Mechanocatalytic depolymerization of cellulose combined with hydrogenolysis as a highly efficient pathway to sugar alcohols†
Jakob
Hilgert
,
Niklas
Meine
,
Roberto
Rinaldi
* and
Ferdi
Schüth
*
Max-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470 Mülheim an der Ruhr, Germany. E-mail: rinaldi@kofo.mpg.de; schueth@kofo.mpg.de; Fax: +49 208 306 2995; Tel: +49 208 306 2373
First published on 18th October 2012
Abstract
Cellulose is both insoluble in water and resistant against hydrolysis. These features pose major problems for its conversion into platform chemicals. Herein, we demonstrate that mechanocatalytic, solid-state depolymerization combined with hydrogenolysis, in the presence of Ru/C in water, provides a highly efficient pathway for the production of sugar alcohols. This novel approach leads to yields of hexitols up to 94% at 150 °C in an overall process time of 4 h.
Broader contextUnlike conventional mechanical pre-treatments, which are applied to cellulose to reduce its crystallinity and to improve its accessibility to cellulases or acid catalysts, the mechanocatalytic depolymerization of acid-impregnated cellulose leads to a quantitative yield of oligosaccharides soluble in water. In aqueous solution, these products are converted into platform chemicals (e.g., sugar alcohols) under low-severity conditions, as demonstrated here. The significance of processing solutions of oligosaccharides, instead of cellulose slurries, lies also in the prevention of accumulating the highly recalcitrant cellulosic residues throughout several reuses of the solid catalyst. Overall, the mechanocatalytic approach could well hold the key to a better use of the mechanical forces in biorefining, providing an environmentally friendly entry-point process into lignocellulosic biorefinery schemes. |
The conversion of cellulose into sugar alcohols occurs in a stepwise process, involving (i) hydrolysis of the biopolymer and, subsequently, (ii) hydrogenation of soluble sugars.1–3 The recent advances in this field can be grouped into three main research lines. In the first, bifunctional catalysts, tailor-made for better interaction with solid cellulose, have proven advantageous for facilitating the hydrolytic step.4–8 In the second line of research, Ru catalysts are rationally used for both generating in situ H+ species and for hydrogenating the soluble sugars derived from cellulose hydrolysis.9–12 The main advantage of this group of processes is that no neutralization step is required because the formation of H+ species is a reversible process. In the third approach, a molecular acid catalyst is used in combination with a solid hydrogenation catalyst.13–15 These processes are conducted at lower temperatures than those involving in situ generation of H+ species. Despite these advantages, all these approaches still require severe conditions to achieve high, but not full conversion of cellulose. Overall, this fact poses two serious problems. First, the residual solid substrate accumulates throughout the process cycles,2 making the re-utilization of the catalyst cumbersome.16,17 Second, as high temperatures are needed for achieving high conversion of cellulose, the yields of hexitols (mannitol, sorbitol and sorbitan) are reduced.2 This may negatively affect the overall potential of these processes, which could also be part of an integrated approach for the production of liquid transportation fuels upon conversion of sugar alcohols into alkanes.18
To improve the conversion of cellulose, conventional ball-milling has been extensively explored both in enzymatic and chemocatalytic processes.19,20 As a matter of fact, the mechanical pre-treatment reduces the crystallinity of cellulose, improving the accessibility of the biopolymer to cellulases or acid catalysts.19,20
One of the most recent advances in hydrolytic hydrogenation of cellulose has been reported by Sels et al.21 They reported that, in the presence of cesium salts of heteropolyacids and Ru/C, microcrystalline cellulose (MCC), ball milled for 24 h, undergoes hydrolytic hydrogenation at 170 °C under a pressure of 50 bar H2 (r.t.), resulting in yields of up to 70% of C6-alditols and 20% of sorbitan after 48 h. Kobayashi et al. reported the transfer hydrogenation of cellulose, ball milled for 4 days, in the presence of Ru/C and propan-2-ol (H-donor) at 190 °C. Yields of up to 45% of C6-alditols and 1.4% of sorbitan were achieved after 18 h.22 In another report, the hydrolytic hydrogenation of cellulose, ball milled for 2–4 days and reacted at 190 °C under 50 bar H2 (r.t.) for 24 h, resulted in yields of up to 65% of C6-alditols and 10% of sorbitan in the presence of Pt/C.23 Han and Lee reported that cellulose, ball milled for 2–3 days, undergoes hydrolytic hydrogenation with bifunctional Ru/C–SO3H. At 165 °C and under 50 bar H2 (r.t.) for 24 h, 65% yield of C6-alditols was achieved in a neutral aqueous solution.24
While conventional ball-milling enhances the reactivity of cellulose, the above-mentioned examples indicate that the improvement is only achieved upon milling the biopolymer for a long time (e.g., days). Thus, alternative pre-treatments, preferably performed under solvent-free conditions, are required. In this context, Jérôme et al.25,26 reported the partial depolymerization of MCC assisted by a nonthermal atmospheric plasma (NTAP). This pre-treatment improved the acid-catalyzed hydrolysis of cellulose in an aqueous phase.25,26 However, yields of glucose as high as 58% could be produced only upon milling MCC for a time of 48 h prior to the NTAP step.26 Another solvent-free pre-treatment was introduced by Blair et al.27 They described a mechanocatalytic approach for cellulose depolymerization in the presence of solid acids. Using delaminated kaolinite, conversion up to 84% of cellulose into water-soluble products was obtained after 3 h.27 In spite of the good results, the reuse of the solid catalyst is challenging as full conversion of cellulose is not achieved.
Realizing the fact that the mechanical pre-treatment appears to be unavoidable for the efficient conversion of cellulose, we initiated research into solvent-free, mechanocatalytic processes, aiming at the full conversion of the biopolymer into water-soluble oligosaccharides. We found that the impregnation of cellulosic fibers with catalytic amounts of a strong acid (e.g., HCl, H2SO4) holds the key for the high efficiency.28 This strategy circumvents the contact problems experienced in the mechanocatalytic process using solid acids.27 As a result, oligosaccharides soluble in water are produced in a quantitative yield upon milling for 2 h.28
In the following, we demonstrate that this approach offers a unique entry-point for the efficient production of sugar alcohols with the Ru/C catalyst in water.
To assess the extent of depolymerization achieved by the conventional ball-milling of cellulosic substrates (MCC and α-cellulose) and that achieved by the mechanocatalytic depolymerization of H2SO4-impregnated substrates,29 the milled substrates were derivatized with phenylisocyanate, making the phenyl carbanylate derivatives soluble in THF, which were analyzed by gel-permeation chromatography (GPC).
Fig. 1 shows the distribution of degree of polymerization (DP) for the substrates studied here. The unprocessed MCC and α-cellulose display a weight average DP of 400 and 2210 anhydroglucose units (AGU), respectively. In contrast, MCC and α-cellulose, milled for 2 h, show a DP of 140 and 460 AGU, respectively. These substrates are insoluble in water.
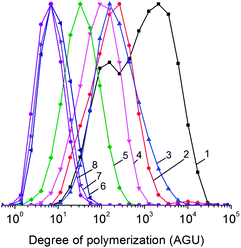 | ||
| Fig. 1 Distribution of apparent DP for the phenyl carbanylate derivatives of α-cellulose (1), MCC (2), α-cellulose ball milled for 2 h (3), MCC ball milled for 2 h (4), IMCC (5), IMCC milled for 1 h (6), 2 h (7) and 3 h (8). | ||
H2SO4-impregnated MCC (IMCC) has a DP of 50 AGU. This result indicates that the substrate undergoes depolymerization upon the impregnation step with H2SO4 for 1 h. However, only upon milling IMCC for 1, 2 and 3 h, the DP was reduced to 11, 9 and 8 AGU, making the products soluble in water. Similar results were obtained by milling H2SO4-impregnated α-cellulose for 2 h. The H2SO4-impregnated substrates, milled for 2 h or longer, were fully soluble in water (for the speciation of the oligosaccharides by GPC in water, see ESI†).
It is important to mention that IMCC was milled directly after the impregnation step. Keeping the acid-impregnated cellulose at the temperature reached during the ball milling (42 °C) does not result in depolymerization to water-soluble products. In contrast, a grayish powder is formed upon aging IMCC for days under these conditions. The grayish product is insoluble in water.
To evaluate the reactivity of the unprocessed and of the ball milled cellulosic substrates, directly after the solid-state reaction, hydrogenolysis of the substrates, suspended or dissolved in water, was performed in the presence of 5 wt% Ru/C catalyst (Aldrich) under 50 bar H2 (r.t.) at a temperature of 160 °C for 1 h.
Table 1 summarizes the results obtained from the experiments with the unprocessed and with the ball milled substrates. Entries 1 and 2 show that the experiments with unprocessed MCC and with MCC ball milled for 2 h, dispersed in water, led to no or negligible formation of hexitols (sorbitol, mannitol and sorbitan). However, processing MCC in a 0.05 M H2SO4 solution resulted in 5.6% yield of hexitols (entry 3). In turn, the experiment with MCC, milled for 2 h (DP 140 AGU), performed in a 0.05 M H2SO4 solution, led to 44.7% yield of hexitols (entry 4). Despite the low DP of IMCC (50 AGU), the conversion of this substrate resulted in only 9.0% yield of hexitols (entry 5). As the same concentration of acid was present in both experiments, the results indicate that the substrate crystallinity has a higher influence than the DP, in the conversion of cellulose.
| Entry | Substrate | Milling time (h) | Reaction medium | Conversionb (%) | Carbon yields (%) | Mass balance (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| C6-Alditols | Sorbitan | Hexitols sum | Xylitol | Othersd | ||||||
| a Reaction conditions: 500 mg substrate, 10 mL water, 100 mg Ru/C, 50 bar H2 (r.t.), 160 °C, 1 h. b Conversion was determined by weight difference. c Substrates were soluble in water, the solution has pH = 1. d The carbon yields of other products are given in the ESI.† | ||||||||||
| 1 | Microcrystalline cellulose (MCC) | 0 | H2O | 8 | 0.0 | 0.0 | 0.0 | 0.0 | 0.8 | 10 |
| 2 | 2 | H2O | 19 | 0.1 | 0.0 | 0.1 | 0.2 | 0.8 | 6 | |
| 3 | 0 | 0.05 M H2SO4 | 13 | 4.8 | 0.8 | 5.6 | 0.9 | 0.7 | 55 | |
| 4 | 2 | 0.05 M H2SO4 | 55 | 41.6 | 3.1 | 44.7 | 3.0 | 0.5 | 88 | |
| 5 | H2SO4-impregnated MCC (IMMC) | 0 | H2O | 32 | 7.8 | 1.2 | 9.0 | 1.3 | 0.8 | 35 |
| 6 | 1 | H2O | 96 | 66.8 | 5.0 | 71.8 | 5.5 | 6.5 | 87 | |
| 7 | 2 | H2O | 100c | 82.0 | 5.6 | 87.6 | 5.3 | 4.3 | 97 | |
| 8 | 3 | H2O | 100c | 73.9 | 5.7 | 79.6 | 6.0 | 3.6 | 89 | |
| 9 | α-Cellulose | 2 | 0.05 M H2SO4 | 62 | 32.0 | 0.0 | 32.0 | 12.6 | 1.6 | 75 |
| 10 | H2SO4-impregnated α-cellulose | 2 | H2O | 100c | 80.3 | 5.6 | 85.9 | 13.7 | 0.4 | 100 |
| 11 | Glucose | 0 | 0.05 M H2SO4 | 100c | 92.6 | 3.7 | 96.3 | 2.1 | 1.6 | 100 |
An unanticipated key finding was revealed by the experiments with the water-soluble oligosaccharides obtained by milling IMCC (entries 6, 7 and 8). Markedly, the experiment with IMCC, milled for 2 h, led to 87.6% yield of hexitols (entry 7). Similar results were obtained with H2SO4-impregnated α-cellulose ball milled for 2 h (entry 10). In this case, the hydrolytic hydrogenation led to 85.9% yield of hexitols.
The experiments with the water-soluble products, obtained by mechanocatalytic reaction, resulted in yields of hexitols quite close to those obtained from glucose (96.3%, entry 11). Since the acid is not destroyed by the mechanical process, the water-soluble oligosaccharides undergo hydrolysis, releasing glucose and xylose (derived from hemicellulose impurities). As already reported by us, this reaction takes place, to the full extent, upon heating the aqueous solution of oligosaccharides at 130 °C for 1 h.28 This demonstrates that the soluble products, derived from the mechanocatalytic depolymerization, display enhanced reactivity toward hydrolysis. This fact accounts for the unprecedented, high yields of hexitols achieved, in the presence of Ru/C, at 160 °C after only 1 h.
Table 1, entries 6–8, shows that the milling time is an important parameter for the optimization of the hexitols yield. With the current milling process, the best results were obtained by processing IMCC for 2 h. In turn, processing IMCC for 3 h reduced the hexitols yield to 79.6% (entry 8). The mechanocatalytic process induces both the depolymerization of cellulose and, to a smaller extent, the non-stereospecific recombination of the oligosaccharides.28 As a result, products comprising linkages other than the 1,4-β-glycosidic bond are formed.28 This could be the reason, why products with slightly different reactivity toward hydrogenolysis are obtained by milling IMCC for 2 and 3 h.
Overall, the mechanocatalytic process requires milling times about 12–48 times shorter than those needed for the conventional milling,21–24 for producing “reactive” substrates for the production of sugar alcohols at high yields. Indeed, the high yield of C6-alditols (mannitol and sorbitol) is another key feature found in the hydrogenation of water-soluble oligosaccharides. In the best examples from Table 1, C6-alditol yields higher than 70%, in two cases even higher than 80%, were achieved (entries 7, 8 and 10).
Sorbitan, a product derived from the dehydration of sorbitol,30 was obtained at yields lower than 6% (Table 1). With regard to the hydrogenation of glucose with Ru/C at 160 °C, a very low yield of xylitol was achieved (3.7%, entry 11). Taken together, these two results indicate that both the dehydration of sorbitol and the hydrogenolysis of C–C bonds are largely suppressed at 160 °C. Accordingly, xylitol (yields in the 5–14% range) derives mainly from xylose (from hemicellulose impurities).28
As the activation of Ru catalysts occurs in the 100–200 °C temperature range,31,32 it is natural to ask whether a pre-activation of the Ru/C catalyst could help to further reduce the reaction temperature for the conversion of water-soluble oligosaccharides into sugar alcohols. To find out whether the pre-activation of Ru/C plays an important role in the process, the Ru/C catalyst was suspended in water and pretreated under 50 bar H2 (r.t.) at 160 °C for 1 h. The pre-activated catalyst is denoted as Ru/C*. The substrate was then added to the catalyst suspension. Experiments were performed in the presence of the Ru/C or Ru/C* catalyst under 50 bar H2 (r.t.) at 140, 150 or 160 °C for 1 h.
Fig. 2 compares the performance of Ru/C and Ru/C* catalysts and reveals a key feature. The activation of Ru/C is the limiting step in the hydrogenation at 140 °C. Indeed, the yield of hexitols, obtained by the experiment at 140 °C for 1 h, markedly increases from 56.0 to 80.5% when using the Ru/C* catalyst. A less pronounced increase in the yield of hexitols was detected for the experiments with Ru/C* performed at 150 and 160 °C for 1 h. However, the best result was obtained using Ru/C* at 150 °C. This experiment led to 94.3% yield of hexitols. The HPLC speciation of the C6-alditols (obtained at 91% yield) revealed a sorbitol-to-mannitol ratio of 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6. At 160 °C, 90.3% yield of hexitols was obtained. The mixture of C6-alditols (produced at 84% yield) displayed a sorbitol-to-mannitol ratio of 88:12.
:6. At 160 °C, 90.3% yield of hexitols was obtained. The mixture of C6-alditols (produced at 84% yield) displayed a sorbitol-to-mannitol ratio of 88:12.
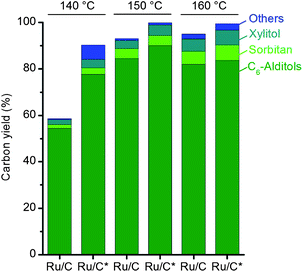 | ||
| Fig. 2 Comparison of the performance of Ru/C and Ru/C* catalysts in hydrolytic hydrogenation of IMCC ball milled for 2 h. Reaction conditions: 500 mg substrate, 10 mL water, 100 mg catalyst, 50 bar H2 (r.t.), 1 h. The reaction temperatures are indicated on the top of the figure. | ||
The significance of processing solutions of oligosaccharides, instead of cellulose slurries, lies in the prevention of accumulating the highly recalcitrant cellulosic residues throughout several reuses of the catalyst. To evaluate the stability of Ru/C in the hydrolytic hydrogenation of IMCC ball-milled for 2 h, six consecutive experiments at 160 °C for 1 h each were performed. Fig. 3 displays the product distribution obtained throughout the recycling experiments.
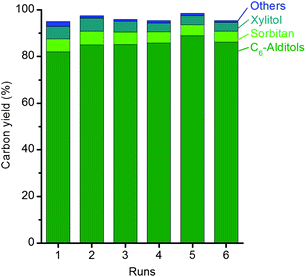 | ||
| Fig. 3 Recycling of Ru/C. Reaction conditions: 500 mg IMCC ball milled for 2 h, 10 mL water, 100 mg Ru/C, 50 bar H2 (r.t.), at 160 °C for 1 h. | ||
The recycling experiments demonstrated that the Ru/C catalyst maintains its activity throughout the six consecutive runs. Fig. 3 shows that the selectivity to hexitols slightly increased throughout the recycling experiments. EDX analysis shows no change in the nominal content of Ru dispersed on carbon in the fresh and spent catalyst (sixth run). TEM analysis of the Ru/C sample (fresh, after one and six runs, Fig. 4) reveals that the size of the Ru nanoparticles increased. The fresh catalyst shows very small particles (1.4 ± 0.3 nm) finely distributed on the support. From the first to the sixth run, Ru nanoparticles became larger and metal agglomeration was detected (first run, 2.0 ± 0.5 nm; sixth run, 2.5 ± 0.6 nm).
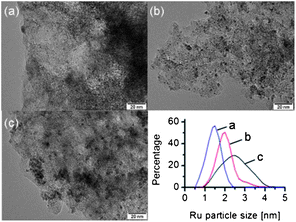 | ||
| Fig. 4 TEM images of the Ru/C catalyst: (a) before reaction, (b) after 1 run and (c) after 6 runs. The inset shows the Ru particle distribution for the catalyst samples. | ||
In conclusion, the mechanocatalytic depolymerization of dry cellulose, impregnated with catalytic quantities of H2SO4, is a unique entry-point process for efficient production of sugar alcohols from cellulosic substrates. Under low-severity conditions, 94.3% yield of hexitols was obtained, in the presence of the pre-activated Ru/C*, by a batch reaction at 150 °C. In addition, such a high yield was achieved in an overall time of only 4 h, that is, 18–27 times faster than the best examples reported so far.21–24 The commercial Ru/C catalyst was fully compatible with the presence of H2SO4 in the reaction medium. Indeed, the catalytic activity of Ru/C was fully maintained throughout six runs at 160 °C for 1 h each. In the recycling experiments, the cumulative productivity achieved about 600 g of hexitols per g of Ru. As the catalyst is still highly active at the sixth reaction run, this value will be much higher in an extended recycling test. Considering also the fact that the oligosaccharides are fully water soluble, the future technical development should progress toward reactions performed in continuous reactors. The downstream acid handling is still a problem. However, there is current industrial development of resins for the recovery of H2SO4 from the downstream of the two-stage H2SO4 process. This technology could also be used to recover the acid used in the mechanocatalytic process. With regard to corrosion problems, both the balls and the mill vial have proven very resistant to corrosion. In fact, no change in the weight of the balls and the mill vial, even after more than 300 experiments, was detected. Apparently, the strong affinity of H2SO4 toward cellulose reduces the acid activity on the stainless steel surface. Overall, the current results provide a real incentive for intensified research into the mechanocatalytic depolymerization of cellulose on a large scale. With advanced technology in place, the impregnation/milling approach could well open up new horizons in cellulose conversion, as demonstrated here.
Acknowledgements
The financial support from the Max Planck Society and by an ERC Advanced Grant to F.S. is acknowledged. R.R. is thankful to the Alexander von Humboldt Foundation for the funds provided in the framework of the Sofja Kovalevskaja Award 2010 endowed by the Federal Ministry of Education and Research. This work was performed as part of the Cluster of Excellence “Tailor-Made Fuels from Biomass”, which is funded by the Excellence Initiative by the German federal and state governments to promote science and research at German universities. We thank A. Deege and H. Hinrichs for the HPLC analyses and B. Spliethoff for the TEM images.Notes and references
- S. Van De Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS.
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS.
- J. A. Geboers, S. Van De Vyver, R. Ooms, B. Op De Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714–726 Search PubMed.
- S. Van de Vyver, J. Geboers, M. Dusselier, H. Schepers, T. Vosch, L. Zhang, G. Van Tendeloo, P. A. Jacobs and B. F. Sels, ChemSusChem, 2010, 3, 698–701 CrossRef CAS.
- S. van de Vyver, J. Geboers, W. Schutyser, M. Dusselier, P. Eloy, E. Dornez, J. W. Seo, C. M. Courtin, E. M. Gaigneaux, P. A. Jacobs and B. F. Sels, ChemSusChem, 2012, 8, 1549–1558 CrossRef.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef CAS.
- L.-N. Ding, A.-Q. Wang, M.-Y. Zheng and T. Zhang, ChemSusChem, 2010, 3, 818–821 CrossRef CAS.
- R. Palkovits, K. Tajvidi, A. M. Ruppert and J. Procelewska, Chem. Commun., 2011, 47, 576–578 RSC.
- P. L. Dhepe and A. Fukuoka, Catal. Surv. Asia, 2007, 11, 186–191 CrossRef CAS.
- P. L. Dhepe and A. Fukuoka, ChemSusChem, 2008, 1, 969–975 CrossRef CAS.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS.
- J. Geboers, S. van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC.
- R. Palkovits, K. Tajvidi, J. Procelewska, R. Rinaldi and A. Ruppert, Green Chem., 2010, 12, 972–978 RSC.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Chem. Commun., 2011, 47, 5590–5592 RSC.
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS.
- R. Rinaldi and F. Schüth, Energy Environ. Sci., 2009, 2, 610–626 CAS.
- N. Li, G. A. Tompsett, T. Zhang, J. Shi, C. E. Wyman and G. W. Huber, Green Chem., 2011, 13, 91–101 RSC.
- P. Kumar, D. M. Barrett, M. J. Delwiche and P. Stroeve, Ind. Eng. Chem. Res., 2009, 48, 3713–3729 CrossRef CAS.
- L. R. Lynd, C. E. Wyman and T. U. Gerngross, Biotechnol. Prog., 1999, 15, 777–793 CrossRef CAS.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Green Chem., 2011, 13, 2167–2174 RSC.
- H. Kobayashi, H. Matsuhashi, T. Komanoya, K. Hara and A. Fukuoka, Chem. Commun., 2011, 47, 2366–2368 RSC.
- H. Kobayashi, Y. Ito, T. Komanoya, Y. Hosaka, P. L. Dhepe, K. Kasai, K. Hara and A. Fukuoka, Green Chem., 2011, 13, 326–333 RSC.
- J. W. Han and H. Lee, Catal. Commun., 2012, 19, 115–118 CrossRef CAS.
- M. Benoit, A. Rodrigues, Q. Zhang, E. Fourré, K. O. Vigier, J. M. Tatibouët and F. Jérôme, Angew. Chem., Int. Ed., 2011, 50, 8964–8967 CrossRef CAS.
- M. Benoit, A. Rodrigues, K. O. Vigier, E. Fourré, J. Barrault, J. M. Tatibouët and F. Jérôme, Green Chem., 2012, 14, 2212–2215 RSC.
- S. M. Hick, C. Griebel, D. T. Restrepo, J. H. Truitt, E. J. Buker, C. Bylda and R. G. Blair, Green Chem., 2010, 12, 468–474 RSC.
- N. Meine, R. Rinaldi and F. Schüth, ChemSusChem, 2012, 8, 1449–1454 CrossRef.
- We chose to impregnate the cellulosic substrates upon dispersion of the substrate in a 0.061 mol L−1 H2SO4 solution in MTBE for 1 h. After removal of the solvent, an H+ loading of 0.9 mmol per gram of substrate was achieved.
- N. Khan, D. Mishra, J.-S. Hwang, Y.-W. Kwak and S. Jhung, Res. Chem. Intermed., 2011, 37, 1231–1238 CrossRef CAS.
- P. G. J. Koopman, A. P. G. Kieboom and H. van Bekkum, Colloids Surf., 1981, 3, 1–12 CrossRef CAS.
- P. G. J. Koopman, A. P. G. Kieboom and H. van Bekkum, J. Catal., 1981, 69, 172–179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental part. See DOI: 10.1039/c2ee23057g |
| This journal is © The Royal Society of Chemistry 2013 |