Amorphous CoSnO3@C nanoboxes with superior lithium storage capability†
Zhiyu
Wang
b,
Zichen
Wang
b,
Wenting
Liu
a,
Wei
Xiao
*a and
Xiong Wen (David)
Lou
*b
aSchool of Resource and Environmental Sciences, Wuhan University, Wuhan 430072, P. R. China. E-mail: gabrielxiao@whu.edu.cn
bSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 62 Nanyang Drive, Singapore 637459, Singapore. E-mail: xwlou@ntu.edu.sg; Web: http://www.ntu.edu.sg/home/xwlou
First published on 4th October 2012
Abstract
Amorphous CoSnO3@C nanoboxes have been synthesized by thermal annealing of CoSn(OH)6 nanoboxes, followed by carbon nanocoating. Benifiting from the unique structure, they exhibit exceptional long-term cycling stability over 400 cycles for highly reversible lithium storage.
Broader contextNext-generation lithium-ion batteries (LIBs) with higher energy/power density are vitally important for electric vehicles and mobile electronics with a market value of billions of dollars. The development of high-performance electrode materials, especially those with long service life, is the key to implement them for practical use. In this contribution, we have rationally designed a multifeatured nanostructure, amorphous CoSnO3@C nanoboxes, for highly reversible lithium storage. This unique structure incorporates several desirable design rationales for long-life anode materials based on hollow nanostructures, carbon nanopainting, mixed conductive matrix and crystalline texture engineering. In favor of the synergy and interplay of the matrix effect and intrinsic structural advantages, the CoSnO3@C nanoboxes exhibit an exceptional cycle life of over 400 cycles and improved high-rate capability when evaluated as an anode material for lithium-ion batteries. |
Rechargeable lithium-ion batteries (LIBs) have been the most popular power source for high-end consumer electronics for many years. The superior advantages such as high energy density, long lifespan, no memory effect and environmental benignity make them the best system for electric energy storage with a market value of billions of dollars.1–6 In addition to the portable electronics, high-energy LIBs are also urgently needed to meet the continuously surging demand in large-scale energy applications such as electric vehicles, renewable power plants and electric grids. This creates a great deal of interest in seeking high-performance electrode materials that can store and deliver more energy efficiently. Among the available anode materials, metal oxides have always been regarded as very appealing candidates because of their much higher capacity than that of commercial graphite anodes (372 mA h g−1), widespread availability, intrinsically enhanced safety and low processing cost.6,7 Basically, they can be classified into three groups based on the different reaction mechanisms towards lithium. The first group of metal oxides such as TiO2 can take lithium into the vacant sites of crystalline structure without bond cleavage.8 Due to the limited number of Li+ ions that can be inserted, this class of metal oxides often exhibits relatively low lithium storage capacities of below 200–300 mA h g−1. The second group, mostly the interstitial-free transitional metal oxides with redox centers, store lithium via a conversion reaction, where the metal oxides are reduced to metallic nanocrystals dispersed in a Li2O matrix upon lithiation and then are reversibly restored to the initial state after delithiation.6,9–11 In general, they can deliver high specific capacities of 600–1000 mA h g−1 as a result of a fully utilized oxidation state. Tin-based metal oxides fall into the third group of metal oxides, of which the lithium storage mainly relies on the reversible alloying–dealloying reaction between lithium and metal nanocrystals generated from the initial irreversible reduction of oxides.12–16 This class of materials generally functions at a relatively low potential of below 1.0 V (vs. Li/Li+) to exhibit very high capacity, and thus can be combined with most high-voltage cathode materials (e.g., LiCoO2, LiFePO4 and LiMn2O4) to make LIBs with high energy density. A major drawback of this class of materials, however, is the severe electrode pulverization caused by the drastic volume variation (up to 200%) during repeated electrochemical cycling, which greatly hampers their long-term cycling stability.
Because of the fundamental electrochemical origin, the mitigation of electrode pulverization has proven to be quite difficult. One commonly used approach to enhance the electrode stability of tin-based metal oxides is to allow the alloying reaction to proceed in a hybrid matrix of distinct material systems, for example the oxides and carbonaceous materials. The confining matrix, which can be either electrochemically active or inactive towards lithium, may effectively buffer the volume changes of the active materials upon cycling while preventing them from aggregation, thus prolonging the cycle life of the electrodes. In practice, the benefit of this concept has been demonstrated in a series of tin-based nanocomposites such as Sn2BPO6, Li2O–CuO–SnO2 multideck-cages, nanostructured Sn–C composites, Sn–metal–C thin films, nanoarchitectured Cu supported Ni3Sn4 alloys, coaxial SnO2@C hollow nanospheres and graphene-confined Sn nanosheets.14,17–24 Another popular way is to pass tin-based metal oxides from bulk to delicate hollow nanostructures with a large surface area, short diffusion path and enhanced reactivity.12–15,25,26 In such structures, not only is the lithium diffusion much easier, but the strain associated with lithium intercalation is better accommodated, resulting in significantly improved electrochemical performance. Despite the appeal of these procedures, each design strategy alone always leads to limited improvement in the electrochemical properties of tin-based metal oxides. In this regard, the fabrication of truly durable tin-based electrodes with satisfactory cycling ability and high capacity is still highly desirable for next-generation LIBs.
In this work, we develop a new type of hybrid nanostructure, carbon coated hollow nanostructure of amorphous complex metal oxides, for highly reversible lithium storage. This unique structure well integrates three main design principles and is anticipated to manifest excellent lithium storage performance because of the superior structural advantages. First, an atomically mixed matrix can be formed within stoichiometric complex metal oxides, which significantly relieves the internal stress of the electrode from the isotropic volume change upon cycling.27,28 The formation of a homogeneously amorphous structure also facilitates the lithium diffusion by enhancing the atomic/ionic mobility within the matrix.29 In this sense, amorphous complex metal oxides are very advantageous over the mechanically mixed heterogeneous powders. Second, the hollow structure provides sufficient tolerance to the volume variation upon Li+ insertion/extraction.12–15,25,26 It also offers a large interfacial area and an extremely reduced path for Li+/electron transport, enabling better diffusion kinetics for lithium storage. Additionally, the robust conductive carbon nanocoating further functions as a physical buffering matrix to reinforce the hollow structure, prevent interparticle agglomeration and enhance the electronic conductivity of the electrodes.14,23,30,31 In this work, we chose CoSnO3 to demonstrate our concept owing to its high lithium storage capacity, amorphous structure and limited success in synthesizing its hollow shells. Amorphous CoSnO3 nanoboxes have been synthesized by annealing CoSn(OH)6 nanoboxes in N2 flow. After carbon nanocoating, they exhibit exceptional stable capacity retention over 400 cycles and improved high-rate performance for highly reversible lithium storage.
The synthesis strategy of carbon-coated CoSnO3 (denoted as CoSnO3@C) nanoboxes is straightforward, as schematically illustrated in Scheme 1. Porous CoSn(OH)6 nanoboxes are first synthesized in aqueous solution by fast stoichiometric co-precipitation of Sn4+, Co2+ in the presence of OH− and followed alkaline etching under ambient conditions.32 After annealing in N2, CoSnO3 nanoboxes with a homogenously amorphous texture and high porosity are obtained by thermal-induced dehydration of CoSn(OH)6. This process can be described by the following formula:
| CoSn(OH)6 → CoSnO3 + 3H2O |
 | ||
| Scheme 1 Schematic illustration for the formation of amorphous CoSnO3 nanoboxes with carbon nanocoating. | ||
The resultant CoSnO3 nanoboxes are further coated with a thin layer of glucose-derived carbon-rich polysaccharide (GCP) by hydrothermal decomposition of glucose in solution. Such GCP contains abundant hydroxyl groups and can be carbonized at a relatively low temperature in inert atmosphere.14,31 As a result, high-quality CoSnO3@C nanoboxes are eventually produced as a high-performance electrode material for LIBs.
Fig. 1a and b show the field emission scanning electron microscopy (FESEM) images of CoSnO3 products obtained by annealing CoSn(OH)6 nanoboxes (Fig. S1, see ESI†) in N2 flow at 500 °C. A panoramic view shows that the sample consists entirely of uniform nanoboxes with an average edge length of around 250 nm. Their interior space can be examined directly by FESEM for cracked nanoboxes, as shown in Fig. 1b. The transmission electron microscope (TEM) analysis indicates that the conversion from CoSn(OH)6 to CoSnO3 boxes undergoes a topotactic transformation process without apparent collapse or fusion of the shells (Fig. 1c–e). The CoSnO3 nanoboxes are highly uniform with a shell thickness of around 30–40 nm, of which the inner cavities are clearly revealed by the sharp contrast between the shells and hollow interiors. The selected area electron diffraction (SAED) pattern of a single nanobox manifests as a dispersed and very ambiguous halo (Fig. 1f), indicating that the nanoboxes are amorphous in nature. The high resolution TEM (HRTEM) analysis further evidences the amorphous texture of CoSnO3 nanoboxes in view of the absence of a crystalline lattice fringe (ESI, Fig. S2†). The energy-dispersive spectroscopy (EDS) measurement confirms the co-existence of Co, Sn and O elements (Fig. 1g), and the elemental mapping based on a single nanobox evidences the homogeneous distribution of all three elements within the nanobox (Fig. 1h). Being consistent with the literature report,33 the X-ray powder diffraction (XRD) analysis further proves the formation of amorphous CoSnO3, as characterized by a plain pattern with a broad peak between 30 and 40° (Fig. S3, see ESI†). No signals from possible impurities such as Co2SnO4 or SnO2 are detected since they can only form at much higher annealing temperatures (e.g., 700 °C, Fig. S3, see ESI†). The nitrogen adsorption/desorption isotherm of CoSnO3 nanoboxes shows a typical type IV curve with a type H4 hysteresis loop, as shown in Fig. S4a (see ESI†). It gives rise a high Brunauer–Emmett–Teller (BET) specific surface area of 124 m2 g−1 due to the high porosity inherited from the CoSn(OH)6 nanoboxes, the dehydration of CoSn(OH)6 and the formation of a less dense amorphous structure.
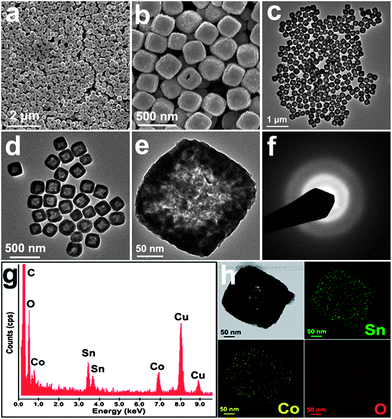 | ||
| Fig. 1 (a and b) FESEM and (c and d) TEM images of CoSnO3 nanoboxes; (e) a free-standing nanobox; (f) SAED pattern showing the amorphous nature of the CoSnO3 nanoboxes; (g) EDX spectrum of CoSnO3 nanoboxes, in which the Cu signal originated from the Cu grid support for TEM observation; and (h) elemental mapping showing the homogeneous distribution of all three elements of Sn, Co and O in the nanobox. | ||
The robust structure of CoSnO3 nanoboxes provides the feasibility of exploiting nanocoating of amorphous carbon around the particle by controlled hydrothermal carbonization of glucose. FESEM examination shows that after carbon coating, there is no apparent change in the morphology of the nanoboxes although their surface becomes slightly rougher (Fig. 2a and b). TEM characterization indicates that the entire surface of the nanoboxes is covered with a continuous amorphous carbon overlayer with uniform thickness of around 10 nm, as shown in Fig. 2c–f. Owing to the significant shrinkage of GCP during carbonization, the carbon shell is tightly attached to the CoSnO3 nanobox, which is beneficial for mechanical reinforcement and enhancing electronic conductivity of the structure.14 After hydrothermal treatment, the amorphous texture of CoSnO3 nanoboxes remains the same because of the relatively low temperature employed for carbon coating, as depicted by SAED and XRD analysis (the inset of Fig. 2d, and Fig. S3 in the ESI†). The carbon content in CoSnO3@C nanoboxes can be readily determined by thermogravimetric analysis (TGA) to be about 19 wt%, as shown in Fig. S5 (see ESI†). Despite the partial physical blocking of pores by the surface coating, CoSnO3@C nanoboxes can still possess a high specific surface area of around 85 m2 g−1 (Fig. S4b, see ESI†), which offers them sufficient interface to facilitate the electrochemical reactions with respect to the bulk materials.
 | ||
| Fig. 2 (a and b) FESEM and (c and d) TEM images of CoSnO3@C nanoboxes, of which the SAED pattern is shown in the inset of (d); (e) a free-standing CoSnO3@C nanobox; and (f) a uniform carbon coating layer on the surface of CoSnO3 nanoboxes. | ||
We have investigated the potential use of these CoSnO3@C nanoboxes as an anode material for LIBs. In the range of 0.01–1.5 V, the cyclic voltammogram (CV) curves of the CoSnO3@C electrode are almost identical to that of tin oxides (ESI, Fig. S6†). In this sense, Sn may contribute most of the capacity for lithium storage in the CoSnO3@C structure. Although electrochemically inactive, Co also determines the electrochemical properties of CoSnO3@C nanoboxes. It is not only indispensable for the formation of the amorphous Co–Sn–O phase but also serves as a buffer component against the volume change of the active material. Fig. 3a shows the representative discharge/charge voltage profiles of CoSnO3@C nanoboxes at a current density of 200 mA g−1 within a cut-off voltage window of 0.01–1.5 V. High initial discharge and charge capacities of around 1410 and 480 mA h g−1 can be delivered, respectively. The large capacity loss in the first cycle is mainly attributed to the initial irreversible formation of Li2O, and other irreversible processes such as trapping of some lithium in the lattice, inevitable formation of a solid electrolyte interface (SEI) layer and electrolyte decomposition, which are all quite common for most anode materials.28,33–38 Also, it is partly because the electrode is charged up to 1.5 V only in the present work. This drawback can be efficiently minimized by pre-lithiation of as-prepared electrodes at room temperature, which reduces the irreversible capacity to be less than 100 mA h g−1 (Fig. S7, see ESI†).39 From the second cycle onwards, CoSnO3@C nanoboxes exhibit a stable capacity retention of nearly 100% for 200 cycles with an initial capacity of around 530 mA h g−1. Encouragingly, their calendar life can be extended to as long as 400 cycles with a high capacity of over 450 mA h g−1. This value is much higher than the theoretical capacity of commercial graphite electrodes (372 mA h g−1) and is superior to many other tin-based ternary metal oxide electrodes.28,33–38 After the deep cycling, we have carried out the post-mortem study by TEM and FESEM examination. Remarkably, the original textural properties of CoSnO3@C nanoboxes can be well retained in terms of shape, size, and structural integrity, indicating the good structural stability of this material (Fig. S8, see ESI†). In favor of the unique structure, the CoSnO3@C nanoboxes also exhibit greatly improved cycling response to a continuously varying current density although tin-based metal oxides are generally suffering from sluggish kinetics for lithium storage. While cycling at high rates of 400–1000 mA g−1, capacities of 310–450 mA h g−1 can still be retained, as shown in Fig. 3d. Evidently, the CoSnO3@C nanoboxes have exhibited excellent long-term stability for lithium storage, perhaps benefiting from their unique structural characteristics.
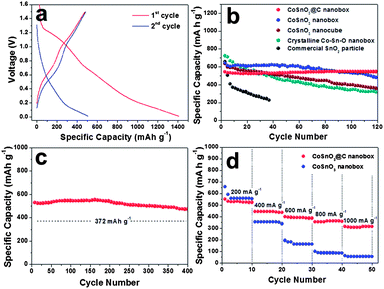 | ||
| Fig. 3 (a) Discharge/charge voltage profiles of CoSnO3@C nanoboxes for the first two cycles; (b) comparative cycling performance of CoSnO3 nanoboxes with and without carbon coating, CoSnO3 nanocubes, crystalline Co–Sn–O nanoboxes and commercial SnO2 particles; and (c) long-term cycling stability of CoSnO3@C nanoboxes. All these tests are conducted at a current density of 200 mA g−1 between 0.01 and 1.5 V. (d) Rate capability of CoSnO3 nanoboxes with and without carbon coating, which is obtained between 0.01 and 1.5 V at various current densities. | ||
To demonstrate the advantages of CoSnO3@C nanoboxes for lithium storage, the cycling performance of CoSnO3 nanoboxes without carbon coating, crystalline Co–Sn–O (Co2SnO4/SnO2, Fig. S3, see ESI†) nanoboxes, and commercial SnO2 nanoparticles is also investigated under identical test conditions, as shown in Fig. 3b. Without carbon coating, the CoSnO3 nanoboxes exhibit deteriorated cycling stability (around 480 mA h g−1 is left after 120 cycles at 200 mA g−1) and poor capacity retention at higher current densities. Apparently, not only the structural stability, but also the lithium storage kinetics of the CoSnO3 is enhanced in the presence of the robust conductive carbon nanocoating that can alleviate material pulverization, preventing particle aggregation, enhance electrode conductivity and stabilize the SEI film.40 Although not as good as that of CoSnO3@C nanoboxes, the cycling performance of CoSnO3 nanoboxes is still much better than that of CoSnO3 nanocubes with the same size (Fig. 3b and S9 in the ESI†) and it clearly indicates the beneficial effect of the hollow nanostructures on their electrochemical properties, namely, the highly flexible structure, large electrode–electrolyte interface and extremely reduced diffusion path.25,26 For crystalline Co–Sn–O nanoboxes and commercial SnO2 nanoparticles (50–100 nm), they lose almost all the capacity within 40–120 cycles. In this regard, the atomically mixed, homogeneously amorphous texture of CoSnO3 may also contribute to their lithium storage performance because the volume change upon cycling can be partly mitigated in an isotropic, loose dense structure with high atomic/ionic mobility.29,41,42
In summary, we have rationally designed a multifeatured nanostructure, amorphous CoSnO3@C nanoboxes, for highly reversible lithium storage. This unique structure incorporates several desirable design rationales for long-life anode materials based on hollow nanostructures, carbon nanopainting, mixed conductive matrix and crystalline texture engineering. In favor of the synergy and interplay of the matrix effect and intrinsic structural advantages, the CoSnO3@C nanoboxes exhibit an exceptional cycle life of over 400 cycles and improved high-rate capability when evaluated as an anode material for lithium-ion batteries. With rational and delicate design, this class of functional materials may hold great promise for the development of high-performance lithium-ion batteries.
Notes and references
- F. Y. Cheng, Z. L. Tao, J. Liang and J. Chen, Chem. Mater., 2008, 20, 667–681 CrossRef CAS
.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS
- B. Scrosati, J. Hassoun and Y. K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 CAS
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS
- L. W. Ji, Z. Lin, M. Alcoutlabi and X. W. Zhang, Energy Environ. Sci., 2011, 4, 2682–2699 CAS
- Z. Y. Wang, L. Zhou and X. W. Lou, Adv. Mater., 2012, 24, 1903 CrossRef CAS
- J. F. Ye, W. Liu, J. G. Cai, S. Chen, X. W. Zhao, H. H. Zhou and L. M. Qi, J. Am. Chem. Soc., 2011, 133, 933–940 CrossRef CAS
- J. Chen, L. N. Xu, W. Y. Li and X. L. Gou, Adv. Mater., 2005, 17, 582–586 CrossRef CAS
- Y. F. Shi, B. K. Guo, S. A. Corr, Q. H. Shi, Y. S. Hu, K. R. Heier, L. Q. Chen, R. Seshadri and G. D. Stucky, Nano Lett., 2009, 9, 4215–4220 CrossRef CAS
- J. C. Park, J. Kim, H. Kwon and H. Song, Adv. Mater., 2009, 21, 803–807 CrossRef CAS
- J. F. Ye, H. J. Zhang, R. Yang, X. G. Li and L. M. Qi, Small, 2010, 6, 296–306 CrossRef CAS
- X. W. Lou, Y. Wang, C. Yuan, J. Y. Lee and L. A. Archer, Adv. Mater., 2006, 18, 2325–2329 CrossRef CAS
- X. W. Lou, C. M. Li and L. A. Archer, Adv. Mater., 2009, 21, 2536–2539 CrossRef CAS
- J. S. Chen, L. A. Archer and X. W. Lou, J. Mater. Chem., 2011, 21, 9912–9924 RSC
- C. Wang, Y. Zhou, M. Y. Ge, X. B. Xu, Z. L. Zhang and J. Z. Jiang, J. Am. Chem. Soc., 2010, 132, 46–47 CrossRef CAS
- Y. Idota, T. Kubota, A. Matsufuji, Y. Maekawa and T. Miyasaka, Science, 1997, 276, 1395–1397 CrossRef CAS
- Y. Yu, C. H. Chen and Y. Shi, Adv. Mater., 2007, 19, 993–997 CrossRef CAS
- G. Derrien, J. Hassoun, S. Panero and B. Scrosati, Adv. Mater., 2007, 19, 2336–2340 CrossRef CAS
- J. Hassoun, S. Panero, P. Simon, P. L. Taberna and B. Scrosati, Adv. Mater., 2007, 19, 1632–1635 CrossRef CAS
- K. T. Lee, Y. S. Jung and S. M. Oh, J. Am. Chem. Soc., 2003, 125, 5652–5653 CrossRef CAS
- L. Y. Beaulieu and J. R. Dahn, J. Electrochem. Soc., 2000, 3237–3241 CrossRef CAS
- W. M. Zhang, J. S. Hu, Y. G. Guo, S. F. Zheng, L. S. Zhong, W. G. Song and L. J. Wan, Adv. Mater., 2008, 20, 1160–1165 CrossRef CAS
- B. Luo, B. Wang, X. L. Li, Y. Y. Jia, M. H. Liang and L. J. Zhi, Adv. Mater., 2012, 24, 3538–3543 CrossRef CAS
- X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS
- Z. Y. Wang, L. Zhou and X. W. Lou, Adv. Mater., 2012, 24, 1903–1911 CrossRef CAS
- D. W. Kim, Y. D. Ko, J. G. Park and B. K. Kim, Angew. Chem., Int. Ed., 2007, 46, 6654–6657 CrossRef CAS
- Y. Sharma, N. Sharma, G. V. Subba Rao and B. V. R. Chowdari, Adv. Funct. Mater., 2007, 17, 2855–2861 CrossRef CAS
- H. Li, P. Balaya and J. Maier, J. Electrochem. Soc., 2004, 151, A1878–A1885 CrossRef CAS
- Z. Y. Wang, D. Y. Luan, S. Madhavi, Y. Hu and X. W. Lou, Energy Environ. Sci., 2012, 5, 5252–5256 CAS
- X. M. Sun, J. F. Liu and Y. D. Li, Chem. Mater., 2006, 18, 3486–3494 CrossRef CAS
- Z. Y. Wang, Z. C. Wang and X. W. Lou, 2012, submitted.
- F. Huang, Z. Y. Yuan, H. Zhan, Y. H. Zhou and J. T. Sun, Mater. Lett., 2003, 57, 3341–3345 CrossRef CAS
- N. Sharma, K. M. Shaju, G. V. Subba Rao and B. V. R. Chowdari, Electrochem. Commun., 2002, 4, 947–952 CrossRef CAS
- A. Rong, X. P. Gao, G. R. Li, T. Y. Yan, H. Y. Zhu, J. Q. Qu and D. Y. Song, J. Phys. Chem. B, 2006, 110, 14754–14760 CrossRef CAS
- G. Wang, X. P. Gao and P. W. Shen, J. Power Sources, 2009, 192, 719–723 CrossRef CAS
- F. M. Courtel, H. Duncan, Y. Abu-Lebdeh and I. J. Davidson, J. Mater. Chem., 2011, 21, 10206–10218 RSC
- Y. Qi, N. Du, H. Zhang, P. Wu and D. R. Yang, J. Power Sources, 2011, 196, 10234–10239 CrossRef CAS
- J. Hassoun, K. Lee, Y. Sun and B. Scrosati, J. Am. Chem. Soc., 2011, 133, 3139–3143 CrossRef CAS
- H. Q. Li and H. S. Zhou, Chem. Commun., 2012, 48, 1201 RSC
- M. H. Seo, M. Park, K. T. Lee, K. Kim, J. Kim and J. Cho, Energy Environ. Sci., 2011, 4, 425–428 CAS
- L. Y. Beaulieu, K. W. Eberman, R. L. Turner, L. J. Krause and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A137–A140 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, XRD, BET and more FESEM/TEM images, as well as electrochemical data. See DOI: 10.1039/c2ee23330d |
| This journal is © The Royal Society of Chemistry 2013 |