Phagocytes mediate targeting of iron oxide nanoparticles to tumors for cancer therapy†
Seiko
Toraya-Brown‡
a,
Mee Rie
Sheen‡
a,
Jason R.
Baird
a,
Stephen
Barry
b,
Eugene
Demidenko
c,
Mary Jo
Turk
ad,
P. Jack
Hoopes
def,
Jose R.
Conejo-Garcia
g and
Steven
Fiering
*adh
aDepartment of Microbiology and Immunology, Geisel School of Medicine at Dartmouth, Hanover, NH 03755, USA. E-mail: Steve.Fiering@Dartmouth.edu
bAspen Medisys, LLC, MA 01752, USA
cDepartment of Community and Family Medicine, Geisel School of Medicine at Dartmouth, Hanover, NH 03755, USA
dNorris Cotton Cancer Center, Lebanon, NH 03756, USA
eDepartment of Surgery, Geisel School of Medicine at Dartmouth, Hanover, NH 03755, USA
fThayer School of Engineering, Dartmouth College, Hanover, NH 03755, USA
gTumor Microenvironment and Metastasis Program, the Wistar Institute, Philadelphia, PA 19104, USA
hDepartment of Genetics, Geisel School of Medicine at Dartmouth, Hanover, NH 03755, USA
First published on 10th August 2012
Abstract
Nanotechnology has great potential to produce novel therapeutic strategies that target malignant cells through the ability of nanoparticles to get access to and be ingested by living cells. However its specificity for accumulation in tumors, which is the key factor that determines its efficacy, has always been a challenge. Here we tested a novel strategy to target and treat ovarian cancer, a representative peritoneal cancer, using iron oxide nanoparticles (IONPs) and an alternating magnetic field (AMF). Peritoneal tumors in general are directly accessible to nanoparticles administered intraperitoneally (IP), as opposed to the more commonly attempted intravenous (IV) administration. In addition, tumor-associated immunosuppressive phagocytes, a predominant cell population in the tumor microenvironment of almost all solid tumors, and cells that are critical for tumor progression, are constantly recruited to the tumor, and therefore could possibly function to bring nanoparticles to tumors. Here we demonstrate that tumor-associated peritoneal phagocytes ingest and carry IONPs specifically to tumors and that these specifically delivered nanoparticles can damage tumor cells after IONP-mediated hyperthermia generated by AMF. This illustrates therapeutic possibilities of intraperitoneal (IP) injection of nanoparticles and subsequent ingestion by tumor-associated phagocytes, to directly impact tumors or stimulate antitumor immune responses. This approach could use IONPs combined with AMF as done here, or other nanoparticles with cytotoxic potential. Overall, the data presented here support IP injection of nanoparticles to utilize peritoneal phagocytes as a delivery vehicle in association with IONP-mediated hyperthermia as therapeutic strategies for ovarian and other peritoneal cancers.
Insight, innovation, integrationNanotechnology has considerable potential to contribute to novel and improved cancer therapy but efficacy generally depends on concentrating nanoparticles in tumors rather than normal tissues. We observed that tumor-associated phagocytes recruited by tumors mediate nanoparticle delivery into ovarian tumors after intraperitoneal injection of the nanoparticles. Therefore, engulfment of nanoparticles by phagocytes was utilized to increase nanoparticle accumulation into ovarian tumors in our study. Therapeutic potential of phagocyte-mediated IONPs delivery and associated hyperthermia by AMF to damage tumor cells without harming normal tissue was evaluated by histochemical assay for cell death. This work illustrates the potential of phagocytes to deliver nanoparticles to tumors where they may be used to directly kill tumor cells or stimulate antitumor immune responses in peritoneal cancers. |
Introduction
Primary tumors and relatively localized secondary tumors found at early stages can be treated with radiation or surgery and generally are not lethal if caught prior to metastasis. However as cancer progresses and multiple metastases occur, it is difficult to precisely locate all metastases and therefore removal by surgery or radiation therapy is impossible. Chemotherapy can act on tumors that have not been identified, but its lack of specificity to cancer cells causes severe side effects due to damage of normal tissues. There is an enormous demand for a method that enables targeting tumors specifically without knowing the exact location of the tumors.Cancer nanotechnology is a relatively new and growing field that aims to solve this specificity problem. One of the major strategies taken is to conjugate a chemotherapeutic drug or treatment material in a package of some sort on the size scale of 10–200 nanometers (nm), to molecules such as monoclonal antibodies that bind to proteins expressed specifically or preferentially on cancer cells, such as HER2.1 However, this strategy has been challenging for many reasons, of which the most difficult problem that hinders all such targeting regardless of the nanoparticle platform is having the nanoparticles preferentially located in the tumor rather than other normal tissues.2 When nanoparticles with or without antibody conjugates were administered intravenously (IV), the majority of the injection dose ends up in the spleen and liver where it is sequestered in phagocytes that reside in those organs.3,4 There are techniques like adding polyethylene glycol to the surface of the particle that increase the half-life of the particle by inhibiting uptake by phagocytes, but this generally has little impact on how much of the administered dose accumulates in the tumor.5
Many of the cancers with the highest mortality are largely confined, even at late stages, predominantly to the peritoneal region. Such cancers include ovarian, pancreatic, colon, liver, and stomach cancers. The peritoneal cavity is accessible by IP injection and it is possible that IP injection is a route of administration that improves the partitioning of therapeutic nanoparticles between the tumor and normal tissues.
ID8-Defb29/Vegf-a is an aggressive mouse metastatic ovarian cancer model,6 derived from the ID8 murine ovarian cancer cell line,7 which accurately recapitulates characteristics of metastatic human ovarian cancer in C57BL/6 mice. Four weeks post IP inoculation of ID8-Defb29/Vegf-a cells (i) tumor masses can be found all over the peritoneal cavity, (ii) ascites develops, and (iii) great numbers of tumor-associated immunosuppressive phagocytes are recruited to the ascites. Interestingly, those phagocytes have potential to be further recruited to the tumor and therefore are suggested to have a potential to function as a vehicle to carry material that they phagocytose in the peritoneal cavity to the tumor.8 Recruitment of phagocytes to the tumor microenvironment is a fundamental aspect of solid tumors in both mice and humans.9–11
One variety of nanoparticles with cancer therapy potential is IONPs that can be heated with an AMF.12 Therefore, our goal is to generate techniques to target IONPs to peritoneal tumors and develop the therapeutic potential of magnetic IONP-mediated hyperthermia induced by AMF for ovarian cancer and eventually for other peritoneal cancers. This study investigated the ability of IONPs with various coatings to partition selectively into peritoneal tumors and how that may affect the therapeutic possibilities. These results demonstrate that peritoneal phagocytes that are recruited and manipulated by tumors are powerful tools to carry nanoparticles to the tumor without requiring specific conjugation to targeting molecules. With further development, this method could be employed in treating a broad range of peritoneal cancers.
Materials and methods
Animals and tumor cell line
Female C57BL/6 mice were purchased from the National Cancer Institute. All animal procedures were performed in accordance with the Institutional Animal Care and Use Committee of Geisel Medical School at Dartmouth. Murine ovarian cancer cell line, ID8-Defb29/Vegf-a, was cultured in RPMI 1640 medium (Cellgro) containing 10% fetal bovine serum (HyClone), 1X penicillin/streptomycin (Gibco), and 2 mM L-glutamine (Gibco). Mice (6–8 week old) were injected IP with a total of 1.5 × 106 ID8-Defb29/Vegf-a cells to establish tumor masses in the peritoneal cavity. After approximately 4 weeks post tumor inoculation, mice were utilized for experiments.IONPs
BNF-starch coated IONPs with a diameter of 100 nm on average (starch-IONPs) were purchased from Micromod Partikeltechnologie GmbH, Germany. Other IONPs coated with dextran or mannan (dextran-IONPs and mannan-IONPs) with similar composition and size to starch-IONPs were synthesized by Aspen Medisys. All IONPs were analyzed for iron content by inductively coupled plasma mass spectrometry (ICP-MS) before use.IONP distribution
Mice were euthanized using CO2 and perfused with 20 ml of phosphate buffered saline (PBS, Cellgro) from the left ventricle. Peritoneal tumors, spleen, liver, kidney, lung, and heart were harvested in weighed conical tubes. Each tissue was weighed and then subjected to acid digestion using 3–10 ml of trace metal grade acid mixture (HNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HCl = 9:1, Fisher) at 90 °C for an hour in a heating block. 0.5 ml of 30% trace metal grade H2O2 (GFC Chemicals) was added and total volume was made up to 15 or 50 ml with double distilled water. Each digest's iron content was measured using the ICP-MS.
:HCl = 9:1, Fisher) at 90 °C for an hour in a heating block. 0.5 ml of 30% trace metal grade H2O2 (GFC Chemicals) was added and total volume was made up to 15 or 50 ml with double distilled water. Each digest's iron content was measured using the ICP-MS.
Tissue histology
(i) Prussian blue stain. 6 μm paraffin- or cryo-sections were stained for iron using a Gomori Prussian blue iron stain kit (Newcomer Supply) following the manufacturer’s protocol and mounted with Permount (Fisher). (ii) Immunofluorescence. 6 μm cryo-sections were fixed in cold acetone, hydrated, blocked with anti-CD16/32 (eBioscience) for 30 minutes, and stained with Alexa Fluor 488 conjugated anti-mouse CD45 or its isotype (BioLegend) for an hour in a moist chamber. (iii) Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). 6 μm cryo-sections were fixed in formalin, permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate and stained using an in situ cell death detection kit (Roche) following the manufacturer's protocol. For immunofluorescence and TUNEL staining, Hoechst 33342 nucleic acid stain (Molecular Probes) was used to visualize nuclei and slides are mounted with PermaFluor (Thermo Scientific).In vitro uptake of IONPs
ID8-Defb29/Vegf-a cells were placed in 24-well plates at 5 × 105 cells per 0.5 ml of culture media per well and cocultured with starch-, dextran-, or mannan-IONPs (56 μg of iron). Cells were harvested at desired time points and washed twice with PBS to remove free IONPs. Thereafter, 5000 cells were cytospun onto glass slides and stained with Prussian blue to detect iron. Images were taken from random areas (>200 cells in total) and the percentage of iron-positive cells was calculated.Cell sorting
Total peritoneal wash cells were obtained following IP injection of 5–10 ml of PBS. After removal of red blood cells with lysis buffer (8.3 g NH4Cl, 1 g KHCO3, and 0.02 g EDTA in 1 L of dd H2O), retrieved peritoneal wash cells were first blocked with a blocking antibody (eBioscience) for 20 minutes and stained with PerCP-anti-mouse CD45 and PE-anti-mouse CD11b antibodies (BioLegend). Cells were sorted into CD45−, CD45+, CD45+ CD11b−, and CD45+ CD11b+ fractions by fluorescence-activated cell sorting (FACS) FACS-Aria (BD Biosciences). 10,000 cells from each population were cytospun onto glass slides and stained with Prussian blue to detect iron.Peritoneal wash cell transfer
12 Donor mice bearing ID8-Defb29/Vegf-a for 4 weeks were injected IP with starch-IONPs (280 μg of iron). Next day, 5–10 ml of PBS was injected into the peritoneum and this peritoneal wash was collected and red blood cells were lysed. In total 30 × 106 of retrieved peritoneal wash cells were transferred IP into each of four recipient mice (4 week post tumor inoculation). 30 × 106 cells contained 337 μg of iron, as determined by ICP-MS.AMF application
Mice were anesthetized with isoflurane and fiber optic temperature probes (FISCO, Canada and Luxton/LumaSense) were placed in the rectum and peritoneal cavity. Mice were fixed in a 50 ml conical tube set in a solenoid coil attached to a 10 kW generator (Huttinger Elektronik, Germany). Conical tube temperature was maintained at 36.5 °C by blowing air using Bair Hugger (Arizant). During AMF application, temperatures were monitored using the software Evolution (FISCO). An AMF of 600 Oersted (Oe) was given to all mice for 15 minutes (Fig. 6), or AMF field strength was manually adjusted between 550–650 Oe (167.5 kHz) so that the peritoneal temperatures of mice injected with IONPs were maintained at 42 °C for 20 minutes, and the identical AMF was applied to the control mice not injected with IONPs (Fig. 7).Liver toxicity assays
Blood was collected from the posterior vena cava in a plasma separator tube (BD) and centrifuged at 10,000 rpm for 3 minutes. The serum levels of alanine aminotransferase (AST) and aspartate aminotransferase (ALT) were analyzed by Dartmouth Hitchcock Medical Center Pathology Services.Reactive oxygen species (ROS) detection assay
ID8-Defb29/Vegf-a cells were cultured in T25 culture flask at 2 × 106 cells per 5 ml of culture media with or without starch-IONPs (112 μg of iron) overnight. Next day, cells were harvested and washed twice with PBS to remove free IONPs. 1 × 106 cells resuspended in 500 μl PBS were then incubated at either 37 or 42 °C for 20 minutes. After heat treatment, 2 × 105 cells of each sample were placed into a round-bottom 96 well plate and incubated with 7.5 μM CM-H2DCFDA (Invitrogen) at 37 °C in the dark for 30 minutes. Cells were harvested and washed with complete-RPMI media and analyzed by FACS CANTO (BD Biosciences). Cells incubated without CM-H2DCFDA were used as a negative control. Cells incubated with CM-H2DCFDA for 30 minutes and then treated with 100 μM H2O2 (Fisher) for 5 minutes served as a positive control. The results were analyzed with FlowJo 8.6 software (ThreeStar) for the mean fluorescence intensity (MFI).Statistical analysis
Differences between experimental groups were analyzed using two-tailed unpaired Student’s t-test. Regression analysis was done to assess the relationship between the injection dose of IONP and the total amount of iron per tissue or iron per gram of tissue and to determine the relationship between the peritoneal temperature and the amount of iron injected IP. Statistical significance of the regression analyses was assessed via the p-value of the slope coefficient.Results
IP administration localizes IONPs in ovarian tumors
To maximize the effect of IONP-mediated treatment and minimize any possible side effects, selecting an appropriate injection route that concentrates IONPs in the tumor rather than other organs is important. Since tumor masses develop all over the peritoneal cavity in our metastatic ovarian cancer model, we hypothesized that IP administration of IONPs would deliver more IONPs to the tumor and less to other organs than IV administration.Four weeks post tumor inoculation, mice were injected with starch-IONPs (280 μg of iron) either IP or IV, and tumors and major organs were collected the following day (Fig. 1A). IP administration increased the total amount of iron in the tumors (Fig. 1B left) and the amount of iron per gram of tumor (Fig. 1B right) but did not increase iron in the spleen or liver. IV administration also increased iron in the tumor, but the increase was significantly smaller than IP, and IV administration greatly increased the iron level in the liver. Accumulation of IONPs in the tumor but not in major organs by IP injection was further confirmed by Prussian blue staining (Fig. 1C). These results demonstrate that IP is the superior administration route for concentrating IONPs in peritoneal ovarian tumors, therefore all subsequent in vivo experiments utilized IP injection.
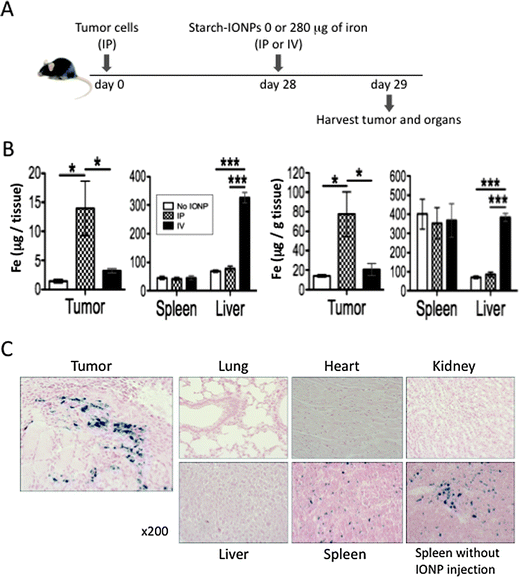 | ||
| Fig. 1 IP administration localizes IONPs in ovarian tumors. (A) Experimental design to compare IONP distribution by IP and IV administration. (B) Total amount of iron per tissue (left) and amount of iron per gram of tissue (right) in the tumor, spleen, and liver. Data obtained by ICP-MS. 4–5 mice per group. (C) Representative pictures of the tumor and major organs (lung, heart, kidney, liver, and spleen) from mice administered IP with starch-IONPs stained for iron with Prussian blue. A picture of spleen without IONP injection is shown lower right for reference. Error bars in B are the mean ± standard error of the mean (SEM). *P < 0.05, ***P < 0.001. | ||
IONP sequestration in the tumor is directly dependent on the total dose administered and independent of the number of aliquots administered
We next compared the effect of different doses and schedules of IONP administration (Fig. 2A). When the dose was increased 4-fold from 280 μg to 1,120 μg, roughly 4-fold more iron was sequestered in the tumor with no significant change in iron levels in the liver or spleen as compared to uninjected control animals (Fig. 2B). Regression analysis of this data (Fig. 2C) shows that the increase in the total amount of iron per tissue and the amount of iron per gram of tissue in the tumor was dose dependent, with the increase rates of 23% and 20% per 100 μg of iron respectively. Iron in the kidney also increased dose dependently, but the increase rates were much lower (7% per 100 μg iron for both total amount of iron per tissue and amount of iron per gram of tissue) than those of tumor. No other tissue had a dose-dependent increase in iron. In order to see whether multiple injections with smaller doses of IONP would be better sequestered in the tumor, tumor-bearing animals were injected with 280 μg of iron on 4 consecutive days to give the 1120 μg dose. The distribution of iron was similar regardless of whether it was given as a large dose or an equal amount of iron in 4 small doses on consecutive days (Fig. 2B). Representative histology pictures of the tumor from each dosing schedule stained with Prussian blue to highlight iron are shown in Fig. 2D.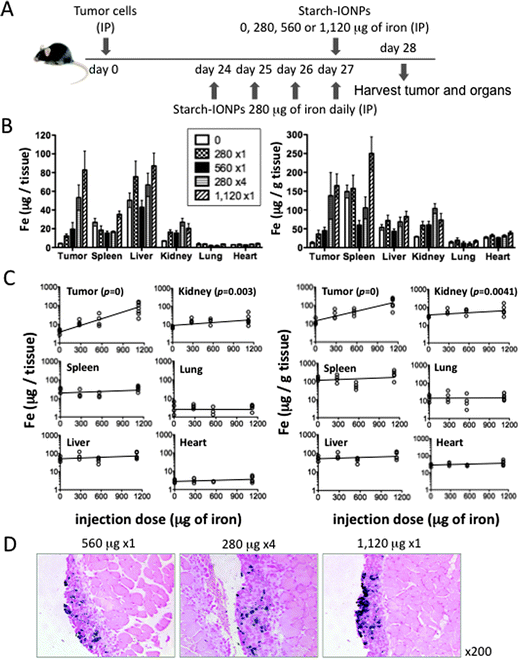 | ||
| Fig. 2 IONP sequestration in the tumor is directly dependent on the dose administered and independent of the number of aliquots administered. (A) Experimental design to compare different IONP administration schedules. (B) Total amount of iron per tissue (left) and amount of iron per gram of tissue (right) in the tumor and major organs (spleen, liver, kidney, lung, and heart). Data obtained by ICP-MS. 4–7 mice per group. Statistics on iron concentrations in the tumor: 0 μg < any other schedules (* or **), 280 μg × 1 or 560 μg × 1 < 280 μg × 4 or 1120 μg × 1 (* or **), ns between 280 μg × 4 and 1120 μg × 1. Iron concentrations in the kidney: 0 μg < any other schedules (* or ** or ***). Iron concentrations in other organs: none was significantly higher than 0 μg animals. (C) Regression analysis showing the relation between the injection dose and the amount of iron per tissue (left) or per gram of tissue (right). (D) Representative pictures of the tumor from mice administered with IONPs using different schedules. Error bars in B are the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant. | ||
Difference in surface coating affects IONP uptake by tumor cells in vitro but not in vivo
Different nanoparticle preparations are taken up differentially by various types of cells.13,14 Here we tested whether the surface coating of nanoparticles affects in vitro uptake of IONPs by tumor cells using IONPs coated with either starch, dextran, or mannan. The experiments were performed by exposing the ID8-Defb29/Vegf-a cells to a standard level of each IONP and monitoring the percentage of iron-positive cells that stained with Prussian blue. Starch-IONPs were efficiently bound or taken up by tumor cells and almost all cells were stained with Prussian blue after 26 hours incubation (Fig. 3A), while almost no cells took up dextran-coated IONPs and only 25% of cells took up IONPs coated with mannan (Fig. 3A and B).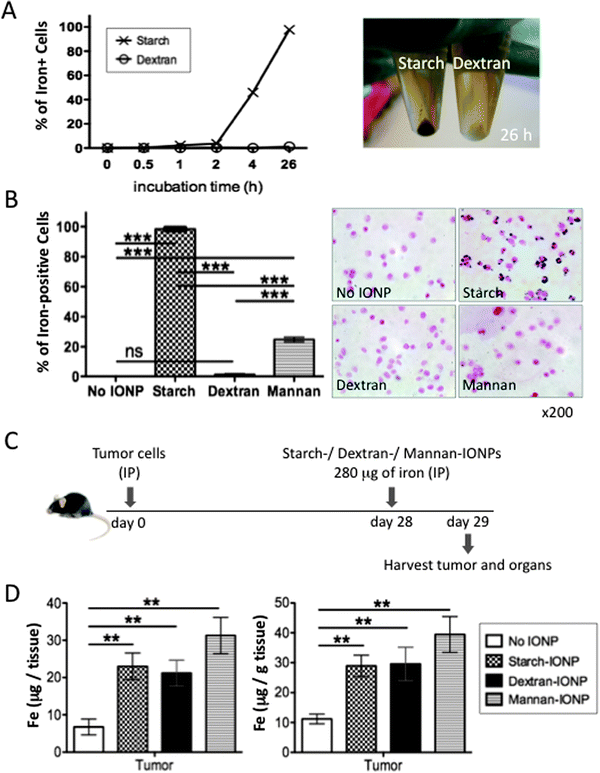 | ||
| Fig. 3 Difference in surface coating affects IONP uptake by tumor cells in vitro but not in vivo. (A) 5 × 105 ID8-Defb29/Vegf-a tumor cells were cultured with starch- or dextran-coated IONPs (280 μg of iron) in 0.5 ml culture media. Cells harvested at desired time points were stained for iron with Prussian blue, and the percentage of iron-positive cells was calculated (left). Photo of these cells harvested after 26 hours incubation visually showing iron accumulation in starch-IONP exposed cells (right). (B) The percentage of iron-positive cells by Prussian blue stain after 26 hours incubation with starch-, dextran-, or mannan-coated IONPs using the same conditions as in A (left). Representative pictures of cells after 26 hours incubation stained for iron with Prussian blue (right). (C) Experimental design to compare in vivo distribution of IONPs with different coatings. (D) Total amount of iron per total tumors (left) and amount of iron per gram of tumor (right). Data obtained by ICP-MS. 4 mice per group. Error bars in B and D are the mean ± SEM. **P < 0.01, ***P < 0.001, ns = not significant. | ||
These results (Fig. 3A and B) suggested that starch-IONPs would be sequestered preferentially in tumor cells as compared to similar particles with either dextran or mannan coating when injected IP into tumor-bearing mice. We next sought to test if the coating difference also makes a difference in IONP accumulation in the tumor in vivo. Four weeks post tumor inoculation, mice were injected with either starch-, dextran-, or mannan-coated IONPs (280 μg of iron) and tumor and major organs were harvested the following day for measuring iron content by ICP-MS (Fig. 3C). IONPs with all three coatings significantly accumulated in the tumor (Fig. 3D) but not in major organs (Fig. S1, ESI†). Surprisingly, there was no significant difference in IONP accumulation in the tumor between the three different coatings. The fact that although there was a dramatic difference due to the coating in IONP uptake by tumor cells in vitro but not in the tumor in vivo suggests that the mechanism of IONP accumulation in the tumor in vivo is not mainly by direct binding to or uptake by tumor cells.
Peritoneal phagocytes take up and carry IONPs specifically to tumors
Since uptake of IONPs by tumor cells in vivo was not affected by the particle coating as it was in vitro, we hypothesized that the IONPs did not enter tumors by direct interaction with tumor cells and instead ovarian tumor-associated peritoneal phagocytes take up and carry IONPs specifically into tumors in vivo. To address our hypothesis, we first determined whether peritoneal leukocytes in the tumor microenvironment take up and retain IONPs. All leukocytes express the CD45 antigen, so peritoneal cells were assayed for CD45 expression to differentiate leukocytes from tumor cells. At day 29 post tumor cell inoculation, peritoneal cells are composed of a mixed population of approximately 46% CD45− and approximately 54% CD45+ cells (Fig. 4B). In Fig. 4C, 18 hours after IP injection of IONPs, peritoneal wash cells from tumor-bearing mice were sorted into CD45− and CD45+ fractions by FACS and each population was stained for iron with Prussian blue. Iron-positive cells were only found within the CD45+ population (Fig. 4C), which suggests that only leukocytes are ingesting nanoparticles and in vivo tumor cells are not taking up IONPs. When CD45+ cells were further sorted into CD11b− and CD11b+ populations, IONPs were only found in the CD45+CD11b+ population (Fig. 4C). Quantitative analysis of Prussian blue stained cells is plotted in Fig. 4C. When tumors from mice injected IP with starch-IONPs were stained for CD45, IONPs were concentrated in CD45+ cells (Fig. 4D). These data (Fig. 4C and D) provide strong evidence that the IONPs are carried into the tumors by phagocytes following engulfment in the peritoneal lumen.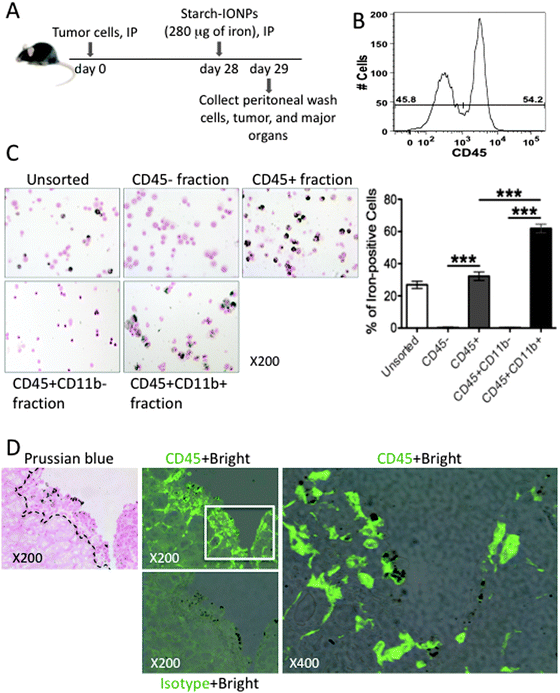 | ||
| Fig. 4 CD45+ CD11b+ peritoneal phagocytes take up IONPs and IONPs colocalize with CD45+ cells in the tumor. (A) Diagram of experimental strategy, IONPs administered IP into 28 day tumor-bearing mice, then peritoneal wash cells, tumors, and major organs were collected after 18 hours. (B) The percentage of CD45− and CD45+ cells in peritoneal wash cells. Peritoneal wash cells were stained with anti-CD45 and analyzed by FACS. Data are representative of 6 independent experiments. (C) Prussian blue staining of IONP-loaded peritoneal wash cells without sorting or after cell sorting of CD45−, CD45+, CD45+ CD11b−, and CD45+ CD11b+ fractions to examine iron content. Data are representative of 3 independent experiments. The percentage of iron-positive stained cells in unsorted, CD45−, CD45+, CD45+ CD11b−, and CD45+ CD11b+ fractions were plotted. Error bars are presented as the mean ± SEM. Two-tailed unpaired t test was used for statistical analysis. **P < 0.005, ***P < 0.001. (D) Representative histology pictures showing that IONPs accumulated in tumor were colocalized with CD45. Dashed lines indicate the boundary of normal tissue (muscle) and tumor on Prussian blue stained section. CD45+ bright are images of CD45 fluorescence staining overlaid by bright field which reveals iron. Isotype control antibody was used to reveal non-specific binding. | ||
In order to directly test whether IONP-loaded phagocytes traffic into the tumor, we performed a cell transfer experiment. Unfractionated peritoneal wash cells harvested from tumor-bearing mice injected IP with starch-IONPs were transferred into recipient tumor-bearing mice by IP injection (Fig. 5A). Tumor and major organs were collected from recipient mice one day post transfer, and IONP accumulation was analyzed with Prussian Blue stain. The histology data showed specific IONP accumulation in the recipient tumors (Fig. 5B), but not in any normal tissues (Fig. S2, ESI†). Importantly, although all tumors are formed as relatively thin coatings on the surface of tissues, the iron is deeply incorporated into the tumor, indicating that the phagocytes carrying the IONPs are actively entering the tumors and not simply adhering to the tumor surface. Taken together these results indicate that CD45+ peritoneal phagocytes carry IONPs specifically into tumor.
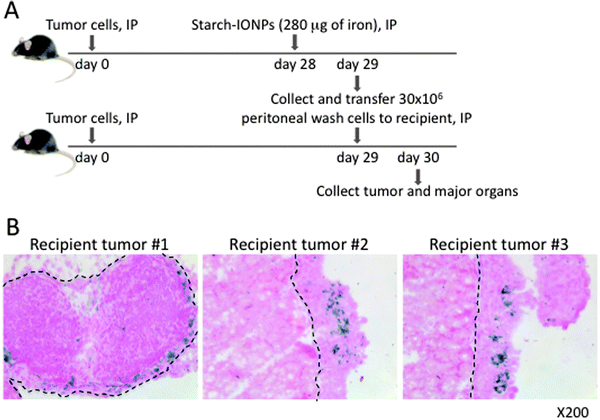 | ||
| Fig. 5 CD45+ peritoneal phagocytes carry IONPs specifically to tumors. (A) Diagram of experimental strategy, IONPs were injected IP 28 days post tumor cell inoculation. 18 hours later, peritoneal wash cells were collected and 30 × 106 unsorted cells were injected IP into tumor-bearing recipient mice. (B) Prussian blue staining of tumor sections to test the homing of IONP-loaded peritoneal cells into tumor. Iron-positive cells were found in recipient tumors. Dashed lines indicate the boundary between normal tissues (pancreas in the left and muscles in the center and right) and tumor burden. Data are representative of 3 independent experiments using 4 mice each time. | ||
IONPs injected IP respond to AMF and heat the peritoneal cavity
Our long-term goal is to define the potential of treating ovarian cancer with magnetic IONP-mediated hyperthermia. The approach depends upon (a) the ability to specifically target IONPs to tumors in a sufficient amount to achieve hyperthermia when an AMF is applied, and (b) the ability to deliver high heat doses to tumors, while maintaining lower temperatures in other normal tissues.To approach this goal we first tested the possibility of specific and significant heat delivery within the peritoneum by magnetic IONP-mediated hyperthermia in a dose-dependent manner. Tumor-bearing mice were exposed to different amounts of IONPs by IP injection, and subjected to an AMF 18 hours post IONPs injection, as outlined in Fig. 6A. Upon application of an AMF field of 600 Oe, peritoneal temperatures were taken with fiber optic probes and monitored for 15 minutes. As shown in Fig. 6B, the peritoneal temperature increased in a dose-dependent manner with the increase in iron, indicating that although at 600 Oe the thermal heat generated directly by the AMF device causes heating (the magnetic inducing coil itself gets quite warm), the amount of IONPs utilized are causing significant heating to the peritoneal region when activated with an AMF, despite the dispersal of the IONPs. The peritoneal temperature increased in a manner linearly proportional to the increase in iron concentration (Fig. 6C). The linear relationship modeled using regression analysis was statistically significant with the p value of the slope <0.0001.
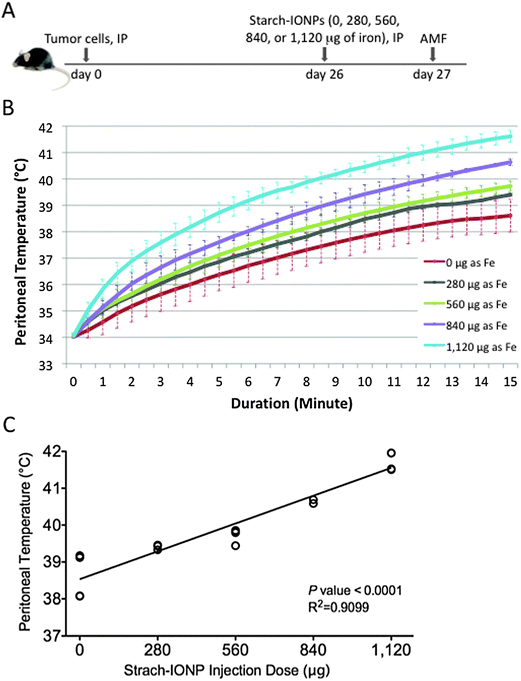 | ||
| Fig. 6 IONPs injected IP respond to AMF and heat the peritoneal cavity. (A) Diagram of experimental strategy, different amounts of starch-IONPs were injected IP into 26 day tumor-bearing mice. Mice were treated with AMF 18 hours post IONP injection. (B) Graph of peritoneal heating history of each group upon application of an AMF field of 600 Oe. Error bars are standard deviation of the mean. (C) Regression analysis demonstrating the linear dose-dependent relationship between the peritoneal temperature 15 minutes after AMF application and the IONP dose. P < 0.001, 4 mice per group. | ||
Magnetic IONP/AMF-mediated hyperthermia induces cell death in tumors
The ability to assay localized heating in the peritoneum is limited, so the temperature in the tumors was not determined. However, if the temperature of the tumor is higher than the temperature of the peritoneum we would expect to see specific damage to tumors. We investigated whether magnetic IONP-mediated hyperthermia induced by the AMF could damage tumor cells and induce cell death. To detect cell death after hyperthermia, we employed the TUNEL method that specifically recognizes cells whose DNA is fragmented because they are undergoing apoptosis or necrosis. As described in Fig. 7A, 18 hours post IP injection of IONPs, tumor-bearing mice were subjected to varying combinations of AMF by adjusting field strength to maintain peritoneal temperature at 42 °C for 20 minutes for the IONP + AMF group, and the identical AMF field strength was used to treat the AMF only group. The graph in Fig. 7B represents a peritoneal heating history. It should be noted that the peritoneal temperature without IONPs was on average approximately 3 °C lower than that with IONPs, while both began at 34 °C (Fig. 7B). Rectal temperatures in both groups were maintained between 36.6–38.5 °C throughout the AMF treatment time. To test for potential liver damage, serum levels of liver enzymes, AST and ALT, were assayed 0.5 hours and 24 hours after treatment. AST and ALT levels after AMF were not outside of the normal range for mice15 although within the experimental groups there were very minor elevations of AST 0.5 hours after AMF treatment (Fig. 7C). By 24 hours after the treatment there were no statistically significant differences in AST or ALT. These data show that any liver damage that may have occurred was biologically inconsequential. Tumors and major organs were collected from mice 0.5 or 24 hours after treatment and stained with TUNEL in conjunction with H&E stain. TUNEL staining showed that 0.5 hours post treatment with AMF, there was cell death in tumor regions in which magnetic IONPs were embedded (Fig. 7D) but not in major organs (data not shown), including the spleen which has an endogenous iron level detectable with Prussian blue staining (as shown in Fig. 1C). TUNEL staining was visible in tumors after 24 hours but was less pronounced (data not shown). AMF treatment alone did not induce any TUNEL staining and IONP alone induced a very weak level of TUNEL staining visible at 0.5 hours (Fig. 7D). TUNEL staining in IONP/AMF treated tumors broadly coincided with iron that was visible on bright field (Fig. 7D).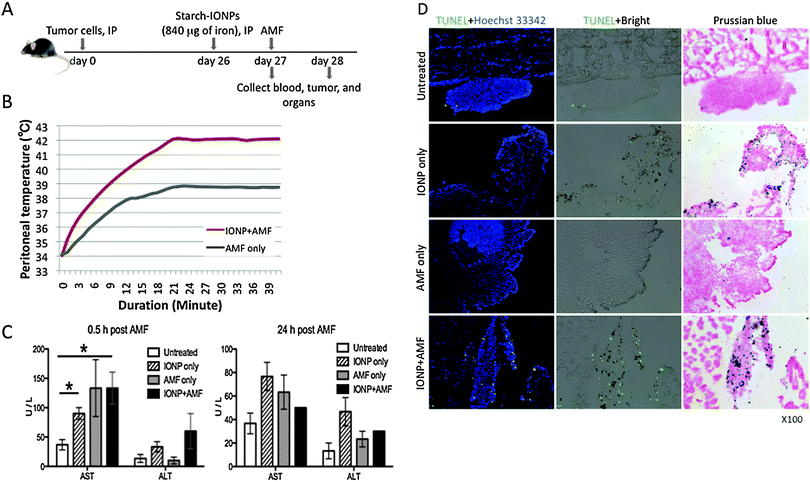 | ||
| Fig. 7 Magnetic IONP/AMF-mediated hyperthermia induces cell death in tumors. (A) Diagram of experimental strategy, starch-IONPs (840 μg of iron) were administered IP to 26 day tumor-bearing mice. 18 hours after IONP injection, mice were subjected to varying AMF to maintain peritoneal temperature at 42 °C for 20 minutes for the IONP+ AMF group, and the identical AMF field strength was applied to the AMF only group. (B) The peritoneal heating history of each group upon application of the AMF, showing higher peritoneal temperature in the IONP+ AMF group compared to the AMF only group. Data are representative of one pair from both groups in 3 independent experiments. (C) Serum clinical chemistry of mice after treatment. Serum AST and ALT levels were plotted and all bar graphs are presented as the mean ± SEM. Two-tailed unpaired t test was used for statistical analysis. *P < 0.05. (D) Representative histology pictures of tumors showing that IONP/AMF treatment induced cell death. Tumors and major organs were collected 0.5 hours after AMF treatment to evaluate cell death. TUNEL detects DNA fragmentation and is revealed as a bright green spot. Tumor sections were stained with Hoechst 33342 to visualize nuclei. IONPs are visualized as brown spots in bright field images and as blue spots in images of Prussian blue staining. TUNEL + Hoechst 33342 are images of TUNEL fluorescence staining merged with Hoechst 33342 fluorescence staining. TUNEL + Bright are images of TUNEL fluorescence staining overlaid by bright field. Data are representative of 2 independent experiments using 3 mice each time. Pictures were taken with X100 magnification. | ||
It has been demonstrated that some nanoparticles (ambient ultrafine particles and cationic polystyrene nanospheres) induce spontaneous ROS production and interaction of cellular components with nanoparticles generates toxic oxidative stress involving mitochondrial injury.16 It has also been demonstrated that hyperthermia can induce cell apoptosis and a significantly higher accumulation of ROS when human or rat cells are exposed to 43°–44 °C for 1 hour.17,18 To check the potential of starch-IONPs, heat, or starch-IONPs and heat to catalyze the production of ROS we compared the cellular ROS production level in ID8-Defb29/Vegf-a cells cocultured with or without starch-IONPs followed by maintenance at 42 °C (heat) or 37 °C for 20 minutes. The ROS level was assayed as the MFI 0.5 hours after heat treatment by using a DCFDA assay and flow cytometry analysis. Prussian blue staining demonstrated that 100% of tumor cells took up starch-IONPs after 22 hours of coculture (Fig. S3A, ESI†). However, neither IONPs alone, heat alone, nor IONPs with heat treatment increased ROS production significantly (Fig. S3B, ESI†). These data suggest that cell death observed after IONP/AMF treatment is not from oxidative cell death.
Discussion
The outcomes for ovarian cancer patients have not improved over the last 30 years and new therapeutic strategies are needed. Hyperthermia as a cancer treatment has been studied extensively for decades and has some efficacy for ovarian cancer when used to sensitize tumors to chemotherapy.19,20 However, since the heat is not localized to the tumor, whole peritoneal hyperthermia also generally increases the sensitivity of normal tissues to damage by chemotherapy, which generates limited improvement in the therapeutic index. The therapeutic index of hyperthermia would be improved if hyperthermia is targeted to the tumors more than normal tissues.Using nanoscale size reagents permits ingestion of the particles by cells, and iron nanoparticles, which have minimal toxicity, have been used safely as an MRI contrast agent. The central problem with IV administration of nanoparticles is sequestration by phagocytic cells in the liver and spleen. The ovarian cancer microenvironment recruits large numbers of tumor-associated phagocytes into the peritoneal cavity from which they traffic to the tumors. Ovarian cancer that is metastatic in the peritoneum is directly accessible through the peritoneal cavity. So we utilized the ingestion of nanoparticles by phagocytes and the IP route of administration to improve, rather than limit, partitioning of nanoparticles into tumors instead of the liver or spleen and we show that the IP administration dramatically increases the accumulation of IONPs in the peritoneal tumor as compared to other organs. This study used an orthotopic ovarian cancer model that closely recapitulates the clinical condition of human ovarian cancer in order to generate the most relevant preclinical data. Although not tested here, other nanoparticles are as likely as IONPs to have improved targeting into peritoneal tumors by IP injection. Other peritoneal cancers such as pancreatic, colon, or liver are also likely to be better targeted with nanoparticles via IP injection rather than IV injection.
Most cancers actively recruit large numbers of tumor-associated immunosuppressive phagocytes into the tumor microenvironment. The experiments reported here show that specific delivery of IONPs into tumors was mediated by peritoneal phagocytes, and this illustrates the potential for using phagocytes to deliver nanoparticles to peritoneal tumors. These tumor infiltrating phagocytes are critical for tumor progression because the selective depletion of these cells impairs the growth of peritoneal ovarian cancer.21 Such phagocytes could be manipulated by nanoparticle technology in three ways (Fig. 8). First, phagocytes ingest and carry nanoparticles specifically into tumors and can thus deliver either a cytotoxic heat dose with an AMF, or cytotoxic agents in nanoparticles, to kill tumor cells. In addition to the direct value of tumor cell killing, death of tumor cells would be immunostimulatory through release of tumor antigens and generation of inflammatory signals associated with cell death. Second, if the tumor-associated immunosuppressive phagocytes themselves can be eliminated by engulfment of nanoparticles through hyperthermia or cytotoxic agent delivery, the tumor microenvironment could shift from immunosuppressive to immunostimulatory, and thereby contribute to enhanced anti-tumor immune responses.6,10,21 Third is hyperthermia stimulation of immune response. Hyperthermia induces expression of heat shock proteins (HSPs) that are thought to mediate immune activation (Fig. 8).22,23 HSPs increase antitumor immune response both locally and systemically via enhancement of the cross-presentation of HSP-chaperoned endogenously synthesized antigens on the Class I major histocompatibility complex.24,25 HSPs also induce antigen-presenting cell (APC) maturation and cytokine secretion26 resulting in the activation of cytotoxic CD8+ T cells. In association with these heat-dependent immunological reactions, direct killing of tumor cells and deletion of tumor-associated immunosuppressive phagocytes may provide a therapeutic path for stimulating antitumor immune response.
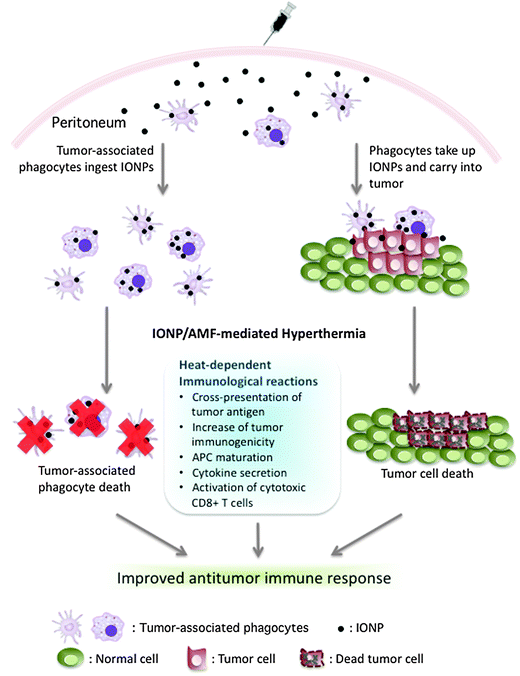 | ||
| Fig. 8 Overview of the therapeutic potential of IONP delivery by phagocytes in ovarian cancer. On the left, tumor-associated phagocytes in the peritoneum ingest IONPs and can be eliminated by IONP/AMF-mediated hyperthermia. On the right, phagocytes in the peritoneum take up and carry IONPs specifically to tumor, and IONP/AMF-mediated hyperthermia damages tumor cells. Noted are reported heat-dependent effects on immune responses. The combined effect of direct killing of tumor cells, elimination of tumor-associated immunosuppressive phagocytes, and heat-dependent immunological effects may improve antitumor immunity. | ||
Efficacy of these three therapeutic possibilities using IONPs and AMF to generate hyperthermia depends on significant cytotoxic heat dose delivery to tumors or tumor-associated phagocytes, while maintaining lower non-cytotoxic temperatures in normal tissues. As reported here, heating of IONPs embedded in tumors by an AMF is dependent on the dose of IONPs. The heat generated by IONPs with AMF causes damage specifically in tumors, which was revealed by histochemical assay for detection of apoptotic and necrotic cells. With the parameters utilized here for IONPs and AMF application, damage to other organs was negligible and this is true as long as the global peritoneal temperature is kept at or below 42 °C. This suggests that it is possible to increase the amount of IONPs further to accomplish thermoablation more efficiently without significantly harming normal organs. In addition, targeting nanoparticles by conjugating cell-surface receptor ligands or monoclonal antibodies that bind specifically to molecules found on the surfaces of cancer cells, such as the high-affinity folate receptor27 or receptors for luteinizing hormone releasing hormone28 could possibly improve partitioning of nanoparticles into tumors by adding direct binding to tumor surface molecules to uptake and delivery by phagocytes. As demonstrated here, it should also be possible to mix IONPs with phagocytes cultured briefly in vitro followed by IP administration of these nanoparticle-carrying phagocytes to further increase the specific targeting of IONPs into peritoneal cancers.
In summary, we have achieved our key objective of establishing the potential of IP injection of IONPs to deliver nanoparticles preferentially to tumors by phagocyte-mediated delivery. When combined with an appropriate AMF, the IONPs deliver a sufficient amount of heat safely and specifically into tumors for achieving thermoablation. This study provides support for strategies in which rather than striving to keep nanoparticles out of phagocytes, the tendency of phagocytes to ingest nanoparticles and the recruitment of phagocytes to tumors by specific mechanisms is therapeutically exploited. Eventually, use of IP injection of nanoparticles to achieve the combined effects of direct killing of tumor cells and elimination of tumor-associated phagocytes may also be used synergistically in combinatorial therapies with other treatment modalities.
Abbreviations
| IONP | Iron oxide nanoparticle; |
| AMF | Alternating magnetic field; |
| IP | Intraperitoneal/intraperitoneally; |
| IV | Intravenous/intravenously; |
| ICP-MS | Inductively coupled plasma mass spectrometry; |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling; |
| FACS | Fluorescence-activated cell sorting; |
| Oe | Oersted; |
| AST | Alanine aminotransferase; |
| ALT | Aspartate aminotransferase; |
| ROS | Reactive oxygen species; |
| MFI | Mean fluorescence intensity; |
| HSP | Heat shock protein; |
| APC | Antigen-presenting cell |
Acknowledgements
We would like to acknowledge Alan Foreman for assistance with the AMF generator, Paul Spear for technical help with the ROS detection assay, DartLab for flow cytometry, Dartmouth Trace Element Core for ICP-MS, Toxicology, Pathology and Biodistribution Core of Dartmouth Center of Cancer Nanotechnology Excellence for pathology and pharmacokinetics assistance, Dartmouth Hitchcock Medical Center Pathology Services for AST/ALT assays. Funding was provided by NIH, NCI 1 U54 CA151662-01, SBIR RFP PHS2010-1 topic 295 and NCI grant#CA157664.References
- A. Chavez-Blanco, V. Perez-Sanchez and A. Gonzalez-Fierro, et al., HER2 expression in cervical cancer as a potential therapeutic target, BMC Cancer, 2004, 4, 59 CrossRef.
- D. B. Kirpotin, D. C. Drummond and Y. Shao, et al., Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models, Cancer Res., 2006, 66, 6732–6740 CrossRef CAS.
- M. H. Mendonca Dias and P. C. Lauterbur, Ferromagnetic particles as contrast agents for magnetic resonance imaging of liver and spleen, Magn. Reson. Med., 1986, 3, 328–330 CrossRef CAS.
- S. Fisher, W. B. Riley and C. D. Shorey, Studies on the mechanism of increased splenic particle uptake in mice after the injection of some finely divided agents, J. Pathol. Bacteriol., 1968, 96, 463–471 CrossRef CAS.
- R. L. Hong, C. J. Huang and Y. L. Tseng, et al., Direct comparison of liposomal doxorubicin with or without polyethylene glycol coating in C-26 tumor-bearing mice: is surface coating with polyethylene glycol beneficial?, Clin. Cancer Res., 1999, 5, 3645–3652 CAS.
- J. R. Conejo-Garcia, F. Benencia and M. C. Courreges, et al., Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A, Nat. Med. (N. Y.), 2004, 10, 950–958 CAS.
- K. F. Roby, C. C. Taylor and J. P. Sweetwood, et al., Development of a syngeneic mouse model for events related to ovarian cancer, Carcinogenesis, 2000, 21, 585–591 CrossRef CAS.
- J. R. Cubillos-Ruiz, X. Engle and U. K. Scarlett, et al., Polyethylenimine-based siRNA nanocomplexes reprogram tumor-associated dendritic cells via TLR5 to elicit therapeutic antitumor immunity, J. Clin. Invest., 2009, 119, 2231–2244 CAS.
- K. D. Phipps, A. P. Surette, P. A. O’Connell and D. M. Waisman, Plasminogen receptor S100A10 is essential for the migration of tumor-promoting macrophages into tumor sites, Cancer Res., 2011, 71, 6676–6683 CrossRef CAS.
- P. M. Kelly, R. S. Davison, E. Bliss and J. O. McGee, Macrophages in human breast disease: a quantitative immunohistochemical study, Br. J. Cancer, 1988, 57, 174–177 CrossRef CAS.
- C. Murdoch, A. Giannoudis and C. E. Lewis, Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues, Blood, 2004, 104, 2224–2234 CrossRef CAS.
- R. Ivkov, S. J. DeNardo and W. Daum, et al., Application of high amplitude alternating magnetic fields for heat induction of nanoparticles localized in cancer, Clin. Cancer Res., 2005, 11, 7093s–7103s CrossRef CAS.
- W. H. De Jong and P. J. Borm, Drug delivery and nanoparticles:applications and hazards, Int. J. Nanomed., 2008, 3, 133–149 CrossRef CAS.
- M. Settles, M. Etzrodt and K. Kosanke, et al., Different capacity of monocyte subsets to phagocytose iron-oxide nanoparticles, PLoS One, 2011, 6, e25197 CAS.
- C. R. Patra, S. S. Abdel Moneim and E. Wang, et al., In vivo toxicity studies of europium hydroxide nanorods in mice, Toxicol. Appl. Pharmacol., 2009, 240, 88–98 CrossRef CAS.
- T. Xia, M. Kovochich and J. Brant, et al., Comparison of the abilities of ambient and manufactured nanoparticles to induce cellular toxicity according to an oxidative stress paradigm, Nano Lett., 2006, 6, 1794–1807 CrossRef CAS.
- D. M. Katschinski, K. Boos, S. G. Schindler and J. Fandrey, Pivotal role of reactive oxygen species as intracellular mediators of hyperthermia-induced apoptosis, J. Biol. Chem., 2000, 275, 21094–21098 CrossRef CAS.
- J. Frank, D. K. Kelleher, A. Pompella, O. Thews, H. K. Biesalski and P. Vaupel, Enhancement of oxidative cell injury and antitumor effects of localized 44 degrees C hyperthermia upon combination with respiratory hyperoxia and xanthine oxidase, Cancer Res., 1998, 58, 2693–2698 CAS.
- C. W. Helm and J. C. States, Enhancing the efficacy of cisplatin in ovarian cancer treatment – could arsenic have a role, J. Ovarian Res., 2009, 2, 2 CrossRef.
- P. Wust, B. Hildebrandt and G. Sreenivasa, et al., Hyperthermia in combined treatment of cancer, Lancet Oncol., 2002, 3, 487–497 CrossRef CAS.
- E. Huarte, J. R. Cubillos-Ruiz and Y. C. Nesbeth, et al., Depletion of dendritic cells delays ovarian cancer progression by boosting antitumor immunity, Cancer Res., 2008, 68, 7684–7691 CrossRef CAS.
- Q. Liu, B. Zhai and W. Yang, et al., Abrogation of local cancer recurrence after radiofrequency ablation by dendritic cell-based hyperthermic tumor vaccine, Mol. Ther., 2009, 17, 2049–2057 CrossRef CAS.
- H. Bendz, S. C. Ruhland and M. J. Pandya, et al., Human heat shock protein 70 enhances tumor antigen presentation through complex formation and intracellular antigen delivery without innate immune signaling, J. Biol. Chem., 2007, 282, 31688–31702 CrossRef CAS.
- R. J. Binder and P. K. Srivastava, Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells, Nat. Immunol., 2005, 6, 593–599 CrossRef CAS.
- R. Suto and P. K. Srivastava, A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides, Science, 1995, 269, 1585–1588 CAS.
- M. Breloer, B. Dorner, S. H. More, T. Roderian, B. Fleischer and A. von Bonin, Heat shock proteins as “danger signals”: eukaryotic Hsp60 enhances and accelerates antigen-specific IFN-gamma production in T cells, Eur. J. Immunol., 2001, 31, 2051–2059 CrossRef CAS.
- N. Saltan, H. M. Kutlu, D. Hur, A. Iscan and R. Say, Interaction of cancer cells with magnetic nanoparticles modified by methacrylamido-folic acid, Int. J. Nanomed., 2011, 6, 477–484 CAS.
- C. Leuschner, C. S. Kumar, W. Hansel, W. Soboyejo, J. Zhou and J. Hormes, LHRH-conjugated magnetic iron oxide nanoparticles for detection of breast cancer metastases, Breast Cancer Res. Treat., 2006, 99, 163–176 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ib20180a |
| ‡ Equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2013 |