Vault nanoparticles engineered with the protein transduction domain, TAT48, enhances cellular uptake
Jian Yanga, Aswin Srinivasanb, Yang Suna, Jan Mrazeka, Zhanyong Shuc, Valerie A. Kickhoefera and Leonard H. Rome*ad
aDepartment of Biological Chemistry, David Geffen School of Medicine at UCLA, Los Angeles, California 90095, USA
bDepartment of Molecular Cellular Developmental Biology, David Geffen School of Medicine at UCLA, Los Angeles, California 90095, USA
cSenior Scientist of Protein Science, Abraxis BioScience (Note: Abraxis BioScience and its subsidiaries have become a wholly-owned subsidiary of Celgene Corporation)
dCalifornia NanoSystems Institute at UCLA, Los Angeles, California 90095, USA
First published on 12th July 2012
Abstract
Vaults are naturally-occurring ribonucleoprotein particles found in nearly all eukaryotic cells. They were named for their morphological resemblance to the vaulted ceilings of gothic cathedrals. These ubiquitous nanoparticles are quite abundant with 104–106 copies found in the cytoplasm depending on cell type. The structural shell of the particle can self-assemble from 78 copies of a single protein, the major vault protein. This finding has allowed vaults to be bioengineered, resulting in a variety of new functions and capabilities directed toward overcoming many limitations posed by current gene and drug delivery systems. In this study, we demonstrate that recombinant vaults, with the addition of a cell penetration peptide, TAT, can be rapidly delivered to cells in vitro with significantly elevated binding and uptake efficiency. This TAT-vault nanoparticle could be a valuable tool for improving the retention and penetration of therapeutic drugs at tumor sites.
Insight, innovation, integrationBioengineered vaults are the only nanotechnology based on a naturally-occurring nanoparticle found in most eukaryotic cells. A therapeutic delivery platform based on the vault nanoparticle can overcome safety concerns such as toxicity and environmental impact. Advantages of this technology include: multifunctionality, uniformity, monodispersity and biocompatibility. Systemic delivery of nanoparticles can be an inefficient process as particles often don't accumulate in the tissue of interest. Consequently other delivery methods including direct tumoral injection and inhalation have been developed. The current study was initiated to design a vault nanoparticle with enhanced retention and penetration at the site of solid tumors following direct intra-tumoral injection. |
Introduction
Cancer is the second leading cause of death worldwide, with millions of newly diagnosed cases each year.1 It has been estimated that over half a million cancer related deaths occurred in the United States in 2011.2 Although the first line of treatment for most cancers is chemotherapy, radiation therapy, and/or surgery, newer and novel options are being developed every year.3 The field of nanomedicine, which integrates nanotechnology into the design of therapeutic drugs, is a discipline that has been rapidly progressing to solve many limitations faced by conventional drugs.4 Nanomedicine is poised to attack persistent problems with conventional drugs, such as non-specific targeting, lack of water solubility, poor oral bio-availability, delivery to intracellular sites of action and degradation during circulation.5,6 Novel nanoparticles such as liposomes, dendrimers, polymeric micelles and mesoporis silica have been developed as drug delivery systems to address many of these problems.7,8Over the past 50 years, chemotherapeutic drugs have been primarily delivered intravenously. The fate of these drugs is dependent on a variety of factors that depend on their chemical properties. The circulatory system will disperse drugs systemically where they are usually excreted into the urine, however, their wide distribution can result in non-specific “off-target” tissue damage. With the rapid advance of biomedical tools with precision guided technologies, new methods of delivering drugs directly to the site of the cancer (local treatment), rather than systematically, have been developed and are gaining traction. For example, recent clinical trials on patients with glioblastoma multiforme (GBM) have shown that the injection of the chemotherapeutic drug carboplatin by convection-enhanced delivery (CED) directly into the peritumoral region provoked a significant GBM cell kill at concentrations that are not toxic to normal brain.9,10 A clinical trial on patients with ductal carcinoma in situ breast cancer, found that injection of a low dose of doxorubicin directly into their mammary ducts resulted in tumors less than half the size of those receiving the drug intravenously.11 In a recent study, doxorubicin-loaded nanoparticles (NPs) were delivered to the lungs of animals with non-small cell lung carcinoma by inhalation.12 The results showed a highly significant improvement in survival compared to all control groups with lower side effects.12 Our laboratory has engineered a vault nanoparticle that packages a chemokine, CCL21.13 The CCL21-vault was shown to retain chemoattractant activity, and using an animal model for lung cancer, intratumoral injection of recombinant CCL21-vaults led to infiltration of activated dendritic cells and Th1 cytokine expressing lymphocytes in the tumor, that significantly altered the cytokine milieu in the tumor and induced potent antitumor activity.13 Together, these newer methods of delivering drugs locally to various organs appear to be an effective way of treating cancers while avoiding the intravenous systematic route.
Vaults are the largest cytoplasmic ribonucleoprotein particle (RNP) found in eukaryotic cells, ranging from 104 to 106 particles per cell.14 These highly conserved nanoparticles exhibit a distinct barrel shaped (∼40 × 40 × 70 nm) structure with an estimated molecular mass of 13 MDa.14 Interestingly, native vaults are formed from multiple copies of just three proteins (major vault protein (MVP), VPARP, and TEP1) and a small untranslated RNA termed vRNA.15–20 A definitive vault function has yet to be elucidated, although numerous putative functions have been proposed including roles in intracellular transport, multifaceted signaling pathways, innate immunity, and multidrug resistance.21 In 2001, we demonstrated that the expression of MVP alone in insect cells resulted in the formation of fully assembled recombinant vaults that were morphologically indistinguishable from that of native endogenous vaults.22 As insects are one of the few eukaryotes lacking vaults, these recombinant particles formed in the absence of the other vault components (VPARP, TEP1 and vRNA) and thus were essentially hollow capsules. Three strategies have been utilized to engineer recombinant vaults (Fig. 1A).23 In the first, additional amino acids have been added at the N-termini of MVP which results in these moieties being located on the interior surface of the engineered vault particles at the waist. Second, additional amino acids have been added at the C-termini of MVP which results in particles with these amino acids located on the exterior surface of the particles at the end of the vault caps. Third, exogenous proteins fused to a protein domain derived from VPARP (aa 1563–1724 termed the INT domain for interaction) results in a protein that, when combined with assembled vaults, packages inside the particles in two rings of density above and below the waist of the vault.23 A number of studies have demonstrated that recombinant vaults are a dynamic structure where they can separate at the waist and are therefore capable of exchanging their contents with the cellular milieu.24–27
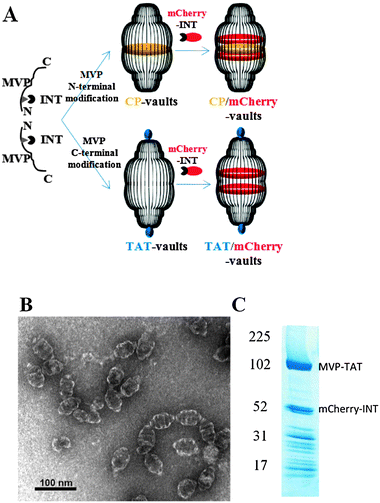 | ||
| Fig. 1 Preparation of recombinant TAT vaults. A. A schematic representation of a TAT-vault and a CP-vault packaged with mCherry-INT fusion proteins. CP peptides (MAGCGCPCGCGA, in yellow) were covalently fused to the N-terminus of the major vault protein and, following expression and self assembly, they are displayed on the inside of recombinant vault nanoparticles. TAT peptides (GRKKRRQRRRAHQ, in blue) were covalently fused to the C-terminus of the major vault protein and, following expression and self assembly, they are displayed on the outside of recombinant vault nanoparticles. mCherry-INT fusion proteins are non-covalently bind to the INT binding side located on the inside of fully assembled vault nanoparticles. B. TEM image showing the negatively stained purified TAT vaults. C. Coomassie stain of purified recombinant TAT vaults containing mCherry-INT fusion proteins. The MVP-TAT (∼100 kDa) and mCherry-INT (∼45 kDa) proteins are indicated on the right and the positions of the molecular weight standards are indicated at the left. | ||
In an effort to create a vault-based drug delivery system for direct, non-systemic applications in cancer therapy, we engineered a cell penetrating peptide (CPP) onto the exterior surface of vaults. CPPs have emerged as a valuable class of short peptide sequences that have the capacity to deliver macromolecular cargos that can be 100-fold higher in molecular weight than the peptides alone.28,29 In the present study, a 13 amino acid peptide (GRKKRRQRRRAHQ), derived from a truncated version of the HIV1 TAT protein,30,31 was bioengineered onto the C-termini of MVP, resulting in localization of this peptide at the end of the vault caps (Fig. 1). Multiple techniques were used to demonstrate that vaults expressing surface TAT peptides (TAT-vaults) showed increased binding to a wide variety of cell types and these bound vaults were internalized at higher efficiencies than vaults lacking the CPPs.
2 Materials and methods
2.1 Construction of recombinant TAT Plasmid
The 13 amino acid sequence of the TAT tag GRKKRRQRRRAHQ was fused to the C-terminus of rat MVP cDNA (rMVP). A reverse primer encoding this peptide, TAT-KpnI-5′-GCCGGTACCTTACTGATGAGCTCTTCGTCGCTGTCTCCGCTTCTTCCTGCCCTTCTGTGCTGG-3′ was synthesized. All primers used in this study were purchased from Invitrogen. The 3′ end of MVP was PCR amplified using a forward primer MVP1812-GCCTCTGTCACCTTTGATGAC, and the TAT-KpnI reverse primer, and the rMVP as the template. The PCR product (containing the TAT sequence fused to the 3′end of the MVP cDNA) was purified on a Qiagen column, digested with XhoI and KpnI, and ligated to XhoI/KpnI digested rat MVP in pFastBac to form MVP-TAT pFastBac. The construct was confirmed by DNA sequence analysis carried out by a commercial sequencing company (Laragen, Los Angeles CA).2.2 Expression and purification of recombinant TAT-vaults and control vaults
Recombinant baculovirus encoding rMVP-TAT was generated according to the Bac-to-Bac protocol (Invitrogen). Approximately 1 gram of Sf9 cells containing rMVP-TAT and 1 gram of Sf9 cells containing mCherry-INT were mixed together and suspended in 10 ml of lysis buffer on ice containing 50 mM Tris-HCl, pH 7.4, 75 mM NaCl, 0.75 mM MgCl2, 1% Triton, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride (PMSF), and a protease inhibitor mixture (2 mg ml−1 aprotinin, 0.5 mM benzamidine, 2 mg ml−1 chymostatin, 5 mM leupeptin, 5 mM pepstatin), incubated on ice for 60 min. Unbroken cells, organelles, and membranes were pelleted by centrifugation at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g (S20) for 20 min at 4 °C. The clear red S20 supernatant was then filtered through a 0.2 μm Acrodisc 25 mm syringe filter (Pall Corporation) and loaded onto a 1 ml Fractogel EMD SO3 column (Strong Cation Exchange, EMD Millipore) pre-equilibrated with a cold buffer solution containing 50 mM Tris-HCl, pH 7.4, 500 mM NaCl. The column was washed with 5 ml of the same cold buffer solution containing 50 mM Tris-HCl, pH 7.4, 500 mM NaCl, and another 5 ml of the cold buffer solution containing 50 mM Tris-HCl, pH 7.4, 1000 mM NaCl. TAT-vaults packaged with mCherry-INT were then eluted from the column using a cold 50 mM Tris-HCl, pH 7.4 buffer containing 2 M NaCl. The protein concentration of the purified TAT-vault preparation was determined using the BCA assay (Pierce), and their purity was analyzed by fractionating on SDS-PAGE gel followed by staining with Coomassie. TAT-vaults packaged with mCherry-INT (TAT/mCherry vaults) were then applied to carbon grids, stained with uranyl acetate, and visualized by transmission electron microscopy (TEM) on a JEM1200-EX microscope in the CNSI Electron Imaging Center for NanoMachines (EICN) as previously described.24
000 × g (S20) for 20 min at 4 °C. The clear red S20 supernatant was then filtered through a 0.2 μm Acrodisc 25 mm syringe filter (Pall Corporation) and loaded onto a 1 ml Fractogel EMD SO3 column (Strong Cation Exchange, EMD Millipore) pre-equilibrated with a cold buffer solution containing 50 mM Tris-HCl, pH 7.4, 500 mM NaCl. The column was washed with 5 ml of the same cold buffer solution containing 50 mM Tris-HCl, pH 7.4, 500 mM NaCl, and another 5 ml of the cold buffer solution containing 50 mM Tris-HCl, pH 7.4, 1000 mM NaCl. TAT-vaults packaged with mCherry-INT were then eluted from the column using a cold 50 mM Tris-HCl, pH 7.4 buffer containing 2 M NaCl. The protein concentration of the purified TAT-vault preparation was determined using the BCA assay (Pierce), and their purity was analyzed by fractionating on SDS-PAGE gel followed by staining with Coomassie. TAT-vaults packaged with mCherry-INT (TAT/mCherry vaults) were then applied to carbon grids, stained with uranyl acetate, and visualized by transmission electron microscopy (TEM) on a JEM1200-EX microscope in the CNSI Electron Imaging Center for NanoMachines (EICN) as previously described.24Control recombinant vaults were produced as previously described using a MVP with a 12 aa cysteine rich peptide (CP) on the N terminus to increase stability.32 These vaults are referred to here as CP-vaults and when packaged with mCherry-INT they are referred to as CP/mCherry vaults.
2.3 Western and confocal microscopy analysis of TAT/mCherry vault uptake into cells
HeLa, Raw264.7, HEK, and U2OS cells were grown and maintained in DMEM supplemented with 10% fetal bovine serum (henceforth referred to as complete DMEM). Prior to addition of TAT/mCherry vaults, the cells (5 × 105 cells/well) were seeded onto 6-well culture plates (Corning Incorporated) for western analysis, or 35 mm glass bottom culture dishes (MatTek Corporation) for confocal microscopic studies, and incubated for 16 h with 2 ml complete DMEM at 37 °C in 5% CO2. Variable amounts of TAT/mCherry vaults were added directly to the complete DMEM medium followed by incubating for various periods of time at either at 37 °C in 5% CO2 or at room temperature in air.Stock solutions of various inhibitors were made according to the manufacturer's instructions before being further diluted in the following assays at the indicated concentrations. Amiloride (Sigma), 5 mM in DMSO; chlorpromazine (Sigma), 28 mM in H2O; methyl-β-cyclodextrin (Sigma), 20 mM in H2O; heparin (Sigma), 6 mg ml−1 in H2O.
For Western blot analysis, the culture medium containing unbound TAT/mCherry vaults was removed, the cells were washed three times with cold PBS, the cells were lysed directly in SDS loading buffer. Denatured whole cell lysate was separated on 4–15% SDS-PAGE gels, transferred to Hybond membrane (Amersham) and probed with a polyclonal rabbit anti-MVP antibody that has been previously described.15,16
Live cell imaging experiments were performed using a Zeiss LSM5 Pascal confocal microscope (Carl Zeiss AG) equipped with a 543 nm HeNe laser (LASOS) for the excitation and a LP560 filter for the mCherry emission. Images were acquired from live cells with a 100 × oil-immersion objective (Zeiss Alpha Plan-Fluar 100 × 1.45) and analyzed with the PASCAL software. Scanning was performed at an image resolution of 1024 × 1024 or 512 × 512 pixels (1.60 μs/pixel), averaged 1 to 2 times using 80% 543 nm laser power. Images were further processed with Adobe Photoshop (Adobe Systems Incorporated).
3 Results and discussion
Recombinant vaults are versatile nanoparticles that are highly amenable for modification, as illustrated in Fig. 1A. Over the past ten years, we have been able to bioengineer recombinant vaults placing a number of different functional groups onto various locations for specific applications.13,23,24,27,33–41 Several studies have suggested that the vault structure is extremely stable, yet they are also able to interact with the cellular milieu.24–27,36,39,41 TAT is a 13 amino acid positively charged peptide tag derived from human immunodeficiency virus-1 (HIV-1). TAT has been shown in many studies to enhance trans-membrane transport across a broad range of cell types.30,31 In this study, we fused the TAT tag onto the C-termini of the structural protein of the vault, MVP, which ultimately results in display of these tags on the exterior surface of the fully assembled particles at the end of the caps (Fig. 1A). Although in the purified TAT-vaults there were ∼312 additional positively charged residues locally concentrated on the outside of the particle caps (∼39 MVP chains per half vault ×8 arginine and lysine residues per TAT peptide), these TAT-vaults were still robustly produced in baculovirus infected insect cells with no decreased yield compared to previously engineered recombinant vaults (data not shown). There were no apparent structural alterations in these TAT-vaults comparing to unmodified vaults when the particles were evaluated by negative stain TEM (Fig. 1B). Occasional end to end vault connections were seen and may have formed by charge-charge interactions from two adjacent TAT-vaults. These aggregates were perhaps mediated by negatively charged impurities derived from cell lysates and were not observed when 2 M NaCl was present (not shown).The fusion protein, mCherry-INT, has been commonly used in our laboratory to fluorescently label recombinant vaults and thus allow monitoring of their sub-cellular distribution.23,33,38 The excellent packaging of mCherry-INT fusion proteins in TAT-vaults, as indicated by the SDS-PAGE gel (Fig. 1C), indicated that the INT binding sites, located on the inside of TAT-vault nanoparticles, were not compromised as a result of the presence of the TAT tags on the outside of the vault caps, further supporting the structural stability of these nanoparticles.
The plasma membrane of cancer cells plays an important role in reducing the efficacy of anticancer drugs by preventing their entry into cells, where these molecules can interact with their intended cytoplasmic targets. In an effort to overcome this biophysical barrier, many therapeutic nanoparticles have been designed to take advantage of cell-penetrating peptides to enhance their cellular uptake.28,29 Unlike systems that randomly decorate the surface of nanoparticles with CPPs, recombinant TAT-vaults offer a unique, regular and reproducible structural alteration where TAT peptides are concentrated at specific locations on the vault surface. The uptake efficiencies of TAT-vaults was compared to control vaults in Hela cells (Fig. 2). Control CP/mCherry vaults that lack external cell penetrating tags were unable to penetrate the cellular membrane of Hela cells. In contrast, the TAT/mCherry vaults showed a significantly higher cell binding, presumably due to the presence of TAT tags on the outside of the particles (Fig. 2A, TAT/mCherry vaults). Confocal microscopic studies confirmed these results, where the red signal from mCherry was only observed in experiments where TAT/mCherry vaults were added to the Hela cells (Fig. 2B). Similar results were seen when U2OS cells (osteosarcoma) were examined (not shown). Western analysis (Fig. 2C) also confirmed a similar pattern of TAT-vault binding to both cell types.
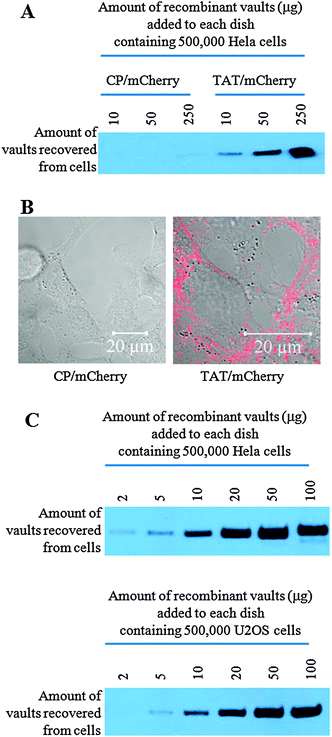 | ||
| Fig. 2 The uptake efficiency of TAT vaults is significantly higher than that of the control vaults in Hela cells. A. Western analysis shows an enhanced uptake of TAT vaults in Hela cells. The indicated amounts of CP/mCherry (control) or TAT/mCherry vaults were incubated with 500000 Hela cells for 16 h. Hela cells were then washed three times with the cold PBS solution. Whole cell lysates were separated on an SDS PAGE gel and probed with anti-MVP antibody. B. Confocal images of live Hela cells (500000 cells/dish) incubated with 10 μg of CP/mCherry (left) or TAT/mCherry (right) vaults for 16 h. The mCherry channel (in red) is overlaid on top of the bright field view. C. Western analysis confirming similar patterns of cell surface binding to both Hela and U2OS cells. A proportional correlation between the amount of TAT/mCherry vaults added to the dish and the amount of TAT/mCherry vaults recovered from Hela and U2OS cells was observed. | ||
Given the enhanced binding of the vault particles displaying TAT tags to cells, we next examined the time dependence for particle association. As shown in Fig. 3A, most of TAT/mCherry vaults were seen to rapidly associate with Hela cells within 2 h after their addition to the culture medium. The amount of recombinant vaults associated with Hela cells was not significantly different between 2 and 16 h based on Western blot analysis (Fig. 3A). However, observation of similarly prepared samples by confocal microscopy revealed significant differences in the subcellular distribution of TAT/mCherry vaults at these two times (Fig. 3B). At 2 h, most of the TAT/mCherry vaults were associated with the outer membrane, as indicated by a red ring in the confocal image (Fig. 3B, left). In comparison, the 16 h confocal image showed many patches of isolated red signals within the membrane boundary of the Hela cells. Upon extended incubation for 64 h after the TAT/mCherry vault addition, no clear red rings could be identified, suggesting all TAT/mCherry vaults that were initially associated with the membrane were internalized into the cytoplasm of the cells (Fig. 3C, Top). The same result was seen when a different cell line HEK (Human Embryonic Kidney) was examined (Fig. 3C, bottom).
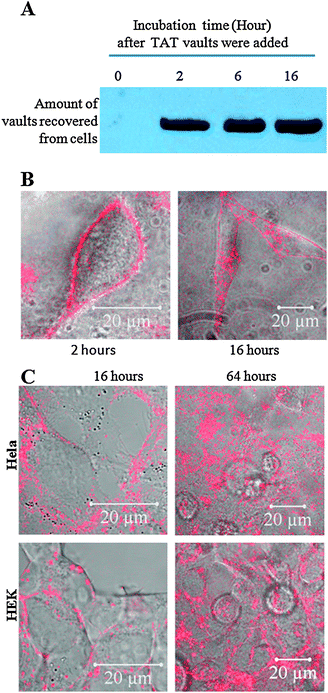 | ||
| Fig. 3 The uptake of TAT-vaults is a two-step process in Hela cells. A. Western analysis showing no significant increase of associated TAT-vaults with Hela cells from two to sixteen hours. TAT/mCherry vaults (10 μg) were added to a dish containing 500000 Hela cells for various amount of time as indicated above the image. After that, Hela cells were washed three times with cold PBS solution, separated on an SDS PAGE gel, and probed by anti-MVP antibody for MVP. B. Confocal images of live Hela cells (500000 cells/dish) incubated with 10 μg TAT/mCherry vaults for 2 (left) and 16 (right) hours. The mCherry channel (in red) is overlaid on top of the bright field view. C. Confocal images of live Hela and HEK cells (500000 cells/dish) incubated with 10 μg TAT/mCherry vaults for 16 (left) and 64 (right) hours. The mCherry channel (in red) is overlaid on top of the bright field view. | ||
Based on these results, the interaction of TAT/mCherry vaults with cells appears to be a two-step process. The first step is a fast, likely ionic, binding between the positively charge TAT tags of the recombinant vaults with the negatively charged cell surface components. While the second step is a relatively slow internalization, requiring hours for the bound TAT/mCherry vaults to be cleared from the plasma membrane possibly driven by normal membrane recycling.
To investigate the uptake mechanism of TAT/mCherry vaults into cells, we attempted to disrupt the presumptive ionic charge-driven rapid binding step by introduction of a highly negatively charged molecule, heparin. Heparin is a highly sulfated glycosaminoglycan with an extremely high negative charge density. Consistent with an ionic charge-driven process, the binding of TAT-vaults to the Hela cells was inhibited by heparin at a concentration of approximately 300 μg L−1, which is similar to the concentration required for heparin to inhibit viral infections (Fig. 4).42 These results may indicate that the TAT-vaults are interacting with negatively charged cell surface molecules such as phospholipids or sulfated proteoglycans e.g. heparin sulfate, a common surface polysaccharide that exists in all animal tissues. In addition to the heparin inhibition at high concentrations, there was a marked potentiation of TAT-vault binding at low heparin concentrations peaking at ∼30 μg L−1. This may be due to the prevention of TAT-vault aggregation by the highly charged polymer thus raising the effective concentration of TAT-vaults.
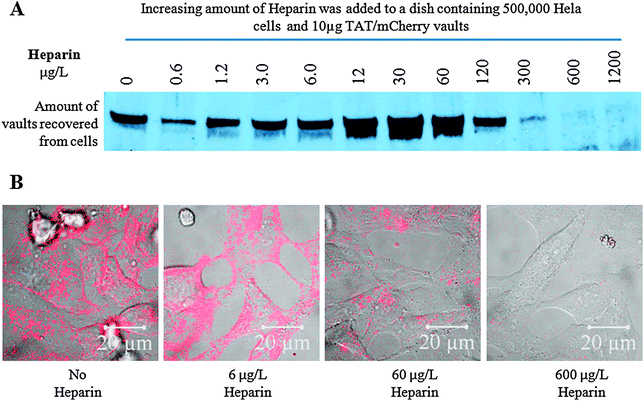 | ||
| Fig. 4 Western (A) and Confocal microscope (B) analysis showing that Heparin is able to block the TAT-vaults from entering the cells, possibly by disrupting the interaction between the positively charged TAT peptides and the negatively charged cell surface. Hela cells were incubated with fixed concentration of TAT/mCherry vaults and increasing concentrations of Heparin for 16 h. | ||
The mechanism by which TAT peptides adhere to and cross the plasma membrane of cells is currently a topic of considerable discussion in the literature, with varied findings being reported.28–31 To further examine the uptake mechanism of TAT-vaults, we investigated the effect of temperature on the uptake rate of the particles (Fig. 5A). As expected, higher temperatures resulted in faster association of the TAT-vaults with cells, likely due to a simple kinetic effect. In addition, we examined the effect of different uptake inhibitors (amiloride, chlorpromazine, and methy-β-cyclodextrin.) in an effort to determine the internalization mechanism of TAT-vaults (Fig. 5B and C). Amiloride is a known inhibitor of macropinocytosis, chloropromazine inhibits clathrin dependent endocytosis, and methyl-β-cyclodextrin blocks caveolae mediated endocytosis.43 Under all circumstances, we did not see any difference in binding of TAT-vaults in comparison to controls without inhibitors. These results tend to support the hypothesis that the TAT tag is binding the vault to the cell surface and the slow internalization may be due to membrane recycling.
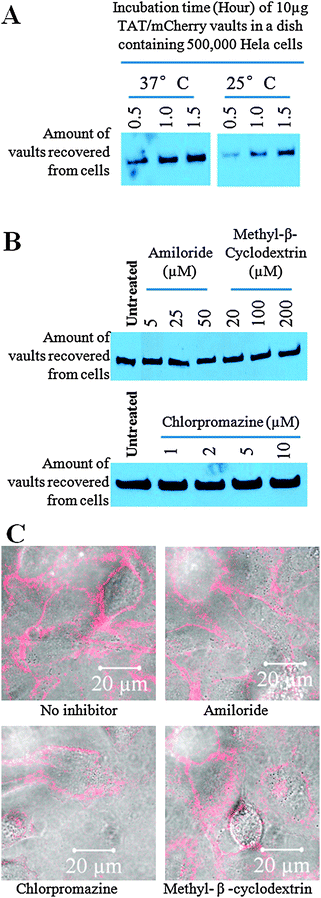 | ||
| Fig. 5 The uptake mechanism of TAT-vaults into Hela cell remains to be resolved. A. Western analysis shows a clear temperature dependence of the TAT-vaults. B. Western analysis did not reveal noticeable difference in the presence of various endocytosis inhibitors. C. Confocal microscopic analysis shows no difference in terms of the uptake pattern of TAT-vaults in Hela cells. | ||
To further test the cell specificity of the TAT-vault nanoparticles we examined binding in a variety of cell types: Raw 264.7 (Mouse leukaemic monocyte macrophage), HEK 293 cells (Human Embryonic Kidney), and U2OS (osteosarcoma). The binding of the control CP vaults (Fig. 6, left side) was highly dependent on the cell type. In contrast, the TAT-vaults were much less selective (Fig. 6, right side). The TAT-vaults could eventually cross the membrane in each of the cell types examined (results not shown).
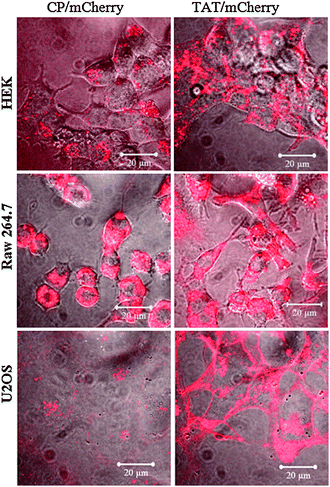 | ||
| Fig. 6 TAT-vaults are capable to interact with a many types of cells than unmodified CP-vaults. Various cells (500000 cells/dish) were incubated with 10 μg CP/mCherry (left) or TAT/mCherry (right) vaults for 16 h. The mCherry channel (in red) is overlaid on top of the bright field view. | ||
Following our successful development of the immune-activating CCL2-vault,13 we were interested in modifying the vault particle to increase its retention at the tumor site, following intra-tumoral delivery. The ability of the TAT-modified vaults to bind to a wide variety of cells predicts that this moiety may be able to potentiate the anti-tumor effects of the CCL21-vault and these experiments are a natural extension of the current studies. Furthermore, the cell-binding properties of the TAT-vault could also be an advantage for applications using intratumoral injection of vaults engineered to contain cytotoxic drugs. We recently described a vault engineered to contain a lipid nanodisc that could bind the lipophilic drugs amphotericin B and all trans retinoic acid.41 Addition of TAT to these nanodisc vaults could lead to an increase in the local concentration of drug at the tumor site following intra-tumoral injection.
4. Conclusions
Cancer is not a single disease, rather it is a broad group of various diseases, all involving unregulated cell growth, often leading to formation of malignant tumors that can invade nearby and distant parts of the body. While it is unlikely that there will ever be a unified therapy for all cancers, there is a possibility to achieve progress on certain types of cancers with careful designs. There are many examples where nanotechnology has been used to deliver therapeutic drugs non-intravenously. Bioengineered vaults are flexible nanoparticles with the capability to integrate a wide variety of functional groups without disrupting the structural integrity of the particle. Several advantages, such as multifunctionality, nanotoxicity, uniformity, and biocompatibility, make the vault a logical choice for therapeutic applications. In this paper, recombinant vaults expressing the TAT CPP have been produced and shown to bind to a wide variety of cell types. The TAT-vault cell interaction occurs in two steps, rapid binding followed by a slow uptake of particles that remain associated with cells for a prolonged period of time. The initial binding is a rapid ionic interaction between the positively charged TAT tags of the vault nanoparticles and the negatively charged cell surface. The TAT-vaults remain associated with the cell membrane for hours before finally being taken inside by a mechanism that is not well understood but might occur during normal membrane recycling. We see this interaction as a unique feature of the TAT-vault that has the potential to alter the bio-distribution profile of packaged drugs at the tumor sites. Our next goal is to test the effectiveness of this TAT-vault in animal models with therapeutic drugs packaged inside.Acknowledgements
This work was supported by the UC Discovery Grant Program in collaboration with our corporate sponsor, Abraxis Biosciences, Inc (Award# BIO07-10671) and by the Mather's Charitable Foundation (Grant# 04095186) and NIH/NIBIB Award R01 EB004553. We would like to thank Dr Kayvan Niazi and Dr Shahrooz Rabizadeh of Abraxis BioScience, for their technical expertise and scientific insights.References
- B. W. Stewart and P. Kleihues, World Cancer Report, World Health Organization Press, Geneva, 2003 Search PubMed.
- American Cancer Society, Cancer Facts & Figures, American Cancer Society, Atlanta, 2011 Search PubMed.
- C. M. Hu and L. Zhang, Therapeutic nanoparticles to combat cancer drug resistance, Curr. Drug Metab., 2009, 10(8), 836–41 CrossRef CAS.
- D. Peer, et al., Nanocarriers as an emerging platform for cancer therapy, Nat. Nanotechnol., 2007, 2(12), 751–760 CrossRef CAS.
- V. Wagner, et al., The emerging nanomedicine landscape, Nat. Biotechnol., 2006, 24(10), 1211–1217 CrossRef CAS.
- M. E. Davis, Z. Chen and D. M. Shin, Nanoparticle therapeutics: an emerging treatment modality for cancer, Nat. Rev. Drug Discovery, 2008, 7(9), 771–782 CrossRef CAS.
- A. Z. Wang, R. Langer and O. C. Farokhzad, Nanoparticle delivery of cancer drugs, Annu. Rev. Med., 2012, 63, 185–98 CrossRef CAS.
- M. Ferrari, Cancer nanotechnology: opportunities and challenges, Nat. Rev. Cancer, 2005, 5(3), 161–71 CrossRef CAS.
- E. White, et al., A phase I trial of carboplatin administered by convection-enhanced delivery to patients with recurrent/progressive glioblastoma multiforme, Contemporary Clinical Trials, 2012, 33(2), 320–31 CrossRef CAS.
- E. White, et al., An evaluation of the safety and feasibility of convection-enhanced delivery of carboplatin into the white matter as a potential treatment for high-grade glioma, J. Neuro-Oncol., 2012, 108(1), 77–88 CrossRef CAS.
- S. Murata, et al., Ductal access for prevention and therapy of mammary tumors, Cancer Res., 2006, 66(2), 638–45 CrossRef CAS.
- W. H. Roa, et al., Inhalable nanoparticles, a non-invasive approach to treat lung cancer in a mouse model, J. Controlled Release, 2011, 150(1), 49–55 CrossRef CAS.
- U. K. Kar, et al., Novel CCL21-Vault Nanocapsule Intratumoral Delivery Inhibits Lung Cancer Growth, PLoS One, 2011, 6(5), e18758 CAS.
- K. A. Suprenant, Vault ribonucleoprotein particles: sarcophagi, gondolas, or safety deposit boxes?, Biochemistry, 2002, 41(49), 14447–54 CrossRef CAS.
- V. A. Kickhoefer, et al., Vaults and telomerase share a common subunit, TEP1, J. Biol. Chem., 1999, 274(46), 32712–7 CrossRef CAS.
- V. A. Kickhoefer, et al., The 193-kD vault protein, VPARP, is a novel poly(ADP-ribose) polymerase, J. Cell Biol., 1999, 146(5), 917–28 CrossRef CAS.
- N. L. Kedersha, et al., Vaults. III. Vault ribonucleoprotein particles open into flower-like structures with octagonal symmetry, J. Cell Biol., 1991, 112(2), 225–35 CrossRef CAS.
- V. A. Kickhoefer, et al., Vault ribonucleoprotein particles from rat and bullfrog contain a related small RNA that is transcribed by RNA polymerase III, J. Biol. Chem., 1993, 268(11), 7868–73 CAS.
- V. A. Kickhoefer, et al., Vaults are up-regulated in multidrug-resistant cancer cell lines, J. Biol. Chem., 1998, 273(15), 8971–4 CrossRef CAS.
- A. van Zon, et al., Multiple human vault RNAs. Expression and association with the vault complexm, J. Biol. Chem., 2001, 276(40), 37715–21 CrossRef CAS.
- W. Berger, et al., Vaults and the major vault protein: novel roles in signal pathway regulation and immunity, Cell. Mol. Life Sci., 2009, 66(1), 43–61 CrossRef CAS.
- A. G. Stephen, et al., Assembly of vault-like particles in insect cells expressing only the major vault protein, J. Biol. Chem., 2001, 276(26), 23217–20 CrossRef CAS.
- M. Han, et al., Targeted Vault Nanoparticles Engineered with an Endosomolytic Peptide Deliver Biomolecules to the Cytoplasm, ACS Nano, 2011 Search PubMed.
- M. J. Poderycki, et al., The vault exterior shell is a dynamic structure that allows incorporation of vault-associated proteins into its interior, Biochemistry, 2006, 45(39), 12184–93 CrossRef CAS.
- L. E. Goldsmith, et al., Vault nanocapsule dissociation into halves triggered at low pH, Biochemistry, 2007, 46(10), 2865–75 CrossRef CAS.
- R. Esfandiary, et al., Structural stability of vault particles, J. Pharm. Sci., 2009, 98(4), 1376–86 CrossRef CAS.
- J. Yang, et al., Vaults are dynamically unconstrained cytoplasmic nanoparticles capable of half vault exchange, ACS Nano, 2010, 4(12), 7229–40 CrossRef CAS.
- K. M. Stewart, K. L. Horton and S. O. Kelley, Cell-penetrating peptides as delivery vehicles for biology and medicine, Org. Biomol. Chem., 2008, 6(13), 2242–2255 CAS.
- L. N. Patel, J. L. Zaro and W. C. Shen, Cell penetrating peptides: Intracellular pathways and pharmaceutical perspectives, Pharm. Res., 2007, 24(11), 1977–1992 CrossRef CAS.
- V. P. Torchilin, Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers, Adv. Drug Delivery Rev., 2008, 60(4–5), 548–558 CrossRef CAS.
- X. K. Zhang, X. Zhang and F. S. Wang, Intracellular transduction and potential of Tat PTD and its analogs: from basic drug delivery mechanism to application, Expert Opin. Drug Delivery, 2012, 9(4), 457–472 CrossRef CAS.
- Y. Mikyas, et al., Cryoelectron microscopy imaging of recombinant and tissue derived vaults: localization of the MVP N termini and VPARP, J. Mol. Biol., 2004, 344(1), 91–105 CrossRef CAS.
- V. A. Kickhoefer, et al., Engineering of vault nanocapsules with enzymatic and fluorescent properties, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(12), 4348–52 CrossRef CAS.
- D. H. Anderson, et al., Draft crystal structure of the vault shell at 9-angstrom resolution, PLoS Biol., 2007, 5(11), 2661–2670 CAS.
- B. C. Ng, et al., Encapsulation of semiconducting polymers in vault protein cages, Nano Lett., 2008, 8(10), 3503–9 CrossRef CAS.
- C. I. Champion, et al., A vault nanoparticle vaccine induces protective mucosal immunity, PLoS One, 2009, 4(4), e5409 Search PubMed.
- L. E. Goldsmith, et al., Utilization of a protein “shuttle” to load vault nanocapsules with gold probes and proteins, ACS Nano, 2009, 3(10), 3175–83 CrossRef CAS.
- V. A. Kickhoefer, et al., Targeting vault nanoparticles to specific cell surface receptors, ACS Nano, 2009, 3(1), 27–36 CrossRef CAS.
- C. Y. Lai, et al., Vault nanoparticles containing an adenovirus-derived membrane lytic protein facilitate toxin and gene transfer, ACS Nano, 2009, 3(3), 691–9 CrossRef CAS.
- Y. Xia, et al., Immobilization of recombinant vault nanoparticles on solid substratesm, ACS Nano, 2010, 4(3), 1417–24 CrossRef CAS.
- D. C. Buehler, et al., Vaults engineered for hydrophobic drug delivery, Small, 2011, 7(10), 1432–9 CrossRef CAS.
- Y. L. Lin, et al., Heparin inhibits dengue-2 virus infection of five human liver cell lines, Antiviral Res., 2002, 56(1), 93–96 CrossRef CAS.
- J. A. Gomez, et al., Cell-Penetrating Penta-Peptides (CPP5s): Measurement of Cell Entry and Protein-Transduction Activity, Pharmaceuticals (Basel), 2010, 3(12), 3594–3613 CAS.
| This journal is © The Royal Society of Chemistry 2013 |