Modulation of amyloid-β 1-42 structure and toxicity by proline-rich whey peptides
Prashant
Bharadwaj
ac,
Richard
Head
b,
Ralph
Martins
c,
Vincent
Raussens
d,
Rabia
Sarroukh
d,
Hema
Jegasothy
e,
Lynne
Waddington
a and
Louise
Bennett
*e
aCSIRO Preventative Health Flagship, Material Science and Engineering, 343 Royal Parade, Parkville, Victoria 3052, Australia
bCSIRO Preventative Health National Research Flagship, PO Box 10041, Adelaide, BC, SA 5000, Australia
cCentre of Excellence for Alzheimer's Disease Research & Care, School of Exercise, Biomedical & Health Sciences, Edith Cowan University, WA 6027, Australia
dCentre de Biologie Structurale et de Bioinformatique Faculte des Sciences, Universite Libre de Bruxelles Boulevard du Triomphe, Bat. BC, Niveau 4, Zip: 1050 Brussels, Belgium
eCSIRO Preventative Health Flagship, Food and Nutritional Sciences, 671 Sneydes Road, Werribee, Victoria 3030, Australia. E-mail: louise.bennett@csiro.au
First published on 7th September 2012
Abstract
A proline-rich peptide product prepared from bovine whey protein that was enriched in several hydrophobic amino acids including proline (whey proline-rich peptide, wPRP) was shown to modulate the folding pathway of human amyloid beta peptide 1–42 (Aβ42) into oligomers. Concentration-dependent changes in ThT-binding to Ab42 by wPRP indicated suppression of oligomerisation, that was supported by Transmission Electron Microscopy. Suppression of β-sheet and specifically, anti-parallel β-sheet structures by wPRP was demonstrated by ATR-FTIR spectroscopy, where evidence for capacity of wPRP to dissociate pre-existing β-sheet structures in Aβ42 was also apparent. Suppression of anti-parallel β-sheets of oligomeric Aβ42 was associated with rescue of yeast and SH-SY5Y neuronal cells providing important evidence for the association between anti-parallel β-sheet structure and oligomer toxicity. It was proposed that the interaction of wPRP with Aβ42 interfered with the anti-parallel folding pathway of oligomeric Aβ42 and ultimately produced ‘off-pathway’ structures of lowered total β-sheet content, with attenuated cellular toxicity.
Introduction
In 2010, cases of dementia including Alzheimer's disease (AD) affected 35.6 million people worldwide with predicted increase of 65.7 million to 115.4 million from 2030 to 2050, according to Alzheimer's Disease International, the worldwide federation of Alzheimer's Associations. The substantial projected social and economic burden of AD world-wide has driven the need for evaluation of the costs of the illness, the cost-effectiveness of interventions and ultimately, the implementation of public policies and services.1 Disease-modifying therapies remain a high priority for development and use in AD care, however the current absence of effective disease-modifying therapies reflects the significant challenge posed by this goal. Furthermore, as preventative measures are likely to be most effective in pre-symptomatic stages of disease, diet and lifestyle interventions are favoured at this stage. Of relevance to this study is the precedent set by the proline-rich peptide extract prepared from ovine colostrums known as ‘Colostrinin’2 for which anti-fibril and other neuroprotective bioactivities have been extensively reported.Amyloid beta (Aβ) is a 40 or 42 amino acid cleavage product of Amyloid Precursor Protein (APP) produced in low levels in the normal ageing brain. It is the major component of senile extra-cellular plaques in the brain of AD patients and increased levels of Aβ42 and its self-aggregation in the brain are key events thought to be responsible for the progressive cognitive decline associated with AD.3 In addition to the heterogeneity and toxic variability of extra-cellular amyloid structures,4 disease progression has also been attributed to the retention of intra-cellular Aβ42.5 Amyloid fibrils comprise highly ordered, cross-β-sheet arrays that elongate into long fibrils and aggregate as tangled plaques. Oligomeric Aβ42 represents a low mass, soluble form of Aβ42 produced by an early stage folding pathway,6 that can be isolated and is reported to exhibit high stability.7 Oligomeric Aβ42 exhibits higher relative toxicity compared with amyloid fibrils8,9 and is now considered the more important target for diagnostic detection and therapeutic intervention in AD.10 However, detailed structural characterization of oligomers is uncertain, with the consensus that the transition of Aβ42 monomers to oligomers is accompanied by β-sheet organization.11,12 Further, using NMR methods, Yu et al, (2009),12 showed that oligomers of Aβ42 comprised β-sheet in mixed parallel and anti-parallel orientation and evidence from the crystal structure of a structurally ‘trapped’ form of Aβ42 also suggested that the fundamental oligomer building block may comprise of a pair of Aβ42 dimers associated in a non-parallel orientation.13 Recent reports of structural characterization of soluble forms of Aβ40 (ref. 14) and Aβ42 (ref. 15 and 16) suggested that conformational transition to β-sheet may be important in toxicity. It was also suggested that oligomer structural ‘polymorphism’ may account for variability in toxicity to different cell types reflecting differentiated interactions with their membranes.17 Agents that can perturb the early self-assembly of oligomers and show capacity to regulate toxicity, such as effects of plasmalogen ethanolamine vesicles,18 can assist in the challenge of understanding important aspects of relationships between structure and toxicity in oligomeric Aβ42.
A wide range of inhibitors and activators of Aβ42 aggregation, in addition to β-sheet ‘breakers’ that dissociate aggregates, have been shown to regulate morphology and toxicity of Aβ42 structures both in vitro and in vivo.9,10,19 These species fall into peptide, protein and small molecular classes of natural and synthetic origin.10 The chemical diversity of these inhibitory species reflects the multiple surfaces and opportunities for potential interaction with the core two-layered β-sheet platform of Aβ42 (ref. 20). For example, a series of protein mimetics were able to selectively drive either fibrillar or oligomeric pathways of Aβ42 self-assembly.7 Similarly, the small molecule DC-AB1 prevented β-sheet stacking and fibril assembly by maintaining Aβ42 secondary structure in α-helix.21 Anti-fibril peptides were designed to block the N terminal region of Aβ1–28 using a cyclised form of Aβ1–28 linking Lys17 and Asp21 side chains.22 Short peptide sequences homologous to the central region of Aβ42 were derivatized with putrescine for improved blood brain barrier permeability and included D-amino acids for proteolytic stability, which inhibited and dissociated pre-formed fibrils.23 A range of other synthetic peptide derivatives have been shown to inhibit Aβ42 fibrillisation with favourable solubility, membrane permeability and proteolytic resistance.24,25
Specific milk proteins are also reported for their fibril inhibitory activity. The micellar structure of the bovine milk caseins represents a thermodynamically stable architecture that accommodates the amphiphilic casein proteins and colloidal calcium phosphate components of milk.26 When isolated, two of the four proteins of the casein micelle, κ-casein (κCn) and αS2 casein readily form fibrils under either reducing or non-reducing conditions, respectively27,28 but in milk, this behaviour is suppressed by αS1-casein and β-casein, present in stoichiometric excess and organised in a micellar structure.27,28 By extension, we have shown that hydrolysates prepared from selected milk proteins exhibited anti-fibril properties29 using reduced, carboxy-methylated κCn (RCM-κCn) for fibril inhibitor activity screening.30 In this study, we describe dose-dependence effects of a proline-rich whey protein-derived peptide hydrolysate (wPRP) on structure and toxicity of Aβ42. wPRP was prepared using enzymatic processing, optional fractionation by C18 solid phase extraction (products eluted in either 40% or 100% solvent) and contained small peptides (<6 kDa) enriched in proline and other hydrophobic amino acids. We have characterised the modulation of Aβ42 structures by wPRP using in vitro and physico-chemical methods and cell-based assays of Aβ42-mediated toxicity. We report that wPRP exhibited interesting dose-dependent capacity for modulation of Aβ42 fibril morphology and toxicity and propose key secondary structural features of Aβ42 associated with toxic effects.
Results
Characterisation of whey proline-rich peptide (wPRP)
The active form of the whey protein hydrolysate was obtained by preparative SPE with peptide fractions eluted in either 40% acetonitrile (SPE40 product), followed by 100% acetonitrile (SPE100 product) elution, or a ‘total’ peptide fraction eluted in 100% acetonitrile (wPRP). HPLC profiles indicated significant retention of peptides by the C18 SPE media and overlap between SPE40 and SPE100 peptide assemblages (Fig. 1a). The SPE40 contained a relatively higher proportion of species eluting in the 15 to 22 min range (Fig. 1a). The ratio of solids eluted by 40% and 100% acetonitrile was approximately 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. This means that the wPRP product (used due to diminished supplies of SPE100) contained only 5% of the most hydrophobic peptides, thought to be the most bioactive species for regulating self-assembly of Aβ42.
:1. This means that the wPRP product (used due to diminished supplies of SPE100) contained only 5% of the most hydrophobic peptides, thought to be the most bioactive species for regulating self-assembly of Aβ42.
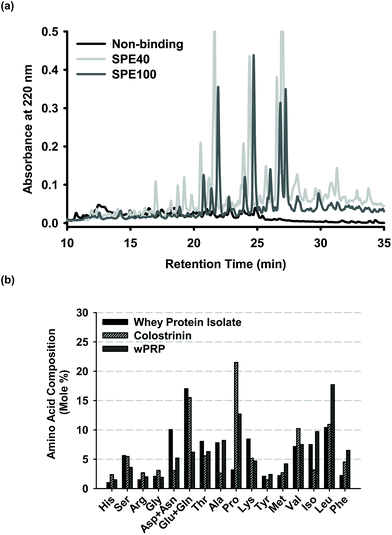 | ||
| Fig. 1 (a) Reverse phase HPLC profiles of sub-fractions of wPRP prepared by solid phase extraction (SPE) showing non-binding fraction (pooled void plus wash fractions) and fractions eluted with 40% and remaining bound species with 100 % acetonitrile, designated SPE40 and SPE100, respectively. The ratio of SPE40 to SPE100 present in wPRP was approximately 19:1 and profiles have been standardised for mass of solids analysed. (b) Amino acid analysis of wPRP compared with the whey protein isolate substrate and ‘Colostrinin' prepared from ovine colostrum.73 | ||
The amino acid composition of the wPRP was compared with the whey protein isolate (WPI) substrate from which it was prepared and Colostrinin, which is a peptide hydrolysate derived from ovine colostrum, reported to exhibit many comparable functional properties to wPRP. Colostrinin was significantly richer in mole percentage of glutamic acid, glutamine and proline whereas wPRP was relatively richer in the non-polar amino acids, alanine and leucine. wPRP was significantly enriched in proline, leucine and phenylalanine in comparison with both WPI and Colostrinin.
Modulation of Aβ42 self-assembly
The following studies were unavoidably conducted using Aβ42 from different suppliers and some differences in preparatory methods, however the essential common feature of all methods was the use of HFIP and either filtration or centrifugation to maximise the aggregate-free character of the amyloidogenic peptide at the zero timepoint.The wPRP product exhibited concentration-dependent modulation of Aβ42 self-assembly using ‘aggregate-free’ Aβ42, intended to model the earliest stages of oligomerisation. wPRP exhibited interesting capacity for apparent ‘promotion’ of fibril assembly at low concentrations (∼2 μg ml−1Fig. 2), as monitored by ThT binding capacity and fluorescence, which extended to higher concentrations of wPRP if the Aβ42 was allowed to self-aggregate (by pre-incubating at 22 °C for 1 h) before introducing the wPRP product (Fig. 2). However, at higher concentrations of wPRP (>11 μg ml−1), ThT fluorescence decreased compared with control, indicating inhibition of Aβ42 oligomerisation (Fig. 2). The inhibition curve shifted to higher concentrations of wPRP as a consequence of pre-incubation of Aβ42, suggesting that higher concentration of wPRP was necessary to suppress self-assembly of pre-existing oligomeric structures.
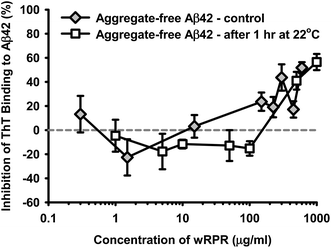 | ||
| Fig. 2 Concentration-dependent change in percentage inhibition of ThT fluorescence for aggregate-free Aβ42 (30 μg ml−1, 6.7 μM) in the presence of increasing concentration of wPRP. Results are shown for ‘aggregate-free’ Aβ42 used in the assay either immediately (control) or after 1 h incubation at 22 °C. Subsequent assay incubation period was 37 °C for 24 h and data represent the mean of triplicate analyses and error bars showing standard deviation. | ||
Modulation of Aβ42 secondary structure in the presence of increasing concentrations of wPRP was further studied using ATR-FTIR spectroscopy. Results are shown after subtraction of the contribution of wPRP and Aβ42 at zero time (Fig. 3) or after subtraction of the contribution of wPRP only (Fig. 4). FTIR characteristics of wPRP control indicated that wPRP alone did not absorb in designated β-sheet regions either at 1695 or 1629 cm−1, and exhibited no change in secondary structure over the concentration range studied (not shown).
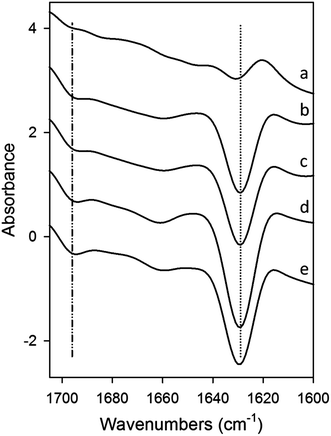 | ||
| Fig. 3 ATR-FTIR difference spectra of mixtures of Aβ42 (10 μM) and wPRP after subtraction of Aβ42 and wPRP controls at each concentration, showing structural changes in Aβ42 peptide compared with Aβ42 control, resulting from the presence of wPRP. Spectral subtraction was applied between 4000 and 800 cm−1 and changes mainly reflected effects of wPRP on Aβ42 secondary structure in the amide I band region (1700–1600 cm−1). wPRP concentrations were (a) 0.001, (b) 0.005, (c) 0.01, (d) 0.05, (e) 0.1 mg ml−1 and samples were incubated at 30 °C for 20 h. Each spectrum represents the mean of independent samples analysed in triplicate. | ||
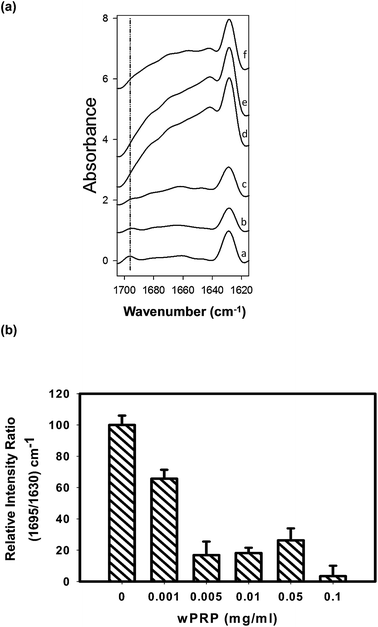 | ||
| Fig. 4 (a) ATR-FTIR difference spectra of Aβ42 incubated at 30 °C for 20 h in the presence of (a) 0, (b) 0.001, (c) 0.005, (d) 0.01, (e) 0.05, (f) 0.1 mg ml−1 of wPRP, after subtraction of respective wPRP controls at each concentration, showing structural changes in Aβ42 resulting from the presence of wPRP. Each spectrum represents the mean of independent samples analysed in triplicate. (b) Ratio of 1695/1630 cm−1 mean intensities normalized to control Aβ42 (100%) and plotted as a function of the concentration of wPRP. Error bars represent standard deviation. | ||
Effects of wPRP on modulating β-sheet character of Aβ42 were significant. After incubation for 20 h at 30 °C in the absence of wPRP, Aβ42 exhibited anti-parallel β-sheet structure associated with the presence of oligomers (Fig. 4a, spectrum (a)) as previously shown.16 In the absence of wPRP, Aβ42 exhibited almost exclusively total β-sheet (1629 cm−1) with a significant anti-parallel contribution (1695 cm−1), and low α-helical content, as previously described.16
The wPRP induced FTIR spectral changes to Aβ42 in the amide I region (1700–1600 cm−1) reflecting effects on extent of Aβ42 self-assembly (Fig. 3). Specifically, FTIR residual spectra indicated that wPRP-concentration-dependent structural changes occurred in the β-sheet (1613–1629 and 1695 cm−1) and to a lesser extent, in the α-helix and/or random coil structure regions, clustering between 1620 and 1705 cm−1. Increasing concentration of wPRP produced strong suppression of β-sheet (1629 cm−1) and anti-parallel β-sheet (1695 cm−1) suggesting interference of Aβ42 self-assembly into cross-β-sheet configuration (Fig. 3). Difference spectra showing changes in Aβ42 structure indicated that the presence of wPRP induced both suppression of anti-parallel β-sheet and of the total β-sheet content (1629 cm−1 peak becoming more negative. Fig. 3) in soluble aggregates of Aβ42.
Difference spectra produced after subtraction of wPRP at each concentration permitted evaluation of progressive structural changes in Aβ42 in comparison with the unmodified control representing oligomeric Aβ42 formed after the 20 h incubation period (Fig. 4a). The ratio of intensities at 1695/1630 cm−1 permits the changes in ratio of anti-parallel to total β-sheet to be quantified,16 and highlighted the effect of wPRP specifically on formation of oligomeric Aβ42. By this method, the oligomer content of Aβ42 was found to be significantly lowered by wPRP in a concentration-dependent manner (Fig. 4b) with the concomitant development of random coil and/or helical structures at 1650–1660 cm−1 (Fig. 4a). Overall, these results suggested that wPRP progressively inhibited the self-assembly of Aβ42 into oligomers and perturbed existing β-sheet structures favouring re-organisation into random coil and/or helical structures (Fig. 4a).
To further study the effects on Aβ42 structure and oligomerization, Aβ42 and SPE100 mixtures were co-incubated (under comparable conditions to samples prepared for FTIR studies) and studied by TEM and Western blot analysis. Aβ42 alone exhibited large globular oligomeric (50–60 nm in diameter) and proto-fibrillar structures (100–150 nm in length, Fig. 5a). The SPE100 product alone was characterised by small globular particles of approximately 10 nm (Fig. 5b). However, Aβ42 co-incubated with SPE100 also comprised small globular structures (10–20 nm in diameter, Fig. 5b) similar to the SPE100 control, and was devoid of larger oligomeric and fibrillar structures.
 | ||
| Fig. 5 Transmission electron micrographs of Aβ42 peptide (10 μM) incubated at 30 °C for 20 h alone (a), SPE100 alone (0.1 mg ml−1), (b) or Aβ42 with SPE100 (0.1 mg ml−1), (c). | ||
Where TEM analysis involved SPE100, which appeared to produce visible aggregates (Fig. 5b), FTIR analysis was conducted with wPRP containing a significant proportion of less hydrophobic peptide species and did not self-aggregate according to FTIR control experiments (not shown). In addition, the apparent aggregation of SPE100 was more likely to be observed in TEM experiments because the concentration of SPE100 during incubation was 0.5 mg ml−1, before dilution for TEM imaging and was therefore 5-fold higher than the highest concentration of wPRP studied by FTIR, where self-aggregation was not observed. The TEM images of Aβ42 incubated with the SPE100 reflected strong suppression of oligomer/protofibril development (Fig. 5c) with structures of reduced size to the control (Fig. 5a). These results supported effects observed by FTIR which was conducted with the relatively more hydrophilic and less self aggregation-prone wPRP.
SPE100 was also found to interfere with assembly of Aβ42 oligomers by Western blot analysis (Fig. 6). The SDS-stable structures of Aβ42 peptide evident by Western blot comprised low (dimers and trimers) initially (0 h timepoint) and additional higher-order oligomers (∼50–110 kDa) after 24 and 48 h incubations (Fig. 6). Higher mass oligomeric Aβ42 (∼50–110 kDa range) products produced at 24 and 48 h were suppressed with increasing concentrations of SPE100, in particular at concentration ratios at and below 200, but not at 1000. This demonstrated that a mass ratio >200 was required for inhibition of self-assembly of higher mass oligomers. However, no change was detected with the intensity of low mass Aβ42 oligomer (∼7–20 kDa range) and monomer bands. This also supported the previous FTIR evidence for suppression of β-sheet content and anti-parallel β-sheets associated specifically with oligomers.
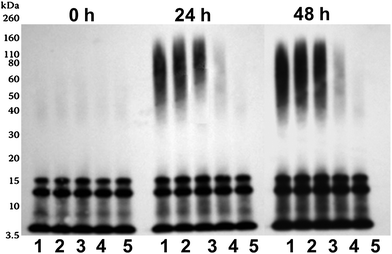 | ||
| Fig. 6 Western blot analysis of Aβ42 peptide (10 μM) incubated at 22 °C for either 0, 24 or 48 h with SPE100 (lane 1–5: 0, 0.01, 0.025, 0.05 and 0.1 mg ml−1), before immunoblotting using WO2 anti-body (anti-Aβ-3–10). | ||
Modulation of Aβ42 toxicity
SPE40 and SPE100 were tested in Aβ42 challenge assays with both yeast and neuronal cells. Aβ42, prepared so as to favour oligomer formation over the incubation period, was toxic to both yeast and neuronal cells in a dose-dependent manner with the concentration of 10 μM chosen for subsequent challenge experiments (Fig. 7a). Yeast cells challenged with Aβ42 were protected in the presence of both SPE40 and SPE100 in a dose-dependent manner (Fig. 7b) that appeared to mimic the concentration dependence on Aβ42 fibril inhibition (Fig. 2). That is, both SPE40 and SPE100 promoted a structure of Aβ42 that was apparently more toxic (concentration ratio range ratio 10000 to 200) before becoming protective at the concentration ratio of 100 (Fig. 7b). Yeast challenge experiments indicated that SPE100 was more protective than SPE40 at 0.05 and 0.1 mg ml−1 (Fig. 7b) and subsequent studies with neuronal cells focussed on effects of SPE100 only. For neuronal cells, the SPE100 product also exhibited concentration-dependent rescue of cell viability, measured by either LDH or MTS methods, respectively (Fig. 7c) although did not appear to transition through a stage of enhanced toxicity, as observed for yeast. This may reflect the difference in incubation time adopted for yeast (20 h) versus neuronal cells (3 days) or the different methods of measurement of cell viability for yeast compared with indirect and less sensitive methods used for neuronal cells. In both cases, SPE40 and SPE100 were protective against toxicity at the highest SPE concentration ranges.
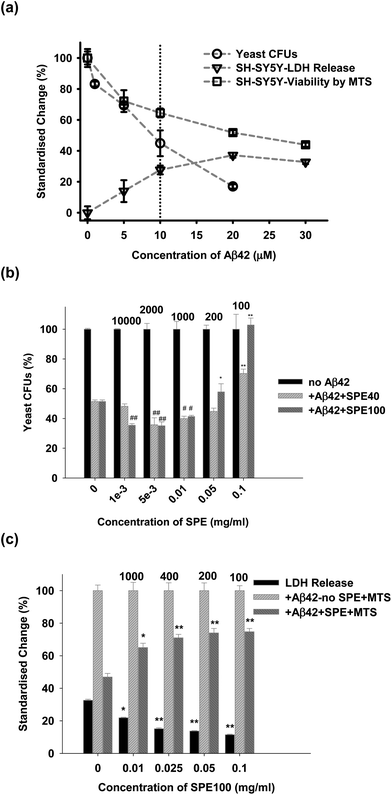 | ||
| Fig. 7 (a) Dose-dependent effects of Aβ42 on viability of either yeast (by colony forming units count, CFU) or SHSY5Y cells, measured by LDH and MTS methods. Subsequent challenge experiments were conducted using 10 μM Aβ42. (b) Dose-dependent effects on viability of SPE40 and SPE100 on exponentially growing yeast cells incubated at 30 °C for 20 h with Aβ42 (10 μM) with or without SPE40 and SPE100 (0.001 to 0.1 mg ml−1), and reported as changes in CFU. (c) Dose-dependent effects on viability of SPE100 (0.01 to 0.1 mg ml−1) on SHSY5Y cells. Cell viability was determined by the ratio of colony numbers in the absence and presence of SPE samples, and reported by LDH and MTS assay methods. The data were reported as the percentage standardized change compared with sample-free controls, after correction for reagent blanks. Results represent the mean and SEM of triplicate determinations at each concentration with significance of differences to control (*, P < 0.05; **, P < 0.001 for protective effects on toxicity; #, P < 0.05; ##, P < 0.001 for negative effects on toxicity) determined by Student’s t-test. The ratios of Aβ42 (in μM) to SPE product concentration (in mg ml−1), are shown above bars to permit comparison with other results. Some batch to batch variation in Aβ42 toxicity may have occurred between dose response and yeast and neuronal cell challenge experiments. | ||
Discussion
Self assembling and chaperone-dependent folding pathways of Aβ42
The self-assembly of proteins into elongate fibrils based on cross β-sheet ‘laminae’ of amyloidogenic polypeptides, is proposed to represent a common folding pathway of many proteins.31 Furthermore, ‘amyloid’ structuring of proteins has been described as a detoxification strategy to mask the promiscuous surface of the oligomeric building block.32 Aβ42 fibril structure has been described as a planar ‘laminate’ of up to six parallel β-sheets, stacking and elongating the fibril perpendicular to the laminate plane.33 However, formation of meta-stable oligomeric Aβ42 represents an early competing folding pathway6 characterised by anti-parallel β-sheet structure.34 In this study, Aβ42 preparations modelled the earliest stages of oligomer assembly from aggregate-free stocks16 over a standard incubation period of 20–24 h.Widely differentiated behaviour in propensities for self-aggregation into oligomers and fibrils is observed in naturally secreted forms of Aβ (e.g., Aβ37–42, Aβ40) and also in forms of Aβ generated by genetically controlled mutations of APP in the brain.35–37 Underlying these effects is the key role of primary sequence in permitting β-sheet organisation. Due to its natural tendency to self-associate via multiple folding pathways, it is likely that amyloid structures in the brain are highly heterogeneous, incorporating truncated forms of Aβ42,38 and adventitious ligands may affect morphology and regulate proteostasis.39
Molecular ligands that facilitate or prevent assembly of either the β-sheet laminate or the intra-laminate assembly are likely to catalyse or inhibit, respectively, the self-assembly of Aβ42 into oligomers and fibrils. The development of peptide-based inhibitors of Aβ42 aggregation has focused significantly on active domains of chaperone proteins. For example, transgenically expressed human Aβ42 in C. elegans elicited the expression of known heat shock proteins that were subsequently immuno-precipitated with Aβ42,40,41 and which regulated the folding of Aβ42 towards less toxic pathways.41 Similarly, α1-anti-chymotrypsin (ACT), which is present in AD brain plaque, drives Aβ42 along either amorphous aggregation or fibril pathways, depending on the molar ratio.42 A peptide fragment of the chaperone protein α-crystallin also inhibited fibrillisation of Aβ42 (ref. 43) and its toxicity to PC12 cells.44 By analogy, mixture studies with different ratios of Aβ42 in the presence of Aβ40, show that the presence of Aβ40 can inhibit mature fibril development when Aβ40 approaches equi-molar ratios to Aβ42, with corresponding attenuation of cell toxicity.45 Dissociation of intact cross β-sheet structures (β-sheet ‘breakers’), have been successfully designed from peptoid and retro-peptoid analogues of an amyloidogenic peptide such as amylin.46 There are many ways by which chaperone-mediated interactions can alter the morphology and toxicity of fibrils40 and supports that fibrillisation pathways might be strategically manipulated by exogenous ligands.
In this study, we have demonstrated the perturbing effects of wPRP on beta-amyloid protein (Aβ42) folding pathways and cellular toxicity. The bioactivity of wPRP correlates with behaviours reported for proline-rich polypeptides isolated from ovine colostrum, ‘Colostrinin’, for related activity47 and other mammalian sources of colostrum.48 Peptides with capacity for fibril regulation, derived from bovine dairy sources including whey, casein and lactoferrin, have also been reported.29 The Colostrinin peptide complex was shown to inhibit and disrupt β-sheets of amyloid fibrils2 and exert several other bioactive properties49,50 that translated to proven neuroprotective bioactivity against AD.51 We have further shown that wPRP, while manifesting some apparently similar properties to Colostrinin, exerts significant structural modulation of Aβ42 oligomers, with important consequences for toxicity.
Oligomer-regulating effects of wPRP and implication of oligomeric intermediates of Aβ42 in toxicity
The oligomer-regulating effects of wPRP on Aβ42 fibrillisation and morphology were examined by ThT assay and FTIR spectroscopy, respectively. The modulation of ThT-binding to Aβ42 by wPRP implicated structural transitions from more fluorescent anti-parallel to less fluorescent parallel β-sheet fibril structures52 at lower concentrations (0.01 to 1.0 mg ml−1) before exerting inhibition of further fibril development at higher concentrations (>1.0 mg ml−1, Fig. 2). The phenomena of enhancement of ThT fluorescence was also reported for mixtures of Aβ40 with Aβ42.53ATR-FTIR spectroscopy has proven particularly useful for distinguishing secondary structures of oligomeric and fibrillar aggregates in β2-microglobulin,54 Gerstmann–Straussler–Scheinker disease peptide55 and HET-s (218–289) prion peptide.52 In addition, the technique has been successfully applied to evaluate the ratio of anti-parallel (oligomers) to parallel (fibrils) β-sheet content for Aβ42 along the aggregation process.16,34 In support of the current findings, it has been shown that anti-parallel β-sheet structure could also be the signature of toxicity for HET-s(218–289) prion peptide.52
FTIR monitoring of Aβ42 self-assembly in the presence of wPRP indicated that the Aβ42-wPRP complex (at high mass ratio of wPRP to Aβ42) formed structures of net lower β-sheet content and specifically lower anti-parallel β-sheet (Fig. 3 and 4b). The concomitantly protective effects against Aβ42 toxicity in yeast (SPE40 and SPE100 (>0.1 mg ml−1, Fig. 7b) and neuronal cells (SPE100 > 0.1 mg ml−1, Fig. 7c), were clearly evident. Low mass ratio of SPE40 and SPE100 to Aβ42 produced enhanced toxicity in yeast (Fig. 7b), which correlated with concentrations where anti-parallel β-sheet was present (Fig. 4a, a–c). Likewise, the suppression of anti-parallel β-sheet species by wPRP observed by FTIR (Fig. 4d–f), correlated with neuronal cell protection (Fig. 7) while some residual toxicity to yeast was still present (Fig. 7b). FTIR results did not correlate exactly with toxicity for yeast experiments at low mass ratios of SPE40 and SPE100 to Aβ42, suggesting that different Aβ42 isoforms might possess differentiated toxicity profiles in yeast compared with neuronal cells.
The simplest explanation for the ‘enhanced’ toxicity phase of Aβ42 + SPE40 (and SPE100) to yeast cells (Fig. 7b), is that low doses of SPE permitted the formation of toxic oligomers of Aβ42 in anti-parallel conformation compared with control and that high doses of SPE eventually diverted the anti-parallel β-sheet oligomers into less toxic β-sheet proto-fibrils, evident as β-sheet ‘breaking’ activity. This hypothesis is supported by the shifting of the ThT inhibition curve to higher concentrations of wPRP in the presence of pre-existing oligomers. Alternatively, it is also possible that low-dose SPE induced formation of ‘off pathway’ soluble complexes with Aβ42 of enhanced toxicity. The progressive decrease in β-sheet content of Aβ42 structures in the presence of wPRP (Fig. 3) supported capacity for β-sheet breaking activity by wPRP and SPE products.
Analysis of Aβ42-SPE interactions by TEM (Fig. 5) and Western blot analysis (Fig. 6) clearly showed that SPE inhibited Aβ42 oligomerisation and/or fibrillisation, supporting previous data from FTIR studies. In other words, wPRP (and SPE analogues) modulated all stages of Aβ42 self-assembly. The suppression of oligomeric Aβ42 by wPRP was found to be strongly associated with the decreased Aβ42-mediated toxicity in yeast and neurons (Fig. 7b and c) although FTIR (Fig. 4b) appeared to be more sensitive than Western blot analysis (Fig. 6) at detecting protective levels of suppression of oligomer formation at 0.01 mg ml−1 wPRP product. The disappearance of high mass oligomeric Aβ42 (50–110 kDa) due to SPE100 did not appear to be associated with loss of any low mass forms of Aβ42 (Fig. 6) and there was no evidence for disruption of pre-existing low mass oligomers over time, by Western Blot analysis (Fig. 6). The pattern of loss of high mass oligomers by SPE100 and associated suppression of toxicity was very similar to that seen in the presence of curcumin56 whereas the polyphenolic from olive oil ‘oleocanthal’ generated a laddered size-distribution of ‘off-pathway’ SDS-stable oligomers also with attenuated toxicity.57 Overall, we have shown positive correlation between suppression of anti-parallel β-sheet content by SPE40, SPE100 and wPRP (Fig. 3 and 4) and protection against Aβ42-mediated toxicity to both yeast and neuronal cells (Fig. 7).
Aβ42 folding is known to progress from anti-parallel to parallel β-sheet.34 It can be concluded that wPRP was able to interfere with the ‘natural’ self-assembly of Aβ42 into both toxic oligomeric anti-parallel β-sheet structures, described in ref. 34 as well as preventing parallel β-sheet fibrils structures and ultimately invoke ‘off-pathway’ aggregation of Aβ42 with lower ThT binding capacity (Fig. 2). A schematic representation of the proposed effects of wPRP at concentration ratios adequate to suppress toxicity is shown in Fig. 8. The structural features of oligomeric Aβ42 formed after 20 h incubation in Fig. 8 are supported by evidence from FTIR (Fig. 4a), TEM (Fig. 5a) and Western blot analysis (Fig. 6, lanes 1 at 24 and 48 h), whereas structural features of wPRP-bound Aβ42 are supported by evidence from FTIR (Fig. 4a–f), TEM (Fig. 5c) and Western blot analysis (Fig. 6, lanes 4–5 at 24 and 48 h). In addition, the decreasing content of β-sheet evident by FTIR (Fig. 3) indicated a net reversal of total β-sheet content at all tested concentrations of wPRP and specifically of anti-parallel β-sheet at wPRP concentrations above 0.005 mg ml−1 (Fig. 4b), suggesting that wPRP was able to ‘break’ pre-existing β-sheets.
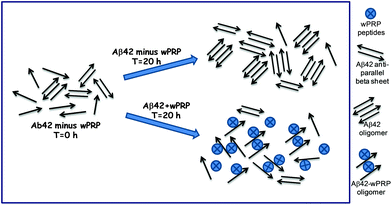 | ||
| Fig. 8 Schematic showing proposed effects of wPRP on structures of soluble forms of Aβ42. | ||
The observation of a generic structure-toxicity relationship was first suggested by Bucciantini et al, (2002) where the cytotoxicity of aggregated proteins nominally unrelated with disease, were proposed to be related to their generic granular or fibrillar structure.58 Further evidence of the structural distinction of Aβ42 oligomers was provided by the selectivity of the A11 conformational antibody59 and recently, anti-parallel organization of β-sheets in Aβ oligomers were characterised by Raussens' group using ATR-FTIR spectroscopy.16,34
Collectively, these results provide important supportive evidence for the direct linkage between toxicity of anti-parallel versus non-toxicity (to cells) of parallel β-sheet structures of Aβ42 and by extension, that the suppression of anti-parallel β-sheet structures is necessary to prevent formation of oligomeric structures linked with toxicity. The lack of toxicity of a mutant form of Aβ12–28,36 in contrast to Aβ25–35 and related variants,60 in spite of the presence of aggregates characterised by having β-sheet secondary structure, further supports these observations. Several groups have reported formation of amorphous, ‘off-pathway’ oligomers of Aβ42 that were non-toxic,61,62 as distinct from the toxic effects of ‘classical’ oligomers.63 The evidence that not all oligomers are toxic further supports the important evidence provided here showing that anti-parallel β-sheet structure is necessary for cellular toxicity.
Experimental
Materials
Whey protein isolate was obtained from Murray Goulburn, Natrapro WPI, MG Nutritionals, Brunswick, Australia. Glutaminase was obtained from Daiwa Kasei K.K., Shiga, Japan; Corolase PN-L from AB Enzymes GmbH, Darmstadt, Germany; Alcalase 2.4L, Flavourzyme 1000L and trypsin were from Novozymes, Bagsvaerd, Denmark). Synthetic human Aβ42 was sourced from either GL-Biochem Ltd, Shanghai, China; American Peptide, Sunnyvale, CA, USA or Keck Laboratory, Yale University, New Haven, CT, USA. Tri-ethanolamine, ethanol, DMSO, 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP), uranyl acetate, thioflavinT (ThT), WO2 mouse, anti-Aβ3–10 and rabbit anti-GFP antibodies were sourced from Sigma Chemical Co., St Louis, MO, USA. Phosphate buffer and sodium chloride were obtained from Merck, Damstadt, Germany. Acetonitrile was from Ajax Fine Chemical, NSW, Australia. Eosin was from B.D.H. Laboratory, Poole, England. Horeseradish peroxidase-reactive enhanced chemiluminiscence (ECL) reagent was from GE Healthcare, Waukesha, WI, USA.Preparation and characterisation of wPRP and SPE products
Dairy protein hydrolysate was prepared from bovine whey protein isolate (Murray Goulburn) by dispersing at 10% total protein (w/w) in 10 mM tri-ethanolamine (Sigma), 10% EtOH, and maintaining at pH 7.4 throughout processing. The enzymes: Glutaminase (Daiwa Kasei K.K.), Corolase PN-L (AB Enzymes GmbH), Alcalase (2.4L, Novozymes) and Flavourzyme (1000L, Novozymes), were introduced in sequence, each at a final concentration of 0.5% (w/w) and incubated sequentially at 50 °C for 1 h. Finally, Trypsin (Novozymes, 0.5%, w/w) was added and incubated at 37 °C for 17 h before heating at 90 °C for 30 min to inactivate all enzymes. The molecular size fraction <8 kDa was recovered by dialysis using regenerated cellulose membrane (6–8 kDa molecular weight cut-off, Spectrum Laboratories, Inc., Dominguez, CA) before further processing by ion exchange (IEX) chromatography, using 2 columns (4.6 × 10 cm) connected in series. Column 1 was packed with cation exchange resin (SP Sepharose Big Beads, GE Healthcare, Uppsala, Sweden) and Column 2 was packed with anion exchange resin (Q Sepharose Big Beads, GE Healthcare). Batches of dialysate (400 ml) were loaded onto the pair of IEX columns with 400 ml of eluant containing non-binding peptides recovered. In this case, the IEX-binding fractions were not recovered. Eluates were freeze dried and stored at −20 °C. A single batch of the total hydrolysate was used throughout.The product (containing 12.2% nitrogen) was sub-fractionated using C18 solid phase cartridges (Strata-X 33 μm Polymeric Reverse Phase cartridges (500 mg per 6 ml, Phenomenex, California, USA). After washing with methanol and re-equilibrating with water, sample (100 mg ml−1 total solids in water, 5.0 ml) was loaded and non-binding solids eluted in a further 5.0 ml of water (designated load + void sample). Bound fractions were sequentially eluted with 5.0 ml each of 40% and 100% acetonitrile respectively (Ajax Fine Chem, NSW, Australia) and designated SPE40, and SPE100 respectively. In some cases the bound fraction was eluted entirely into 100% acetonitrile (wPRP). The ratio of solid eluted by 40% to 100% acetonitrile was approximately 19:1. Products were dried by evaporation under vacuum and stored at −20 °C.
SPE products and wPRP were analysed (5 mg ml−1, 20 μl injection) by reverse phase HPLC (Jupiter 5μ C18 300 Å, 250 × 4.6 – Phenomenex, California, USA) under gradient elution (mobile phase A (0.1% TFA in water, Sigma) and B (0.1% TFA in 95% acetonitrile), using a Waters Alliance HPLC with a flow rate of 1.0 ml min−1 and photo-diode array detector at 220 nm. The gradient was programmed for 2 to 50% B over 54 min, then 100% B for 4 min before re-equilibration to starting conditions. The equivalence of batches of SPE40, SPE100 and wPRP prepared for these studies (from the single batch of hydrolysate), was verified by HPLC profiling.
Amino acid analysis of the wPRP was conducted using the High Sensitivity Waters AccQTag Ultra (Milford, MA, USA) chemistry. Results of duplicate analyses were expressed in mole percent of detectable amino acids. Tryptophan was not detectable by this method.
Aβ42 ThT binding assay
Aβ42 (GL-Biochem Ltd) was dispersed at 1.0 mg ml−1 in HFIP (Sigma) which denatures amyloid aggregates, dissociates cross-β-sheet structures, with gentle vortexing (1 min) followed by incubation at 22 °C for 30 min. Vials containing 0.5 ml aliquots (0.5 mg of peptide) were dried under nitrogen (high purity, BOC Gases, Australia) leaving a ‘film’ of Aβ42, and stored at −18 °C until required. Aggregate-free solutions of Aβ42 were prepared according to the method of Broersen et al (2011).64 Vials containing 0.5 mg Aβ-HFIP films were thawed (22 °C for 10 min) before redispersing in 0.5 ml HFIP and redrying under nitrogen as described above. The Aβ-HFIP film was redissolved in 0.5 ml DMSO (Sigma), by the same method as that for HFIP. After 30 min incubation at 22 °C, the solution was loaded onto a desalting column (HiTrap™ Desalting column, GE Healthcare, Upsala, Sweden) previously washed and equilibrated with chilled buffer solution (50 mM phosphate, 100 mM NaCl, pH 7.2). The peptide solution in DMSO (500 μl) was loaded onto the column and washed with buffer (1 ml) which was discarded. Aggregate-free Aβ42 was then eluted in 2 × 0.5 ml aliquots of chilled buffer, into pre-cooled micro-centrifuge tubes which were maintained on ice during adjustment of concentration and pending use in assay, unless stated otherwise. Using the extinction coefficient of 0.33 mg ml M−1 cm−1 at 280 nm,65 the concentration of aggregate-free Aβ42 was adjusted to 0.1 mg ml−1 with chilled buffer and used in the ThT assay within 30 min unless otherwise stated.The assay for measuring Amyloid fibril inhibition by Aβ42 was adapted from.66 Aggregate-free Aβ42 (0.1 mg ml−1 in buffer) was incubated with test samples and controls using black 96-microwell plates (Ooti-Plate-96F, Perkin Elmer, Shelton, USA) at 37 °C for 24 h, that were sealed to prevent evaporation. wPRP, dissolved in 10% ethanol, was tested over the concentration range from 0.001 to 1 mg ml−1. After incubation, plates were cooled to 22 °C and ThT (Sigma) solution added to a final concentration of 2 μM. Fluorescence was measured using a fluorescence plate reader (VarioSkan, Thermo Scientific, Flash, USA) at excitation and emission wavelengths of 442 and 482 nm, respectively. Positive and negative controls were Eosin (3.4 μM in 10% ethanol, BDH), and buffer containing 10% ethanol, respectively. Percentage fibril inhibition (Fi) was calculated as follows: Fi(%) = 100 − [100 × ((S − C)/(K − B)), where S and C are fluorescence intensities of samples in the presence and absence (control) of Aβ42, respectively. K is the fluorescence intensity of the uninhibited control of Aβ42 and B is the reagent blank. Analysis was conducted in triplicate for all samples.
Fourier transform infra-red spectroscopy
Aggregate-free Aβ1–42 (American Peptide) was prepared as follows: lyophilized Aβ42 was dissolved (2 mg ml−1) in HFIP (Sigma) and the solvent evaporated under nitrogen and vacuum (Speed Vac, Thermo Savant, Thermo Fisher Scientific, USA). Aβ42 was re-dissolved in de-ionised water at 4 °C and contained only low molecular weight species (monomers, trimers and tetramers) by PAGE and 100% anti-parallel β-sheet structure by FTIR.16 Stock solutions of Aβ42 and wPRP prepared in de-ionised water were mixed to desired final concentrations and incubated at 30 °C for 20 h.IR spectra were recorded on an Equinox 55 infrared spectrophotometer (Bruker Optics, Ettlingen, Germany) placed in a thermo-regulated room (21 °C) and equipped with a liquid N2-refrigerated mercury-cadmium-telluride detector. 128 repetitions of triplicate independent samples were recorded at a resolution of 2 cm−1. Other details of sample analysis are described in ref. 16. Fourier self-deconvolution was applied to increase the resolution of spectra in the amide I region, which is that most sensitive to protein secondary structure. The FTIR data were preprocessed as described in ref. 67. Briefly, the water vapor contribution was subtracted with 1956–1935 cm−1 as the reference peak, and then the spectra were baseline-corrected and normalized for equal area between 1700–1500 cm−1. The spectra were finally smoothed at a final resolution of 4 cm−1 by apodization of their Fourier transform using a Gaussian peak shape (full width at half height of 13.33 cm−1) and self-deconvolution was carried out using a Lorentzian peak shape (full width at half height of 20 cm−1). The resolution enhancement factor was therefore 1.5. Extraction of spectral data was conducted using the Kinetics program in Matlab (Mathworks Inc. Natick, MA, USA).
Transmission electron microscopy
Synthetic human Aβ42 (Keck Laboratory). Solutions of aggregate-free Aβ42 were prepared according to the method of Bharadwaj et al, 2008 (ref. 68) with the following modifications. Aβ42 was dissolved in HFIP, before incubating overnight at 22 °C. The HFIP solution was centrifuged (20817 × g, 10 min, Eppendorf 5417R, Hamburg, Germany) and the supernatant recovered. The HFIP was then evaporated and the resulting film dissolved in sterile (0.2 μm-filtered) de-ionised water. The solution was sonicated on ice for 5 min and centrifuged (20817 × g, 10 min, Eppendorf 5417R) before further incubating the supernatant (17 h, 22 °C). The concentration of Aβ42 peptide was adjusted as required using the reported extinction co-efficient at 280 nm.45
Aβ42 solutions were pre-incubated (10 μM, 0.05 mg ml−1 in de-ionised water), with SPE100 product (0.1 mg ml−1 in 10% ethanol) or vehicle (10% ethanol) for 20 h at 22 °C. The samples were diluted by 1/5 with de-ionised, filtered (0.2 μm) water to a final concentration of 2 μM Aβ42 (0.01 mg ml−1) before applying to carbon-coated 400 mesh copper grids, which had been glow discharged in nitrogen. After 1 min adsorption time excess sample was wicked off with filter paper and the sample stained with 2–3 drops of 2% aqueous uranyl acetate (Sigma). The grids were air-dried and examined in a Tecnai 12 Transmission Electron Microscope (FEI, Eindhoeven, The Netherlands) operating at 120 kV. Micrographs are recorded using a Megaview III CCD camera running under AnalySiS imaging software (Olympus Australia, Mt Waverley, Australia).
SDS-page and Western blot analysis
Aβ42 (0.5 mg ml−1, 35 μl, prepared as for TEM studies) and SPE100 (0 to 0.1 mg ml−1, 5 μl) mixture was loaded (50 ng Aβ42) and electrophoretically resolved on 4–12% Bis–Tris gels (Nupage Novex, Invitrogen, Mulgrave, Victoria). The proteins were transferred from the polyacrylamide gel to a nitrocellulose membrane using iBlot (Invitrogen) dry transfer method. Membranes were blocked in TBS solution (5% casein, Tris, 50 mM, NaCl, 150 mM, pH 7.4 for 1 h. Primary antibodies WO2 (mouse, anti-Aβ3–10, Sigma) or rabbit, anti-GFP (Sigma) were diluted in TBST solution (0.5% casein, 0.05% Tween in TBS) at concentrations of 1/3000 and 1/5000 respectively. Incubation (2 h at 22 °C) was followed by three washes with TBST. Secondary antibodies conjugated with horseradish peroxidase (HRP, anti-mouse or anti-rabbit) were diluted by 1/5000 in TBST solution and incubated with membranes for 1 h. After washing with TBST and TBS, membranes were incubated for 2 min with HRP-reactive enhanced chemiluminiscence, ECL) reagent (GE Healthcare, Waukesha, WI, USA). The membranes were developed on films which were scanned using a densitometer.Cell viability assays
Aβ42 toxicity assay in yeast was done according to the method of Bharadwaj et al, 2008 (ref. 69) using the prototrophic yeast strain Candida glabrata ATCC 90030. Cultures of yeast cells were grown in YEPD (1% yeast extract; 2% peptone, 2% dextrose), to exponential growth phase. Cultures containing ∼108 cells per ml, were diluted to ∼5 × 103 cells per ml in sterile, de-ionised water before aliquotting into 96-well plates for sample treatment. SPE40 and SPE100 samples reconstituted in de-ionised water were added to the diluted cell suspension to required final concentrations and constant final volume (125 μl). The plate was sealed with gas-permeable membrane (Diversified Biotech Inc., MA, USA) and incubated at 30 °C with agitation (150 rpm for 20 h). Cell survival was determined by plating aliquots of SPE sample-treated cell suspensions (in triplicate) onto YEPD agar plates (50 g l−1 YEPD, 1.8 g l−1 bacto agar) before counting the number of colony-forming units (CFU). Viability was calculated as the percentage change in CFUs of control to sample-treated samples.The SH-SY5Y human neuroblastoma cell line was maintained and cultured as described in ref. 70. For Aβ42 toxicity experiments, cells were plated in 96-well tissue culture plates at a density of 104 cells per well in DMEM media containing 1% FCS for 20 h. Oligomeric Aβ42 (10 μM) previously co-incubated for 20 h with SPE products (0.001–0.1 mg ml−1) was added to the cells and incubated for 72 h at 37 °C. Lactate dehydrogenase (LDH) released into the media as a result of Aβ42 toxicity, was measured in cell supernatants using the CytoTox 96R Cytotoxicity assay (Promega, Alexandria, NSW, Australia) as described in ref. 71. The cells were then incubated (4 h at 37 °C) with fresh DMEM medium containing 1% (v/v) 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS, Promega) before measuring viability according to ref. 72. The color change at 490 nm was determined using a Fluostar Optima plate reader (BMG Labtech, Victoria, Australia), and results corrected for reagent controls. The data were reported as the percentage standardized change compared with sample-free controls, after correction for reagent blanks.
Statistical analysis
All analyses were performed in at least triplicate unless otherwise stated and errors are given as either standard deviation or standard error of the mean. Analysis of differences between means was conducted using Prism Graphpad (La Jolla, CA, USA), using two-way Student’s t-test and reported at either 95% or 99% confidence levels.Conclusion
A number of molecular species have been shown to regulate the folding pathway of Aβ42. In many cases, the toxicity of products was differentiated from unmodified Aβ42, usually attenuated. Thus, off-pathway modulators of Aβ42 folding may represent useful molecules for development into disease-modifying therapeutics. The results demonstrate the important finding that suppression of anti-parallel β-sheet structures is specifically required for regulation of oligomer toxicity to cells. ATR-FTIR spectroscopy was able to detect these subtle changes in structure and provided important evidence towards understanding wPRP bioactivity. It is unknown the extent to which dietary factors already play a role in perturbing toxicity of Aβ42 in vivo, if at all. It further remains to be determined if bio-available dietary peptides and phytochemicals might contribute to neuroprotection in this way. Successful in vivo neuroprotective studies with Colostrinin, however, suggest that other exogenous peptides such as wPRP, might be also be protective against AD and other amyloidogenic diseases. Finally, wPRP may also be useful for modulating other disease-related proteins displaying the common structural feature of amyloidogenic protein intermediates with anti-parallel β-sheet conformations and physiological toxicity.Abbreviations
| Aβ42 | Amyloid-β1–42; |
| wPRP | Proline-rich dairy peptide product; |
| SPE | Solid phase extraction; |
| ATR-FTIR | Attenuated Total Reflectance Fourier Transform Infra-Red spectroscopy; |
| TEM | Transmission Electron Microscopy; |
| ThT | Thioflavin T; |
| AD | Alzheimer's Disease. |
Acknowledgements
This work was conducted with funding from the CSIRO Preventative Health Flagship. Amino acid analysis was conducted at the Australian Proteome Analysis Facility Ltd using infrastructure provided by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS). V.R. is senior research associate for the National Fund for Scientific Research (FNRS; Brussels, Belgium), R.S. is Research Fellow for the Fund for Research in Industry and Agriculture (Belgium).References
- T. Rapp, J. Nutr., Health Aging, 2010, 14, 630–632 CrossRef CAS.
- D. Schuster, A. Rajendran, S. W. Hui, T. Nicotera, T. Srikrishnan and M. L. Kruzel, Neuropeptides, 2005, 39, 419–426 CrossRef CAS.
- P. R. Bharadwaj, A. K. Dubey, C. L. Masters, R. N. Martins and I. G. Macreadie, J. Cell. Mol. Med., 2009, 13, 412–421 CrossRef CAS.
- V. H. Finder and R. Glockshuber, Neurodegener. Dis., 2007, 4, 13–27 CrossRef CAS.
- F. M. LaFerla, K. N. Green and S. Oddo, Nat. Rev. Neurosci., 2007, 8, 499–509 CrossRef CAS.
- M. Necula, R. Kayed, S. Milton and C. G. Glabe, J. Biol. Chem., 2007, 282, 10311–10324 CrossRef CAS.
- T. Takahashi and H. Mihara, Acc. Chem. Res., 2008, 41, 1309–1318 CrossRef CAS.
- B. A. Yankner and T. Lu, J. Biol. Chem., 2009, 284, 4755–4759 CrossRef CAS.
- C. Nerelius, J. Johansson and A. Sandegren, Front. Biosci., 2009, 14, 1716–U3856 CrossRef CAS.
- L. D. Estrada and C. Soto, Curr. Top. Med. Chem., 2007, 7, 115–126 CrossRef CAS.
- G. Habicht, C. Haupt, R. P. Friedrich, P. Hortschansky, C. Sachse, J. Meinhardt, K. Wieligmann, G. P. Gellermann, M. Brodhun, J. Gotz, K. J. Halbhuber, C. Rocken, U. Horn and M. Fandrich, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19232–19237 CrossRef CAS.
- L. P. Yu, R. Edalji, J. E. Harlan, T. F. Holzman, A. P. Lopez, B. Labkovsky, H. Hillen, S. Barghorn, U. Ebert, P. L. Richardson, L. Miesbauer, L. Solomon, D. Bartley, K. Walter, R. W. Johnson, P. J. Hajduk and E. T. Olejniczak, Biochemistry, 2009, 48, 1870–1877 CrossRef CAS.
- V. A. Streltsov, J. N. Varghese, C. L. Masters and S. D. Nuttall, J. Neurosci., 2011, 31, 1419–1426 CrossRef CAS.
- S. Chimon, M. A. Shaibat, C. R. Jones, D. C. Calero, B. Aizezi and Y. Ishii, Nat. Struct. Mol. Biol., 2007, 14, 1157–1164 CAS.
- A. Sandberg, L. M. Luheshi, S. Sollvander, T. P. de Barros, B. Macao, T. P. J. Knowles, H. Biverstal, C. Lendel, F. Ekholm-Petterson, A. Dubnovitsky, L. Lannfelt, C. M. Dobson and T. Hard, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15595–15600 CrossRef CAS.
- E. Cerf, R. Sarroukh, S. Tamamizu-Kato, L. Breydo, S. Derclaye, Y. F. Dufrene, V. Narayanaswami, E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, Biochem. J., 2009, 421, 415–423 CrossRef CAS.
- M. Stefani, FEBS J., 2010, 277, 4602–4613 CrossRef CAS.
- J. Lee, E. K. Culyba, E. T. Powers and J. W. Kelly, Nat. Chem. Biol., 2011, 7, 602–609 CrossRef CAS.
- L. Dumery, F. Bourdel, Y. Soussan, A. Fialkowsky, S. Viale, P. Nicolas and M. Reboud-Ravaux, Pathol. Biol., 2001, 49, 72–85 CrossRef CAS.
- B. Y. Ma and R. Nussinov, Curr. Opin. Chem. Biol., 2006, 10, 445–452 CrossRef CAS.
- D. X. Liu, Y. C. Xu, Y. Feng, H. Liu, X. Shen, K. X. Chen, J. P. Ma and H. L. Jiang, Biochemistry, 2006, 45, 10963–10972 CrossRef CAS.
- A. Kapurniotu, A. Buck, M. Weber, A. Schmauder, T. Hirsch, J. Bernhagen and M. Tatarek-Nossol, Chem. Biol., 2003, 10, 149–159 CrossRef CAS.
- J. F. Poduslo, G. L. Curran, A. Kumar, B. Frangione and C. Soto, J. Neurobiol., 1999, 39, 371–382 CrossRef CAS.
- N. Kokkoni, K. Stott, H. Amijee, J. M. Mason and A. J. Doig, Biochemistry, 2006, 45, 9906–9918 CrossRef CAS.
- D. J. Gordon, R. Tappe and S. C. Meredith, J. Pept. Res., 2002, 60, 37–55 CrossRef CAS.
- E. Ferrandini, M. Castillo, M. B. Lopez and J. Laencina, J. Anim. Sci., 2005, 83, 143 Search PubMed.
- D. C. Thorn, H. Ecroyd, M. Sunde, S. Poon and J. A. Carver, Biochemistry, 2008, 47, 3926–3936 CrossRef CAS.
- D. C. Thorn, S. Meehan, M. Sunde, A. Rekas, S. L. Gras, C. E. MacPhee, C. M. Dobson, M. R. Wilson and J. A. Carver, Biochemistry, 2005, 44, 17027–17036 CrossRef CAS.
- L. Bennett, R. Williams, H. Ecroyd, Y. Q. Liu, S. Sudharmarajan and J. Carver, Aust. J. Dairy Technol., 2009, 64, 117–121 CAS.
- J. A. Carver, P. J. Duggan, H. Ecroyd, Y. Q. Liu, A. G. Meyer and C. E. Tranberg, Bioorg. Med. Chem., 2010, 18, 222–228 CrossRef CAS.
- M. R. H. Krebs, K. R. Domike, D. Cannon and A. M. Donald, Faraday Discuss., 2008, 139, 265–274 RSC.
- R. W. Carrell, A. Mushunje and A. Zhou, FEBS Lett., 2008, 582, 2537–2541 CrossRef CAS.
- T. S. Burkoth, T. L. S. Benzinger, V. Urban, D. M. Morgan, D. M. Gregory, P. Thiyagarajan, R. E. Botto, S. C. Meredith and D. G. Lynn, J. Am. Chem. Soc., 2000, 122, 7883–7889 CrossRef CAS.
- R. Sarroukh, E. Cerf, S. Derclaye, Y. F. Dufrene, E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, Cell. Mol. Life Sci., 2010 DOI:10.1007/s00018-010-0529-x Search PubMed.
- T. Tomiyama, T. Nagata, H. Shimada, R. Teraoka, A. Fukushima, H. Kanemitsu, H. Takuma, R. Kuwano, M. Imagawa, S. Ataka, Y. Wada, E. Yoshioka, T. Nishizaki, Y. Watanabe and H. Mori, Ann. Neurol., 2008, 63, 377–387 CrossRef CAS.
- A. Peralvarez-Marin, L. Mateos, C. Zhang, S. Singh, A. Cedazo-Minguez, N. Visa, L. Morozova-Roche, A. Graslund and A. Barth, Biophys. J., 2009, 97, 277–285 CrossRef CAS.
- W. B. Stine, S. W. Snyder, U. S. Ladror, W. S. Wade, M. F. Miller, T. J. Perun, T. F. Holzman and G. A. Krafft, J. Protein Chem., 1996, 15, 193–203 CrossRef CAS.
- M. Bibl, B. Mollenhauer, H. Esselmann, P. Lewczuk, H. W. Klafki, K. Sparbier, A. Smirnov, L. Cepek, C. Trenkwalder, E. Ruther, J. Kornhuber, M. Otto and J. Wiltfang, Brain, 2006, 129, 1177–1187 CrossRef.
- C. Voisine, J. S. Pedersen and R. I. Morimoto, Neurobiol. Dis., 2010, 40, 12–20 CrossRef CAS.
- V. Fonte, V. Kapulkin, A. Taft, A. Fluet, D. Friedman and C. D. Link, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9439–9444 CrossRef CAS.
- V. Fonte, D. R. Kipp, J. Yerg, D. Merin, M. Forrestal, E. Wagner, C. M. Roberts and C. D. Link, J. Biol. Chem., 2008, 283, 784–791 CrossRef CAS.
- S. Janciauskiene, H. Rubin, C. M. Lukacs and H. T. Wright, J. Biol. Chem., 1998, 273, 28360–28364 CrossRef CAS.
- B. Raman, T. Ban, M. Sakai, S. Y. Pasta, T. Ramakrishna, H. Naiki, Y. Goto and C. M. Rao, Biochem. J., 2005, 392, 573–581 CrossRef CAS.
- P. Santhoshkumar and K. K. Sharma, Mol. Cell. Biochem., 2004, 267, 147–155 CrossRef CAS.
- A. Jan, O. Gokce, R. Luthi-Carter and H. A. Lashuel, J. Biol. Chem., 2008, 283, 28176–28189 CrossRef CAS.
- R. C. Elgersma, G. E. Mulder, J. A. W. Kruijtzer, G. Posthuma, D. T. S. Rijkers and R. M. J. Liskamp, Bioorg. Med. Chem. Lett., 2007, 17, 1837–1842 CrossRef CAS.
- M. L. Kruzel, M. Janusz, J. Lisowski, R. V. Fischleigh and J. A. Georgiades, J. Mol. Neurosci., 2001, 17, 379–389 CrossRef CAS.
- A. Sokolowska, R. Bednarz, M. Pacewicz, J. A. Georgiades, T. Wilusz and A. Polanowski, Int. Dairy J., 2008, 18, 204–209 CrossRef CAS.
- I. Boldogh and M. L. Kruzel, J. Alzheimer's Dis., 2008, 13, 303–321 CAS.
- M. Zimecki, in Bioactive Components of Milk, Springer-Verlag, Berlin, 2008, vol. 606, pp. 241–250 Search PubMed.
- A. Bilikiewicz and W. Gaus, J. Alzheimers Dis., 2004, 6, 17–26 CAS.
- K. Berthelot, H. P. Ta, J. Gean, S. Lecomte and C. Cullin, J. Mol. Biol., 2011, 412, 137–152 CrossRef CAS.
- Y. Yoshiike, D. H. Chui, T. Akagi, N. Tanaka and A. Takashima, J. Biol. Chem., 2003, 278, 23648–23655 CrossRef CAS.
- H. Fabian, K. Gast, M. Laue, R. Misselwitz, B. Uchanska-Ziegler, A. Ziegler and D. Naumann, Biochemistry, 2008, 47, 6895–6906 CrossRef CAS.
- A. Natalello, V. V. Prokorov, F. Tagliavini, M. Morbin, G. Forloni, M. Beeg, C. Manzoni, L. Colombo, M. Gobbi, M. Salmona and S. M. Doglia, J. Mol. Biol., 2008, 381, 1349–1361 CrossRef CAS.
- F. S. Yang, G. P. Lim, A. N. Begum, O. J. Ubeda, M. R. Simmons, S. S. Ambegaokar, P. P. Chen, R. Kayed, C. G. Glabe, S. A. Frautschy and G. M. Cole, J. Biol. Chem., 2005, 280, 5892–5901 CrossRef CAS.
- J. Pitt, W. Roth, P. Lacor, A. B. Smith, M. Blankenship, P. Velasco, F. De Felice, P. Breslin and W. L. Klein, Toxicol. Appl. Pharmacol., 2009, 240, 189–197 CrossRef CAS.
- M. Bucciantini, E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. S. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson and M. Stefani, Nature, 2002, 416, 507–511 CrossRef CAS.
- R. Kayed and C. G. Glabe, in Amyloid, Prions, and Other Protein Aggregates, Pt C, ed. I.Kheterpal and R. Wetzel, 2006, vol. 413, pp. 326–344 Search PubMed.
- C. J. Pike, A. J. Walencewiczwasserman, J. Kosmoski, D. H. Cribbs, C. G. Glabe and C. W. Cotman, J. Neurochem., 1995, 64, 253–265 CrossRef CAS.
- A. R. A. Ladiwala, M. Mora-Pale, J. C. Lin, S. S. Bale, Z. S. Fishman, J. S. Dordick and P. M. Tessier, ChemBioChem, 2011, 12, 1749–1758 CrossRef CAS.
- S. Rigacci, V. Guidotti, M. Bucciantini, D. Nichino, A. Relini, A. Berti and M. Stefani, Curr. Alzheimer Res., 2011, 841–852 CrossRef CAS.
- M. R. Nilsson, Methods, 2004, 34, 151–160 CrossRef CAS.
- K. Broersen, W. Jonckheere, J. Rozenski, A. Vandersteen, K. Pauwels, A. Pastore, F. Rousseau and J. Schymkowitz, Protein Eng., Des. Sel., 2011, 24, 743–750 CrossRef CAS.
- A. Jan, D. M. Hartley and H. A. Lashuel, Nat. Protoc., 2010, 5, 1186–1209 CrossRef CAS.
- H. Ecroyd, T. Koudelka, D. C. Thorn, D. M. Williams, G. Devlin, P. Hoffmann and J. A. Carver, J. Biol. Chem., 2008, 283, 9012–9022 CrossRef CAS.
- E. Goormaghtigh, V. Raussens and J. M. Ruysschaert, Biochim. Biophys. Acta, Rev. Biomembr., 1999, 1422, 105–185 CrossRef CAS.
- P. Bharadwaj, L. Waddington, J. Varghese and I. G. Macreadie, J. Alzheimer's Dis., 2008, 13, 147–150 CAS.
- P. Bharadwaj, L. Waddington, J. Varghese and I. G. Macreadie, J. Alzheimer's Dis., 2008, 13, 1–4 Search PubMed.
- L. Zhang, H. X. Yu, C. C. Song, X. F. Lin, B. Chen, C. Tan, G. X. Cao and Z. W. Wang, Protein Expression Purif., 2009, 64, 55–62 CrossRef CAS.
- J. Bieschke, J. Russ, R. P. Friedrich, D. E. Ehrnhoefer, H. Wobst, K. Neugebauer and E. E. Wanker, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7710–7715 CrossRef CAS.
- G. D. Ciccotosto, D. J. Tew, S. C. Drew, D. G. Smith, T. Johanssen, V. Lal, T.-L. Lau, K. Perez, C. C. Curtain, J. D. Wade, F. Separovic, C. L. Masters, J. P. Smith, K. J. Barnham and R. Cappai, Neurobiol. Aging, 2009, 32, 235–248 CrossRef.
- J. A. Georgiades, EP1341816-A; WO200246211-A2; AU200252801-A; EP1341816-A2; US2005085422-A1; AU2002252801-A8; US2008085299-A1; EP1935900-A1, 2004.
| This journal is © The Royal Society of Chemistry 2013 |