based on the ultrasonic standing wave action on an immuno-agglutination assay. The sample was inserted into a glass capillary which was placed along the symmetry axis of a cylindrical ultrasonic resonator. The ultrasound was used to boost the bead–bead interaction by concentrating the beads into the pressure nodes of the ultrasonic standing wave. More detailed descriptions of the device design and operation are found in ref. 13 and 14. | ||
It is relevant to ask why a decrease in capture bead concentration leads to higher sensitivity in an agglutination assay. This has been investigated both theoretically and experimentally by Wiklund et al.12 They studied the initial stage of immuno-agglutination and developed a model for predicting the probability of both specific and non-specific agglutination per one collision between individual beads and/or small bead clusters. The model is based on combining two reactions: (I) the binding of antigens, A (which here is considered as the analyte), to the bead-immobilized capture antibodies, Y, and (II) linking of beads or bead aggregates, Li, to other beads or bead aggregates, Lj (where i and j are the number of beads in each aggregate). Each of the two reactions can be described by the rate constants, kon and koff (for the antigen-antibody interaction) and kij (for the bead–bead interaction):
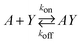 | (1) |
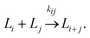 | (2) |
Since eqn (1) is considered as a much faster reaction than eqn (2), the two reactions are treated separately in our model. The antigen–antibody rate constants (kon and koff) in eqn (1) are often expressed in terms of the dissociation constant, KD, as:
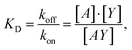 | (3) |
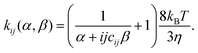 | (4) |
Here, kij is a function of the probabilities of non-specific and specific agglutination at each particle collision, α and β, respectively. Other parameters in eqn (4) are the Boltzmann constant (kB), the temperature (T), the viscosity of the medium (η) and the cluster-surface steric hindrance coefficient (cij). The latter coefficient is defined as 0 < cij < 1, and is introduced in order to take into account that not all the surface of the beads in the cluster is available for contact with another cluster. This coefficient must be numerically determined by averaging all possible geometrical configurations of beads in a cluster.12
The non-specific agglutination probability (α) is assumed to be constant for a given assay protocol. The specific agglutination probability (β), on the other hand, can, for a given antigen–antibody interaction (cf.eqn (1) and (3)), be treated purely geometrically as:18
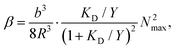 | (5) |
Let us now use eqn (1)–(5) in order to define a theoretical detection limit (i.e. sensitivity) of the agglutination assay. This limit can be defined as the analyte concentration when β = α, i.e., when the amounts of specific and non-specific and agglutinations are equal. If we assume a high-affinity immunoassay (KD ≪ A0 and KD ≪ Y0), this minimum analyte concentration, Amin, can be shown to be:12
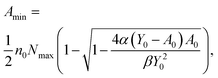 | (6) |
B. Fluorescence assays
An alternative to the immuno-agglutination assay is the immuno-fluorescence assay, see Fig. 1e. This assay also uses antibody-functionalized polystyrene beads (Fig. 1b) for capturing and concentrating the analyte (Fig. 1a). However, instead of relying on bead–bead interaction and clumping, the fluorescence assay uses fluorescently labeled secondary antibodies, or tracer antibodies (Fig. 1c), that bind to another epitope of the analyte. The analyte is then quantified by measuring the fluorescence intensity from each bead. The bead-based immuno-fluorescence assay is particularly suitable for multiplexing. This can be done by encoding the bead with various amounts of two or more fluorophores, a technology called suspension array technology (SAT).19 The read-out instrument for SAT is typically a flow cytometer, and this method has also been commercialized (Luminex Corporation, Austin, Texas, USA). There also exists read-out technology performed in bulk solution, e.g. by the use of confocal fluorescence microscopy20 or two-photon excitation (TPX) technology.21Just as for the immuno-agglutination assay, the sensitivity of a fluorescence assay is dependent on the concentrations of beads in the suspension. This is a consequence of the concentration of capture antibodies attached to the beads, relative the analyte concentration in the sample. In 1989, Ekins proposed the concept of ambient analyte immunoassay,22 for which he demonstrated an increase in sensitivity by using a very low concentration of capture antibodies (<0.01 KD). Ekins applied this concept to microspot array technology,23 where the total concentration of capture antibodies was very small, but locally concentrated to small spots on a solid substrate. The main principle of ambient analyte condition is to increase the fractional occupancy of the capture antibody binding sites by using an excess of analyte and tracer antibodies, resulting in a fluorescence intensity dependent on analyte concentration only.
Ultrasound can be used for improving the sensitivity of bead-based fluorescence assays. However, the improved sensitivity is not a direct consequence of concentrating beads by ultrasound, but rather a consequence of applying the ambient analyte condition to the bead-based immunoassay. Since the ambient analyte condition results in a very low concentration of capture beads (typically a few beads per μL), a method for concentrating the beads and delivering them to the detection site is needed. This has been demonstrated by Wiklund et al.,20 who used ultrasonic standing wave manipulation on a 96-well plate platform combined with confocal fluorescence detection for enhancing the sensitivity in a bead-based immunoassay, see Fig. 4. In their setup, the beads were positioned into a set of planar, compact monolayers matching the horizontal x–y scanning plane of the laser focus of the confocal microscope. These monolayers were positioned into the pressure nodes of the ultrasonic standing wave. Thus, a benefit of this method is the possibility to predict the vertical position of the monolayers (each separated by half the acoustic wavelength), and use a time-efficient scanning procedure. Typically, the device used in ref. 20 used ∼2500 beads of size 3 μm in a 100 μL sample volume in one of the wells in the 96-well plate (corresponding to a volume fraction of 10−7), which were rearranged into 6 monolayer aggregates (each separated by half the acoustic wavelength = 190 μm @ 4 MHz). The bead enrichment efficiency (i.e. the ratio between trapped and total number of beads) was about 10%, corresponding to ∼50 trapped beads in each aggregate. Since the laser focus of the confocal microscope could be tuned to match the size of the beads (3 μm), the local bead concentration in the scanning area was 106–107 times the initial concentration. This is 103–104 times better enrichment than what would be achieved with simple centrifugation or sedimentation to the bottom of the 96-well plate.
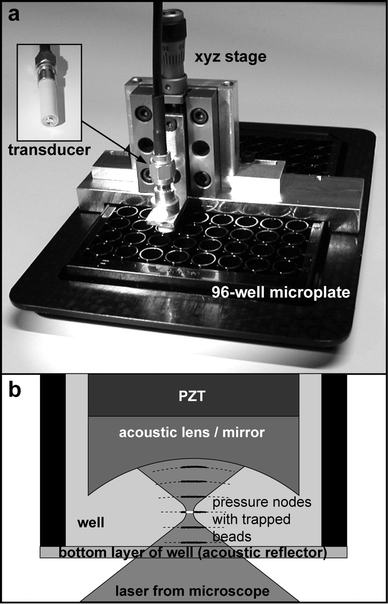 | ||
| Fig. 4 The 96-well microplate platform for ultrasonic enhancement of the bead-based human thyroid stimulating hormone assay developed by Wiklund et al.20 a) The 4 MHz transducer (upper left corner) was submerged into one of the wells of the 96-well plate by a xyz precision translation stage. b) The beads were concentrated into the pressure nodes of the standing wave (dotted lines) formed by reflections in the acoustic lens/mirror (front part of the transducer) and the bottom layer of the well. Each pressure node were then scanned by an inverted confocal microscope. The figure is taken from ref. 20. | ||
The assay used to test the potential of ultrasound-enhanced fluorescence assays was the human thyroid stimulating hormone (hTSH) assay.20 This assay used bead-immobilized capture antibodies and fluorescent tracer antibodies with affinity constants (KA = 1/KD, cf.eqn (1)) of 2 × 1010 M−1 and 1 × 1010 M−1, respectively. These antibodies were directed against two different epitopes of the TSH molecule. The estimated limit of detection was as low as 20 fM when the assay was enhanced by ultrasound. It is interesting to compare this limit with the expected number of analyte molecules bound to each bead. The experiment was carried out with ∼2500 beads in the 100 μL sample, which means that there are ∼480 TSH molecules per bead in the sample at the analyte concentration 20 fM. This number can be compared with the theoretical detection limit12 of the TSH assay, which for the affinity constant (KA = 1010 M−1) and bead capacity (106 capture antibodies per bead) corresponds to ∼130 TSH molecules bound to each bead. This means that about 30% of the analyte is expected to be bound at this limit. This is in good agreement with the detection limit of the confocal fluorescence system used, which was about 100 fluorophores per bead.
III. Ultrasound-enhanced vibrational spectroscopy
The following section deals with the exploitation of ultrasonic particle manipulation in the regime of vibrational spectroscopy sensing applications.Manipulation in the given context means to have control over the spatial distribution of suspended particles in respect to where particles are driven to (and concentrated) when the suspension is exposed to an ultrasonic plane standing wave and on the other hand to generate regions depopulated of particles, i.e. locations where no particles will be found when radiation forces are exerted. Particles used for the various examples were all particles as such, hence solid and therefore denser and harder than the liquid. As a result, the position of agglomeration of the beads, crystals and yeast cells will always be the pressure nodal region of the respective ultrasonic standing wave.2,24 The set-ups used throughout this section were layered resonators, i.e. stacks of parallel sheets of different materials (PZT, glass or aluminium carrier, suspension liquid, reflector). The excitation frequency always was around 2 MHz.
Vibrational spectroscopy25 includes a group of optical measurement techniques increasingly popular in process analytical chemistry because of the ability to directly provide molecular-specific information about a given sample under investigation. The term “vibrational” refers to the measurement of frequencies of periodic motions of atoms in a molecule. The excitement of these modes is governed by quantum mechanics, on a less sophisticated level we see the interaction of the molecule with photons of certain wavenumbers, i.e. the reciprocal of the light frequency, which is specific for a given molecule due to its dependency on atoms or atom groups and their chemical bonds. In a nutshell, the spectrum of light collected after a sample has been exposed to electro-magnetic excitation carries specific information about the sample's chemical composition.
Spectra can be recorded from solids, liquids and gases, providing qualitative as well as quantitative information on the chemical composition of the sample. Spectra can be measured in various ways; hence methods like absorption spectroscopy (in the near or mid-infrared) as well as Raman spectroscopy are pooled in the term vibrational spectroscopy. In the context of real-time process analytical chemistry, advances in the field are of interest as a great amount of the desired (bio)chemical information can be extracted from the recorded spectra.
Both ultrasonic and vibrational spectroscopy techniques are at a stage of development where crucial improvements can be expected in the near future, while at the same time are mature enough to be endorsed in industrial environments. The presented examples of incorporating an ultrasonic standing wave field in different vibrational spectroscopy sensing applications have been successful and will be shown to be advantageous in various respects. The potential of influences due to the combination of the two techniques like cross-sensitivities or cross-talking caused by driving electronics was given close attention, however no adverse effects were observed throughout the experiments.
Agglomeration of crystals for Raman spectroscopy
In Raman spectroscopy,26 a sample is irradiated with a focused laser beam and the rare inelastically scattered photons are recorded. The measured intensities at different wavenumbers make up the Raman spectrum, providing information on vibrational transitions characteristic for the chemical composition of the sample under study. The spatial resolution of Raman micro-spectroscopy in the low-micrometer scale and its ability to probe samples under in vivo conditions allow for new insights into living single cells without the need for fixatives, markers, or stains.27,28However, the quality of measurements is highly dependent on the number of molecules exposed to the incoming light, which is e.g. of special relevance when monitoring reacting suspensions, where Raman spectroscopy holds great promise due to possible non-invasive measurement strategies. It turns out to be difficult for standard on-line Raman spectroscopy to discriminate between Raman photons from the solid matter and signals originating from the pure liquid phase. This problem is of special relevance in the case of low concentration of suspended particles.
Therefore, means to concentrate samples in the light path are regularly used. Acoustic levitation in air for monitoring containerless chemical reactions has been used traditionally in Raman micro-spectroscopy.29 Recently the investigation of red blood cells and micro-organisms with this technique was reported.30
The combination of ultrasonic particle manipulation and confocal Raman micro-spectroscopy is a novel approach to increase selectivity and sensitivity of on-line Raman measurements of suspensions. Aggregating particles in suspension by an ultrasonic standing wave can provide an experimentally simpler approach to immobilize and manipulate micro-particles; they may be deliberately concentrated or removed from the Raman measurement spot, thus allowing to selectively measure the liquid and solid phases, respectively. In addition to the improved selectivity, an increase in sensitivity may be expected due to the local enrichment of the particles.
A resonator comprising a PZT ceramic glued to the side of a small glass cuvette (2 mL) was used to control the spatial distribution of suspended theophylline particles relative to the light path of the Raman microscope, i.e. agglomerates of particles were deliberately positioned within and out of the focus of the instrument.31
The set-up was placed under the Raman microscope with sound propagation direction perpendicular to the light path (Fig. 5, top). Thus, the pressure nodal planes where oriented parallel to the incident beam, allowing to control their locations relative to the light path by slightly changing the excitation frequency. Illustrating the influence of the radiation forces on the spatial distribution of particles, a light micrograph of the agglomeration of theophylline crystals is shown in the enlargement at the bottom of Fig. 5.
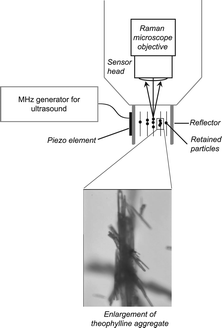 | ||
| Fig. 5 Sketch at the top shows Raman microscope with light path into cuvette resonator. The picture at the bottom shows the theophylline crystals agglomerated by the ultrasonic field. The figure is taken from ref. 31. | ||
The results of investigations of dissolved as well as suspended theophylline are depicted in Fig. 6. The aim was to compare the Raman signal of homogeneously suspended theophylline crystals (black line) with measurements of agglomerates brought about by the ultrasonic standing wave (black with dots). Fig. 6 shows a significant increase (3 to 6-fold) of scatter intensity when the ultrasonic field was applied. Moreover, the data suggest better resolution, e.g. between wavenumbers 1600 and 1700 cm−1 when the theophylline crystals were concentrated by the ultrasonic standing wave.
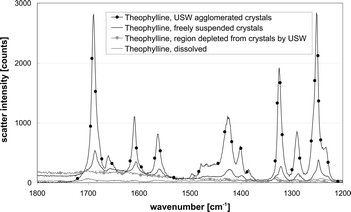 | ||
| Fig. 6 Raman spectra of theophylline solution (grey) and freely suspended theophylline crystals (black) in comparison to theophylline crystals agglomerated by ultrasound (black with dots) and the theophylline solution in a region where the crystals were depleted by the ultrasonic standing wave (grey with dots). The figure is taken from ref. 31. | ||
In contrast, no significant differences were found for regions where no particles are present (in the velocity nodes). The Raman spectrum of a crystal-free theophylline solution (Fig. 6, grey line) was not different from a measurement taken with the optical focus positioned within this depleted region (Fig. 6, grey with dots), hence the very low concentration of particles between the nodal planes enables one to specifically take measurements of the host liquid's composition.
When comparing sediment theophylline crystals, which were supposed to be packed tightly (data not shown) with agglomerates brought about by the ultrasonic radiation forces, a slight increase of scatter intensity was measured when the ultrasonic field was present.
Very similar results were obtained using yeast cells as a model for bio-suspensions, again the Raman measurements indicated that the agglomeration of particles in suspension by the ultrasonic field was a suitable means to achieve higher Raman scatter intensities.
Another study aimed on the strategy of trapping particles by radiation forces in a flow cell allowing for exposure to a variety of reactants and thus the execution of a given set of chemical reactions while being continuously monitored by Raman micro-spectroscopy. This approach as well overcomes drawbacks related to levitation in air like solvent evaporation. The potential of this ultrasonic trapping method to monitor on-bead chemical reactions was investigated with an example of automated generation of a surface-enhanced Raman (SER)-active layer on ultrasonic trapped beads.32 The use of the ultrasonic standing waves for trapping was especially valuable when synthesizing SER-active beads on-line. The process included steps of preparation and subsequent recording of SER spectra of different analytes during which the beads where retained in the focus of the Raman microscope. Furthermore, the discharge of the silver-loaded beads by switching off the ultrasonic field is advantageous in comparison to previously described procedures that also employed beads as carrier for SER substrates.33
A broad variety of assays can be performed with this application in many fields, among which solid-phase synthesis and bead-based bioassays can be envisioned. The robust but gentle handling also allows for prolonged studies on beads as well as on living cells.
Enhancement of stopped flow FT-IR ATR spectroscopy
The growing use of bioprocesses as a manufacturing route for, e.g., antibiotics and other medical compounds, enforces the development of reliable, automated sensors for bioprocess monitoring and control. Such sensors are key for optimal system performance34 as continued analysis is needed in order to control the monitored bioprocess. State-of-the-art sensor systems provide information on physical parameters (pressure, temperature) but only a few chemical parameters like e.g. pH, oxygen and carbon dioxide concentration in liquid and gas phase to mention a few. Fast response times of at least one order of magnitude faster compared to the generation time of the observed microorganism are necessary.35 Real-time information on the chemical composition and on the physiological status of the employed microorganism would be of high diagnostic value. However, due to experimental difficulties such sensors are not available so far. Moreover the development of new sensor designs is triggered by the rapid progress in biotechnology.Fourier transform infrared (FT-IR) spectroscopy is a well-developed method in chemical analysis.36 The incident IR radiation excites parts of the molecules in the sample, therefore, a certain amount of light energy at a given light wavelength is converted into vibrational energy, hence absorbed. The acquisition of an IR absorption spectrum can be conducted in minutes to seconds delivering specific molecular information about the sample in the optical pathway.25 Constantly new devices and concepts for advanced chemical analysis have been developed during recent years.37
FT-IR spectroscopy in combination with attenuated total reflection (ATR) sensing elements is a currently developing, very promising means for process and bioprocess monitoring. The ATR scheme exploits the occurrence of total reflection of light at the interface of two media with different optical densities. FT-IR ATR spectroscopy is a surface sensitive technique, the detection range is only some μm. Any substance covering the said interface influences the incoming light at certain wavenumbers and thus specific information about its chemical composition can be obtained from the absorption spectrum. Due to the exponential decay within the evanescent field the closer the sample is located to the ATR surface the higher its contribution to the recorded spectrum will be, whereas almost no absorption takes place at greater distances. Therefore additional measures are necessary to bring a sufficient amount of sample, e.g. suspended particles, into this region.
The limited detection range is especially advantageous when measuring aqueous samples in the information rich mid-IR spectral range. Due to the strong infrared absorption of water, the optical path must be short (<10 μm) to keep the detection limit low in common FT-IR spectrometers using thermal radiation emitters. Short optical path lengths like this are realized by the ATR technique without putting geometrical constraints on the sample volume.38
The surface sensitivity opens the possibility to measure the spectra of suspended particles (cells) and the supernatant (i.e. the liquid component). The basic idea of the stopped flow technique39 is to keep cells (or other particles) from the horizontal ATR surface by pumping the suspension through a detection cell in bypass while the spectrum of the suspending liquid is measured. Subsequently the throughput is stopped, the cells settle on the surface in the evanescent field of the ATR. Then a spectrum can be taken from this layer of sediment.
A sketch of the used flow cell is shown in Fig. 7 at the top. During the first step of the stopped flow protocol (Fig. 7a) the cells are kept away from the ATR, which allows the determination of dissolved analytes in the supernatant. Upon stopping the flow (Fig. 7b), the cells settle on the ATR surface and thus, coming into reach of the evanescent field, dominate the recorded infrared spectra. Based on this approach, the intracellular composition can be determined. In the case of cells like E. coli, the cleanness and thus the sensitivity of the ATR element could be maintained by periodic rinsing with NaHCO3 (Fig. 7c). Aimed at the enhancement of the stopped flow technique, an ultrasonic transducer was used as lid of the set-up, the ATR element at the bottom represented the reflector. Consequently a standing wave with horizontal nodal planes was built up, enabling the manipulation of particles (cells) within the flow cell. The ultrasonic particle manipulation was applied with two distinct targets:
• When developing a sensor for fermentation monitoring, time resolution is an important issue. The ultrasonic field was used to accelerate the measurement.
• When trying to measure glucose and ethanol in a baker's yeast fermentation with the stopped flow technique, the formation of a bio-film on the horizontal ATR element was observed.40 The ultrasound was used as a means to overcome this detrimental contamination of the sensor.
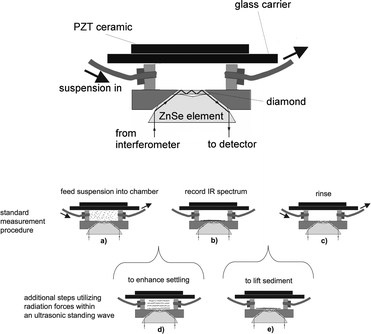 | ||
| Fig. 7 Top: Flow cell comprising the ATR element at the bottom and the PZT-sandwich transducer at the top. Bottom: Stopped flow technique to specifically measure the IR absorbance of suspended particles: the suspension is pumped into the detection volume (a). When the flow is switched off, particles settle onto the ATR surface and the spectrum is recorded (b). After the measurement the cell is rinsed (c). An ultrasonic standing wave was applied to accelerate the measurement time by agglomerating yeast cells prior to the settling (d) and to improve the cleaning by actively lifting the sediment from the ATR prior to the rinse (e). The figure is taken from ref. 42. | ||
In the absence of the ultrasonic standing wave, the suspended yeast cells settled slowly on the ATR surface at a constant rate. A maximum was reached after approximately 185 s signalling a complete coverage of the sensitive surface.
When the ultrasonic field was present during the filling and subsequently switched off when stopping the flow, a different behaviour occurred. For 15 s no cells were detected by the ATR as no material was present at the sensing surface. Following that a strong increase of absorbance was detected, it took some 70 s to reach the same maximum as above. The acceleration due to ultrasound-induced aggregation was therefore increasing the settling rate by a factor of more than two.
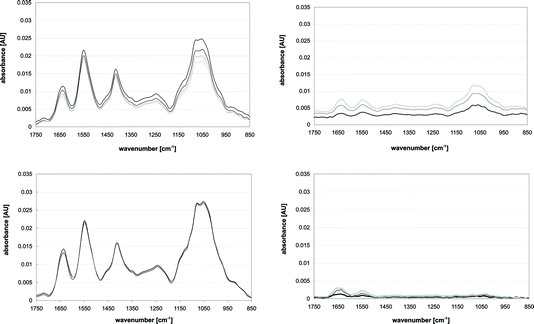 | ||
| Fig. 8 IR Spectra recorded to observe bio-film formation while the ATR was exposed to a yeast suspension. Measurements were taken at 30 (black), 60 (dark grey), 90 (lighter grey) and 120 min (lightest grey). Top left: Result when the basic procedure (cf.Fig. 7a–c) was used. Top right: Measurements representing the bio-film causing a decrease in resolution and sensitivity of the measurements. Bottom left: Result when an ultrasonic standing wave was applied during the rinse (cf.Fig. 7c). Bottom right: Spectra when the ultrasonic standing wave was applied resulting in a significantly reduced bio-film. The figure is adapted from figures in ref. 42. | ||
An obvious measure to remove a bio-film is the application of liquid cleaning agents.43,44 The effects of acids, surfactants and oxidizing agents on the bio-film removal in this flow-cell have been investigated in ref. 45. The detailed comparison of data for protein and carbohydrate removal delivered the best results for sodium hypochlorite (NaOCl), which was able to remove 100% of the protein and 95% of carbohydrates in the bio-film. However using this chemical (i.e. bleach) has constraints due to its aggressive nature on the range of materials that can be used for a sensor. Sodium dodecyl sulfate (SDS), one of the standard surfactants used for cleaning, showed removal abilities of 93% for the protein and 94% for carbohydrates. The downside here was a prolonged removal time, it took 30 min of rinsing until the agent had completely left the chamber.
The approach of interest here was to address the problem by the application of an ultrasonic standing wave as shown in Fig. 7e. The radiation forces exerted on particles were used to actively lift the material covering the horizontal ATR sensor surface during rinsing thus improving its cleanness.
The application of ultrasound performed well (see spectra in Fig. 8, lower graphs) in comparison to the mentioned chemicals. More than 70% of the protein and 90% of carbohydrates could be removed. The reason for this gap in cleaning performance was interpreted to be a result of the different particle sizes. High carbohydrate contents seemed to be associated with large particles like cells or cell debris, while protein molecules might exist in solution or contained in smaller particles, which are significantly less effected by radiation forces.
ATR probe for in-line bioprocess monitoring
In-line sensing approaches, i.e. measurements inside of a reactor, are the preferred option for bioprocess monitoring due to sterility and practicality issues. However, as no recalibration or verification of sensor response is possible, in-line sensing approaches are especially demanding in regard to long-term robustness and calibration stability.In-line ATR sensors connected to the spectrometer by mid-IR fiber optics or optical conduits were exploited in bioprocess monitoring before. A variety of small molecules like sugars, alcohols, organic and amino acids as well as phosphate at concentrations of a few g L−1 were successfully determined in the fermentation broth.46,47 Only chemical information of the supernatant could be assessed with these in-line configurations, as the cells in culture do not reach the ATR's evanescent field. However, from a bioprocess control point of view it would be very desirable to access chemical information about the culture as well.
This demand recently triggered investigations to combine an ultrasonic standing wave with an in-line ATR fiber probe. The ability of ultrasonic standing wave fields to deposit particles on a surface was investigated with functionalised surfaces48 and by optical means.49 The target here was to independently measure the mid-IR spectra of the supernatant and the suspended cells in purposely populating or de-populating the ATR's evanescent field of cells.
To combine the optical and the ultrasonic techniques it was necessary to implement an ATR element in the proximity of an ultrasonic standing wave permitting particle manipulation within the evanescent field.50 The way to accomplish this was to use the ATR element as reflector of an ultrasonic resonator (see Fig. 9). Sound was propagating in the direction of the axis of the ATR fiber probe, hence suspended particles were agglomerated in the nodal planes which were oriented parallel to the evanescent field. The precise location of the agglomerate could therefore be controlled by the ultrasonic frequency.
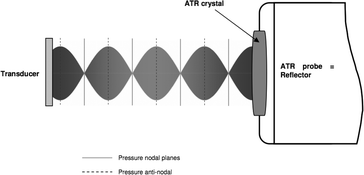 | ||
| Fig. 9 Sketch of the ultrasonic resonator where the ATR probe acts as reflector for the incident acoustic waves. The envelope of the standing wave represents the resonant state like in Fig. 10 left upper picture. Changing the frequency will alter the position of the pressure nodes and therefore the location of particle aggregation. The figure is taken from ref. 50. | ||
Fig. 10 shows four images obtained when polystyrene beads suspended in methanol were used to study the arrangement of particles at the tip of the ATR fiber probe. Bead concentration was kept low to maintain good visual access. The aim was to show that the location of the beads could be contained and manipulated in the proximity of the ATR.
 | ||
| Fig. 10 Images of polystyrene resin beads (100 μm) suspended in methanol and agglomerated in the pressure nodal planes of an ultrasonic standing wave between a transducer (right) and the head of the ATR fibre probe (left). The acoustic path length was adjusted to 3.18 mm. Each image was taken at a distinct ultrasonic field frequency stated below the respective image. Due to an increase of frequency from 1.850 MHz to 1.869 MHz, the suspended beads continuously approached the ATR surface. The figure is taken from ref. 50. | ||
The upper left image in Fig. 10 shows strong alignment of the particles in the gap between the ATR probe head (left) and transducer (right) indicating high acoustic energy densities at the indicated frequency of 1.85 MHz. The visible arrangement reflects the nodal planes half a wavelength apart (sound wavelength 590 μm for methanol at 1.86 MHz). The agglomeration in the pressure nodal planes was established within seconds of the activation of the ultrasonic wave and maintained as long as an ultrasonic field was present.
Next the operating frequency was changed to 1.854 MHz. The result was a slightly less pronounced pattern in respect to the particles' alignment (see upper right image of Fig. 10). A few of the beads in the leftmost nodal plane seemed to touch the diamond ATR surface, but still the majority of the beads remained a short distance from the cone tip. An increase of the frequency to 1.860 MHz further loosened the aggregates (Fig. 10, lower left image). The top plateau was covered more substantially with beads, but still a vast number of beads remained in the nodal plane in front of the ATR without touching it.
Fig. 10 (lower right image) shows the result when the frequency was changed to 1.869 MHz and eventually the major part of the first aggregate came into contact with the diamond. However the strong alignment observed at 1.850 MHz was gone, indicating that the energy density was not very high in the fluid layer at this stage.
The analysis of these images suggests the possibility of manipulating suspended particles onto a smooth, rigid surface which is orientated perpendicular to the sound propagation direction. A broad hint, that particles were actually pushed into the evanescent field was achieved with the results in Fig. 11.51 A setup like that sketched in Fig. 9 was used in a vessel containing a model yeast suspension agitated by a magnetic stirrer. While the respective pushing frequency f2—indicated by the grey colored bar at the bottom of Fig. 11—was applied, a strong increase of the infrared absorption at 1047 cm−1 was observed. This wavenumber is associated with carbohydrates, which are contained in the yeast cells, the data therefore was interpreted as yeast cells entering the sensitive zone of the ATR. Switching to the retracting frequency f1 (blue sections of bar) resulted in a steep decrease back to the base level. Obviously the cells were pulled out of the evanescent field.
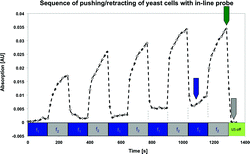 | ||
| Fig. 11 Sequence of repeated applications of the pushing and the retracting frequencies recorded with an in-line probe. The ATR's population/depopulation is indicated by the temporal development of an absorption band (carbohydrates @ 1047 cm−1) of the yeast cells. The figure is taken from ref. 51. | ||
A gradual increase of the absorption peak value over several cycles of switching between the retracting and pushing frequencies was found. It is believed that the agglomerate was slowly growing, i.e. more and more cells were entering the node as the ultrasonic field was applied during the whole experiment. Additionally, the base line was not reached completely after the third cycle for the same reason; the aggregate supposedly was too big to be completely pulled away from the ATR. When finally switching off the field (green bar), the absorption signal fell back to the initial level.
A comparison of the achieved absorption when cells were pushed into the evanescent field by the ultrasonic standing wave with the absorption reached by sedimentation of yeast cells on the ATR (data not shown) was performed. It revealed that approximately 20% of the achievable absorption of the sediment was reached by the in-line probe. The reason for that might be that the radiation forces had to push the cells 135° upwards against gravity in the case of the in-line probe.
The result in Fig. 11 was interpreted as proof of concept. It was shown, that an ultrasonic standing wave can be used to control the spatial distribution of cells (or other particles) in close proximity to an ATR sensor. It was possible to measure the infrared spectrum of the host liquid at times when the retracting frequency was applied. Independently the assessment of the particles' spectrum was accomplished, when the ultrasound frequency was switched to a value at which the radiation forces pushed yeast cells into the evanescent field of the ATR.
IV. Ultrasound-enhanced particle sensors
This section describes in more general terms than Sect. III the phenomena of pushing particles onto a wall by the use of ultrasound. This wall may, like the particles described in Sect. II, be coated with an immuno-selective agent. The term “pushing” is usually used to describe the standing wave force when it acts towards a wall. Nonetheless, here the term “attraction” is used because it is the action of the wall surface that is of interest here, but we are describing the same process. The focus here is on successful designs for the wall rather than details of the mechanism.The phenomenon of attracting particles to a surface
When particles are attracted to a wall by ultrasound, they form clumps in the quite distinctive pattern which is seen in Fig. 12. In this example48 spores have been pushed onto an antibody coated glass microscope slide. The same close arrangement of clumps is seen52 with yeast cells on a 50 μm thick polystyrene film in Fig. 13a, this chamber had no sides so that the water depth could be adjusted. When the water depth was significantly greater than ¼ wavelength another more widely spread pattern of clumps became dominant, and these clumps were suspended in the fluid and were not in contact with any surface. The systems in Fig. 12 and 13 are very different, yet when the water layer thickness was ¼ wavelength or less at the ultrasound frequency used, both produced the distinctive close pattern of clumps against the wall surface. | ||
| Fig. 12 Device B in Table 1. Clumps of Bacillus subtilis var. niger (BG) spores adhered to a microscope slide coated with an anti-BG antibody. White areas are regions where spores have adhered to the surface. The slide formed a half wavelength thick (at 2.9 MHz) attraction surface over a 108 ml−1 suspension of spores, exposed to sound for 5 min. Pictured after the glass slide was removed from the chamber and washed. The figure is taken from Hawkes et al.48 | ||
 | ||
| Fig. 13 Device G in Table 1. Yeast cells (at 107 ml−1) in a thin layer of water between a vibrating (1 MHz) microscope slide and a 50 μm thick polystyrene film (white areas are cell clumps). The water layer thickness was adjusted with a micrometer stage to move the film. The acoustic wavelength (λ) in water at 1 MHz was 0.75 mm. a) Water thickness < λ/4, clumps in contact with the surface. b) Water thickness > λ/4, some clumps in contact with the surface and some free in the suspension. c) Water thickness ≫ λ/4, most clumps free in the suspension. The figure is taken from Hawkes et al.52 | ||
The need for a cell attracting wall in microsystems
For continuous online environmental monitoring for the unlikely appearance of pathogen cells, the addition of highly specific immuno-coated particles for detection as described in Sect. II is not a reasonable option since those particles would need to be selectively retained over long periods, requiring continuous extraction from inhomogeneous environmental samples. A solution is to place the immuno-selective element on the chamber wall. However, without ultrasound this does not form an efficient detector since very few antigen particles in a flow actually come into contact with the wall. A potential application for attracting particles to a surface, is to enhance detection of cells and particles at such a particle sensing surface. Antibody coated, cell specific, “biosensor” surfaces rely on the cells landing on the surface and since, under gravity alone, the terminal velocity of 2 μm diameter cells is in the region of 2 × 10−7 m s−1 small cells often take minutes or hours to make contact with the surface. The lack of sedimentation is exacerbated by Brownian motion and in microfluidic channels the parabolic flow profile carries most cells along the central axis further reducing the probability of wall contacts. It is because small cells in passive systems do not quickly make contact with walls that antibody coated surfaces are rarely used for rapid cell detection. Ultrasound provides a solution; typically attracting cells to a surface in less than a second in sub-mm channels where antibodies can adhere specific cells. After the sound is turned off or switched to a repelling frequency, unattached cells flow away and the remaining cells can be counted.The method has some limitations: when the attraction surface is more than a few millimeters across cells always collect into clumps. The clump separation is near to half a wavelength in the fluid, the pattern is empirically predictable but its physical origin has not been fully investigated. Systems developed to use the phenomenon of attracting particles to a surface must accept this limitation of non-uniform cell coverage. Another limitation which must be appreciated is that currently the smallest cells which can be reliably manipulated with ultrasound standing waves are just over 1 μm diameter due to competing acoustic steaming drag forces. Therefore many, but not all, bacteria can be manipulated by this method.
Device design
Possibly the most important principle of cell manipulation in ultrasound standing waves is that the majority of cell types suspended in aqueous media move to pressure nodes in standing waves. To place a node on one wall of a chamber a water layer thickness less than a half wavelength is usually used and therefore the water layer cannot be simply considered to be an independently resonant element in the system bounded by non moving walls where pressure antinodes are located. For cells to move to a wall, the wall must be carefully designed so that a pressure maximum is not created at the fluid-wall interface. This feat of allowing the wave's velocity antinode to straddle an interface between two materials has however been achieved by several different methods, successful solutions to the problem are illustrated in Table 1 and the properties of each attractor wall are briefly described below (further details are given in ref. 24). With the exception of the first method, all have used a water thickness of less than a ½ wavelength.| Particle attraction walls in experimentally proven systems. (Simplified cross section views) | |||
|---|---|---|---|
| A |

|
E |

|
| Deep channel evanescent wave produced by a flexural plate wave on a 3 μm membrane attractor,53,54 black = SAW type drive electrodes | Semi-flexural wave with a vertical attractor wall59 | ||
| B |

|
F |

|
| ½ wave attractor24,48,56,63 | A rigid particle collection wall61,62 | ||
| C |

|
G |

|
| Absorbing attractor layer57 | Flexural wave and a thin attractor layer52 | ||
| D |

|
||
| Thin attractor layer24,58 | |||
B A half wavelength attractor wall.24,48,56,63 Driving the chamber at the resonance for the attractor wall produces a pressure node at the liquid interface to attract cells. This active pressure release method was demonstrated 10 years ago and has been developed further but it should be noted that the operating frequency is very narrow because of the need for simultaneous resonances in the attractor wall, the PZT and their combination with the water. This has encouraged the search for less demanding alternatives.
C A thick polymer attractor wall. 57 Making use of the sound absorbing properties of polymers, a thick absorbing layer rather than a resonant attractor wall allows some travelling wave components which displace cells towards the, liquid–attractor wall interface. To date this system has been designed for manipulating cells a short distance from the wall, some design alterations would be needed to adapt it to become an attractor wall. The system is transparent and the viewing direction is conveniently parallel to the node.
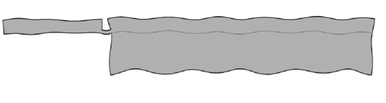 | ||
| Fig. 14 Model of the design by Glynne-Jones et al.59 illustrates that many wave modes can be used in a single system: in the thicker region on the right a symmetric (compression) wave has formed, in the thinner region on the left an anti-symmetric (flexural plate) wave is present. | ||
D A thin attractor wall 24,58like an air interface displacement occurs at the surface. The result is the same as with a resonating half wavelength wall however without the need for resonance it operates over a wider frequency range than the half wavelength wall.
E Introducing the sound from the side,59 introduces new engineering possibilities. A model of this system shown in Fig. 14 illustrates the ability of a system to have both symmetric and anti-symmetric waves in one system.
F A rigid horizontal base, vibrations introduced to the fluid by a non-evanescent flexural plate.60,61 Vertical nodes form and cells sediment down to the base.
G A thin membrane driven by a thicker flexural plate wave,52 this is a combination of the thin wall (D) and the sideways introduction of sound (E). The end-on-drive creates a system where the PZT sound source has only minimal coupling to the chamber, avoiding many interference problems, which always occur when two surfaces are bonded.
1D models have been used to develop some of the systems A–G, these of course cannot be used to explain the clumping patterns observed across the surface (Fig. 12 and 13) and further model development is needed if we are to control the clump locations. This author believes that evanescent acoustic waves may be involved in the attraction of particles towards the walls and the formation of the clump patterns, evanescent waves are certainly possible for the membrane walls: Oberti60 has calculated that at MHz frequencies wavelengths suitable for evanescent fields to occur can be produced when the wall thickness is less than 300 μm which is thicker than membrane walls currently used.
Immuno-based selective cell capture and detection by light
When the attracting wall of the chamber is coated with an antibody, cells or particles with the antigen on their surface become attached when they make contact. After switching the sound off, cells without the antigen are easily washed away. To quantify the number of attached cells the chamber can be taken apart and the attraction plate examined under a microscope.48 This method is cumbersome and the mechanical disturbance risks losing many positive binding events. Here we describe a method to achieve rapid cell identification.The arrival of cells at a surface can be continuously observed with a microscope, but this does not distinguish between cells at a surface and those near a surface. The distinction becomes important when determining whether a cell is bound to an antibody on the surface or is floating just above it. To identify the contact condition evanescent light guides have been integrated with the acoustics. In an evanescent light, the electrical and magnetic components are separated,62 this occurs in waveguides at the angle for total internal reflection where some of the electronic component propagates a very short distance into the adjacent media. Particles on the surface disturb the propagation, destroying the evanescence and displacing light out of the waveguide. The scattered light is easily detectable against a dark background, only particle volumes principally within approximately 1/3 of the light's wavelength from the guide are normally illuminated. The distance can be extended by placing a metal layer on one side of the guide giving normal transmission and reflection on one side which increases the wave component separation on the TIR face, this is known as a metal clad leaky wave (MCLW).62 Laser light can be introduced at the resonant angle to the waveguide through a prism and index matching fluid. BG spores attracted to a surface have been detected with the MCLW guide on a glass microscope slide surface at a concentration down to 103 ml−1.63 Similar results have been obtained by Glynne-Jones et al.56 using potassium ion waveguides whose field penetrates only 100 nm into the fluid, they have also had success by introducing light simply through the roughened edge of a microscope slide which is clearly not a single mode system.
The combination of three technologies; ultrasound, to attract cells to walls; antibodies, to selectively retain particular cell types; and evanescent fields for quantifying the attachments even at low concentrations, has the potential to provide a very rapid cell detection tool. However, this is not yet a commercial system primarily because when a half wavelength attractor wall is used, the narrow band of operating frequencies for the ultrasonic attraction is too highly dependent on temperature and fluid properties for non-specialist use. Recent developments52,58 of plastic and membrane systems have much broader operating frequency ranges and these are likely to be developed into easily used cell detection systems.
The next stage of developments
The three way combination of attracting particles to a surface with ultrasonic standing waves, antibody coated surfaces and evanescent light detection, is a strong contender for the development of label free rapid cell characterization to monitor environmental or medical samples. However, since antibodies provide the selectivity and they have a short life this has taken the developments towards very low cost disposable units,24,52,56 an example of the disposable section is shown in Fig. 15. In the future we expect to see this extended to multi-antibody arrays and controlled cell movement between regions. | ||
| Fig. 15 Very low cost chambers have been made52 using the vacuum forming techniques (also used for low cost blister packaging). Driven by pressing, with a clip, against a transducer, as shown in Table 1G. | ||
V. Conclusions
Ultrasonic standing wave technology for the manipulation of particles and cells has been shown to be suitable for enhancing immunoassays and particle/cell sensors. For immunoassays, the method has been applied to bead-based agglutination assays and bead-based fluorescence assays. Both these assays have been investigated in the homogeneous (wash-free) format. The simplest and most straightforward method is to apply ultrasonic enhancement to latex agglutination tests. Such tests can be improved in speed (readout within a few minutes) and the sensitivity can be improved from the >nM-range to the 100 pM-range. If higher sensitivity is needed, the bead-based fluorescence assay is more suitable. The reason is that the unenhanced format has a typical detection limit in the low pM-range. With ultrasonic enhancement, the sensitivity of the assay was demonstrated to be in the 10–100 fM-range. This is an impressive sensitivity if we take into account that the assay was operated in the homogeneous (wash-free) format. In future, a full implementation of the methods into the lab-on-a-chip format would make them very attractive. It would also be interesting to investigate whether combined acoustic streaming64 and acoustic trapping could be used for enhancing the binding reaction of the fluorescence assay.Another field where ultrasound has been used is to attach (and detach) particles or cells onto a sensor surface. Here, the most mature application is to use ultrasound for enhancing vibrational spectroscopy sensing. This has been demonstrated by using mid-infrared absorption spectroscopy for retrieving molecular-specific information of a certain sample, e.g. for on-line/in-line and real-time monitoring of an in vivo cell suspension. In particular, the use of surface-sensitive optical detection methods based on, e.g., evanescent field and attenuated total reflection technologies show great promise and would be very suitable for integration into the lab-on-a-chip format. Still, as shown in Table 1, designing devices for driving cells or particles denser than the fluid onto a solid surface by ultrasound is not trivial and is also more complicated than designing devices for standard acoustophoretic operation (where particles are driven to pressure nodes in the bulk fluid). Thus, more fundamental investigations are needed and should be combined with modeling in order to verify experimental results and to push forward the development of the promising technique of using ultrasound for enhancing immunoassays and surface-based particle and cell sensors.
References
- M. I. Mohammed and M. P. Y. Desmulliez, Lab Chip, 2011, 11, 569–595 RSC.
- A. Lenshof, M. Evander, T. Laurell and J. Nilsson, Lab Chip, 2012, 12, 684–695 RSC.
- H. Bruus, Lab Chip, 2012, 12, 1014–1021 RSC.
- R. W. Ellis and M. A. Sobanski, J. Med. Microbiol., 2000, 49, 853–859 CAS.
- M. Wiklund and H. M. Hertz, Lab Chip, 2006, 6, 1279–1292 RSC.
- J. M. Singer and C. M. Plotz, Am. J. Med., 1956, 21, 888–892 CrossRef.
- D. Stollar, Can. Med. Assoc. J., 1960, 83, 950–954 Search PubMed.
- J. A. Molina-Bolívar and F. Galisteo-González, J. Macromol. Sci., Part C, 2005, 45, 59–98 Search PubMed.
- W. J. Litchfield, A. R. Craig, W. A. Frey, C. C. Leflar, C. E. Looney and M. A. Luddy, Clin. Chem., 1984, 30, 1489–1493 CAS.
- W. H. Kapmeyer, W. H. Pauly and P. Tuengler, J. Clin. Lab. Anal., 1988, 2, 76–83 CrossRef.
- P. L. Masson, J. Pharm. Biomed. Anal., 1987, 5, 113–117 CrossRef CAS.
- M. Wiklund, O. Nord, R. Gothäll, A. V. Chernyshev, P.-Å. Nygren and H. M. Hertz, Anal. Biochem., 2005, 338, 90–101 CrossRef CAS.
- M. A. Grundy, W. E. Bolek, W. T. Coakley and E. Benes, J. Immunol. Methods, 1993, 165, 47–57 CrossRef CAS.
- M. A. Grundy, K. Moore and W. T. Coakley, J. Immunol. Methods, 1994, 176, 169–177 CrossRef CAS.
- R. J. Porritt, J. L. Mercer and R. Munro, Pathology, 2003, 35, 61–64 CrossRef CAS.
- M. A. Sobanski, R. A. Barnes and W. T. Coakley, Detection of Meningococcal Antigen by Latex Agglutination, In: Meningococcal Disease, Methods in Molecular Medicine, 2001, 67, 41–59 Search PubMed.
- M. Z. Von Smoluchowski, Phys. Chem., 1917, 92, 129–168 CAS.
- I. V. Surovtsev, M. A. Yurkin, A. N. Shvalov, V. M. Nekrasov, G. F. Sivolobov, A. A. Grazhdantseva, V. P. Maltsev and A. V. Chernyshev, Colloids Surf., B, 2003, 32, 245–255 CrossRef CAS.
- J. P. Nolan and L. A. Sklar, Trends Biotechnol., 2002, 20, 9–12 CrossRef CAS.
- M. Wiklund, M. Tirri, J. Toivonen, P. Hänninen and H. M. Hertz, J. Appl. Phys., 2004, 96, 1242–1248 CrossRef CAS.
- P. Hänninen, A. Soini, N. Meltola, J. Soini, J. Soukka and E. Soini, Nat. Biotechnol., 2000, 18, 548–550 CrossRef CAS.
- R. P. Ekins, J. Pharm. Biomed. Anal., 1989, 7, 155–168 CrossRef CAS.
- R. Ekins and F. Chu, Ann. Biol. Clin., 1992, 50, 337–353 Search PubMed.
- P. Glynne-Jones, R. J. Boltryk and M. Hill, Lab Chip, 2012, 12, 1417–1426 RSC.
- J. Chalmers and P. Griffiths, Handbook of vibrational spectroscopy, John Wiley & Sons Inc., New York, Jan. 2002 Search PubMed.
- L. A. Lyon, C. D. Keating, A. P. Fox, B. E. Baker, L. He, S. R. Nicewarner, S. P. Mulvaney and M. J. Natan, Anal. Chem., 1998, 70, 341–362 CrossRef.
- J. R. Baena, J. R. Baena, B. Lendl and B. Lendl, Curr. Opin. Chem. Biol., 2004, 8, 534–539 CrossRef CAS.
- R. Swain and M. Stevens, Biochem. Soc. Trans, 2007, 35, 544–549 CrossRef CAS.
- N. Leopold, M. Haberkorn, T. Laurell, J. Nilsson, J. R. Baena, J. Frank and B. Lendl, Anal. Chem., 2003, 75, 2166–2171 CrossRef CAS.
- L. Puskar, L. Puskar, R. Tuckermann, R. Tuckermann, T. Frosch, T. Frosch, J. Popp, J. Popp, V. Ly, V. Ly, D. McNaughton, D. McNaughton, B. R. Wood and B. R. Wood, Lab Chip, 2007, 7, 1125–1131 RSC.
- S. Radel, J. Schnöller, A. Dominguez, B. Lendl, M. Gröschl and E. Benes, Elektrotech. Informationstech., 2008, 125, 82–85 Search PubMed.
- M. Ruedas-Rama, A. Dominguez-Vidal, S. Radel and B. Lendl, Anal. Chem., 2007, 79, 7853–7857 CrossRef CAS.
- M. J. Ayora Cañada, A. Ruiz Medina, J. Frank and B. Lendl, Analyst, 2002, 127, 1365–1369 RSC.
- P. Harms, Y. Kostov and G. Rao, Curr. Opin. Biotechnol., 2002, 13, 124–127 CrossRef CAS.
- L. Olsson, U. Schulze and J. Nielsen, TrAC, Trends Anal. Chem., 1998, 17, 88–95 CrossRef CAS.
- Aldrich Library of FT-IR Spectra, 2nd Ed., 1997, Sigma-Aldrich, Milwaukee, WI, ISBN-13 978-094163339-0 Search PubMed.
- N. Harrick, Internal Reflection Spectroscopy, John Wiley & Sons Inc., New York, 1967 Search PubMed.
- J. Bertie and Z. Lan, Appl. Spectrosc., 1996, 50, 1047–1057 CAS.
- G. Jarute, A. Kainz, G. Schroll, J. Baena and B. Lendl, Anal. Chem., 2004, 76, 6353–6358 CrossRef CAS.
- J. Schnöller and B. Lendl, Proc. of IEEE Sensors, 2004, 2, 742–745 Search PubMed.
- K. Merritt, V. Hitchins and S. Brown, J. Biomed. Mater. Res., 2000, 53, 131–136 Search PubMed.
- S. Radel, J. Schnöller, B. Lendl, M. Gröschl and E. Benes, Elektrotech. Informationstech., 2008, 125, 76–81 Search PubMed.
- X. Chen and P. Stewart, Water Res., 2000, 34, 4229–4233 Search PubMed.
- B. Meyer, Int. Biodeterior. Biodegrad., 2003, 51, 249–253 Search PubMed.
- S. Radel, J. Schnöller, M. Gröschl, E. Benes and B. Lendl, IEEE Sens. J., 2010, 10, 1615–1622 Search PubMed.
- D. J. Pollard, R. Buccino, N. C. Connors, T. F. Kirschner, R. C. Olewinski, K. Saini and P. M. Salmon, Bioprocess Biosyst. Eng., 2001, 24, 13–24 Search PubMed.
- D. L. Doak and J. A. Phillips, Biotechnol. Prog., 1999, 15, 529–539 CrossRef CAS.
- J. J. Hawkes, M. J. Long, W. T. Coakley and M. B. McDonnell, Biosens. Bioelectron., 2004, 19, 1021–1028 CrossRef CAS.
- P. Glynne-Jones, R. J. Boltryk, M. Hill, F. Zhang, L. Dong, J. Wilkinson, T. Melvin, N. R Harris and T. Brown, Anal. Sci., 2009, 25, 285–291 CrossRef CAS.
- S. Radel, M. Brandstetter and B. Lendl, Ultrasonics, 2010, 50, 240–246 CrossRef CAS.
- S. Radel, M. Brandstetter, C. Koch and L. Bernhard, Proc. of 1st EAA - EuroRegio, Ljubljana, 2010, p. 8 Search PubMed.
- J. J. Hawkes, A. Cabañas Sorando, N. J. Goddard, P. R Fielden, S. Mohr, B. Bastani and M. B. Mcdonnell, Presented at ICU Sept., 2011, Gdańsk.
- R. M. White, Faraday Discuss., 1997, 107, 1–13 RSC.
- J. P. Black, R. M. White and J. W. Grate, Proc. of 2002 IEEE Ultrason. Symp., 2002, 475–479 Search PubMed.
- E. G. Williams, Fourier Acoustic: Sound radiation and nearfield acoustical holography, 1999, Academic Press, Cambridge, United Kingdom Search PubMed.
- P. Glynne-Jones, R. J. Boltryk, M. Hill, F. Zhang, L. Q. Dong, J. S. Wilkinson, T. Melvin, N. R. Harris and T. Brown, Anal. Sci., 2009, 25, 285–291 CrossRef CAS.
- I. Gonzalez, L. J. Fernandez, T. E. Gomez, J. Berganzo, J. L. Soto and A. Carrato, Sens. Actuators, B, 2010, 144, 310–317 CrossRef.
- P. Glynne-Jones, R. J. Boltryk, M. Hill, N. R. Harris and P. Baclet, J. Acoust. Soc. Am., 2009, 126, EL75–EL79 CrossRef CAS.
- P. Glynne-Jones, M. Hill, N. R.Harris, R. J. Townsend and S. Ravula, Proc. of Acoustics 08, Paris, 2008, 5989–5993 Search PubMed.
- S. Oberti, A. Neild and J. Dual, J. Acoust. Soc. Am., 2007, 121, 778–785 CrossRef CAS.
- S. Oberti, PhD Thesis, ETH, Zurich, 2009.
- N. Skivesen, PhD Thesis, Risø National Laboratory, Roskilde, Denmark, 2005.
- M. Zourob, J. J. Hawkes, W. T. Coakley, B. J. Treves Brown, P. Fielden, M. B. McDonnell and N. J. Goddard, Anal. Chem., 2005, 77, 6163–6168 Search PubMed.
- M. Wiklund, R. Green and M. Ohlin, Lab Chip, 2012, 12, 2438–2451 RSC.
| This journal is © The Royal Society of Chemistry 2013 |