Enhanced oxygen diffusion in epitaxial lanthanum–strontium–cobaltite thin film cathodes for micro solid oxide fuel cells†
Ho-Il
Ji
,
Jaeyeon
Hwang
,
Kyung Joong
Yoon
,
Ji-Won
Son
,
Byung-Kook
Kim
,
Hae-Won
Lee
and
Jong-Ho
Lee
*
High-Temperature Energy Materials Research Center, Korea Institute of Science and Technology, Seoul 136-791, Republic of Korea. E-mail: jongho@kist.re.kr; Fax: +82-2-958-5529; Tel: +82-2-958-553
First published on 15th November 2012
Abstract
The chemical diffusion coefficient (Dchem) of epitaxial LSC thin films was measured by a newly designed conductivity relaxation method. Dchem of epitaxial thin films was found to be higher than that of bulk LSC by up to two orders of magnitude, and was found to be enhanced further as the film became thinner.
Broader contextRecently, there have been intensive research efforts to realize the thin film electrode, especially the cathode for micro-solid oxide fuel cells (μ-SOFCs) to lower the ohmic resistance and polarization resistance of the electrode during the fuel cell operation. However, there is still a lack of basic understanding of the oxygen transport phenomena in thin film cathodes due to the many difficulties in characterization of oxygen transport properties such as bulk diffusion coefficient. The major obstacle for the measurement of bulk diffusion coefficient in thin films has been the short diffusion length, which is limited by film thickness. Hence we completely eliminated the limited diffusion length problem by avoiding a situation in which the entire film surface is directly exposed to the atmosphere, thereby limiting the diffusion length to the thickness of the film. This novel method to measure the bulk diffusion coefficient in thin films could eventually afford a great potential for the proper architecturing of a highly efficient electrode for μ-SOFCs and also for other electrochemical devices, such as electrolysis cells, membrane reactors, and gas sensors. |
Micro-solid oxide fuel cells (μ-SOFCs) have received great attention as novel power sources for mobile applications because of their many intriguing properties (high power and energy density, system efficiency, and fuel flexibility).1 Electrode polarization is usually thought to be a main contributor to the performance losses of μ-SOFCs, especially in relation to sluggish kinetics in cathode reactions.2,3 There is still a lack of basic understanding of the physicochemical properties of thin film cathodes. Thus far, most studies on the characterization of thin film cathodes have dealt with the measurement of their surface exchange coefficients (kchem) rather than the bulk diffusion properties (Dchem), because the surface exchange reaction is thought to be a main rate determining step in thin-film cathodes.4–6
There have been only a few studies reporting oxygen diffusion kinetics in SOFC cathode thin films.7,8 The very intricate measurement of the bulk diffusion coefficient (equivalent to the oxygen diffusion coefficient in a thin film cathode) due to its limited diffusion length is another reason for the lack of bulk diffusion studies on thin film cathodes. In a real μ-SOFC system, however, oxygen diffusion kinetics might become very important for cathode reactions because of the extremely complex cathode structure (e.g., gradient structured or multi-layered cathodes based on composite materials).2,9 Hence, the transport properties of thin film cathodes should be thoroughly investigated and clarified to provide a substantial contribution to the development of a high performance cathode for μ-SOFCs.
Here we present the Dchem of La0.6Sr0.4CoO3−δ (LSC) thin film, which has been measured in a rather classical and direct manner by employing a newly designed conductivity relaxation technique. Remarkably, the Dchem of epitaxial LSC thin films was higher than that of the bulk LSC by up to two orders of magnitude, and was found to be enhanced further as the film became thinner. To the best of our knowledge, this is the first proposal for the viable measurement and interpretation of enhanced oxygen diffusion kinetics in thin film cathodes, and we believe that this study can provide an actual insight into the proper implementation of thin film cathodes for high-performance μ-SOFCs.
60 nm and 243 nm thick LSC thin films were deposited on a (100) LaAlO3−δ (LAO) substrate by pulsed laser deposition (PLD). Both LSC and LAO have the same pseudo-cubic perovskite structure and their lattice parameters are a ≈ 3.84 Å (ref. 6 and 10) and a = 3.788 Å (ref. 11), respectively, thus exhibiting a small lattice mismatch with each other. Fig. 1 shows a transmission electron microscopy (TEM) cross-sectional image of the LSC–LAO interface and selected area electron diffraction patterns (SAED) of the LSC–LAO sample. SAEDs of LAO (Fig. 1b), LSC (Fig. 1c), and the LSC–LAO interface (Fig. 1d and e) clearly show that the LSC film was grown epitaxially on the LAO substrate. However, as can be seen in the {001} reflections in Fig. 1d and e, LSC and LAO have different c lattice constants, whereas they have almost the same a (=b) lattice constants. This means that the LSC film was compressed along the lateral a and b directions due to the constraint given by the LAO substrate, which has a smaller lattice constant than LSC, thereby inducing lattice expansion of the LSC film along the c direction as a counteraction to maintain the Poisson ratio. This anisotropic lattice distortion could be gradually released with distance from the interface. The lattice parameter variation along the vertical direction of the LSC thin films, which has been deduced by analyzing the diffraction pattern at the selected region in high-resolution transmission electron microscopy (HRTEM) images via fast Fourier transformations (FFTs), is shown in Fig. S1 (ESI†). The compressive strain given in the lateral direction of the LSC film is fairly released for the 243 nm thick film, whereas that of the 60 nm thick film considerably remains, and thus results in a greater compressive state for the 60 nm thick film than for the 243 nm thick film (Fig. S1, ESI†). The compressive state is expected to become higher in both LSC films at higher temperatures where the Dchem is measured because the thermal expansion coefficient (TEC) of LSC is larger than that of LAO (LSC: 22.3 × 10−6 K−1, LAO: 12.6 × 10−6 K−1).12
![(a) A TEM cross-sectional image of a 60 nm thick LSC film on LAO and (b–e) selected area electron-diffraction patterns along the [110] axis of the LSC–LAO cross-section at (b) the LAO substrate, (c) the LSC thin film, (d and e) the LSC–LAO interface.](/image/article/2013/EE/c2ee21647g/c2ee21647g-f1.gif) | ||
| Fig. 1 (a) A TEM cross-sectional image of a 60 nm thick LSC film on LAO and (b–e) selected area electron-diffraction patterns along the [110] axis of the LSC–LAO cross-section at (b) the LAO substrate, (c) the LSC thin film, (d and e) the LSC–LAO interface. | ||
The Dchem was measured using a conductivity relaxation technique via a DC four-probe method. As previously mentioned, oxygen diffusion inside the film is too fast because of the short diffusion length, which is limited by film thickness; therefore, oxygen transport in the cathode film is generally controlled by the surface exchange reaction.13 Therefore, to accurately measure Dchem, a longer diffusion length should be secured by avoiding a situation in which the entire film surface is directly exposed to the atmosphere, thereby limiting the diffusion length to the thickness of the film. In this study, similar to previously reported way,7,8 the surface of the LSC film was covered with an Al2O3 layer to block oxygen diffusion starting from the entire LSC surface along the vertical direction of the film (Fig. 2), and, thus, only allowing oxygen diffusion along the lateral direction of the film. An additional LAO buffer layer was inserted between the LSC and Al2O3 layers to prevent interfacial reactions between those materials. All these covering layers were fully dense without any cracks and delamination (Fig. S2, ESI†). Considerable leakage current was not observed through the LAO substrate14 during the conductivity measurement because the conductance of the 60 nm thick LSC film was already 500 times larger than that of the LAO substrate at the measurement temperature of 600 °C (Fig. S3, ESI†).
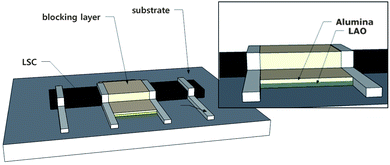 | ||
| Fig. 2 Schematics of the Dchem measurement set-up. | ||
The conductivity relaxation measurements were performed in the temperature range of 550 °C to 650 °C, where the reaction kinetics for the equilibration were sufficiently high, whilst avoiding any film morphology changes caused by thermal aging. The conductivity relaxation curves and their fitting results are shown in Fig. 3a, in which it is shown that the relaxation curves fit well with eqn (1) based on Fick's second law:15,16
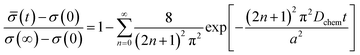 | (1) |
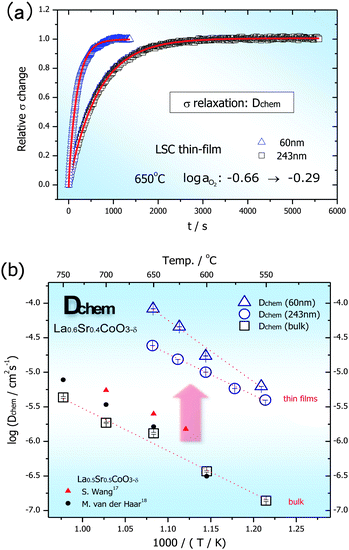 | ||
| Fig. 3 (a) Conductivity relaxations of 60 nm and 243 nm thick films limited by oxygen diffusion inside the films. (b) Dchem of 60 nm and 243 nm thick LSC thin films, and a LSC bulk. Some of previously reported Dchem of LSC bulk samples17,18 are also included for comparison (despite some difference in their composition (La0.5Sr0.5CoO3)). | ||
The Dchem of the 60 nm and 243 nm thick LSC thin films are plotted as a function of temperature in Fig. 3b and compared to those of a LSC bulk. The Dchem of the LSC bulk in Fig. 3b was also extracted from the conductivity relaxation technique, which was well matched with other reports.17,18 Remarkably, the Dchem of the LSC thin films were greater than that of the LSC bulk by two orders of magnitude. From the definition of chemical diffusivity, the Dchem of oxygen in LSC is proportional to the self-diffusivity of oxygen vacancy and electronic transference number ti (ti is equal to 1 in LSC) and is further enhanced by the thermodynamic factor. Therefore, even though the thermodynamic factor (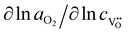 ) of bulk LSC is known to be nearly constant over the oxygen partial pressure (10−1.7 to 1 atm) and temperature range (600–700 °C),17 these enhanced Dchem in LSC films can be attributed in principle either to the increase in oxygen mobility and/or the increase in oxygen vacancy concentration (exactly the increased thermodynamic factor) in LSC thin films. Another interesting observation in Fig. 3b was that the Dchem of the thinner film was larger than that of the thicker one. This increased Dchem of the thinner film can be also explained by the enhanced oxygen mobility and/or the increased thermodynamic factor in the thinner LSC film.
) of bulk LSC is known to be nearly constant over the oxygen partial pressure (10−1.7 to 1 atm) and temperature range (600–700 °C),17 these enhanced Dchem in LSC films can be attributed in principle either to the increase in oxygen mobility and/or the increase in oxygen vacancy concentration (exactly the increased thermodynamic factor) in LSC thin films. Another interesting observation in Fig. 3b was that the Dchem of the thinner film was larger than that of the thicker one. This increased Dchem of the thinner film can be also explained by the enhanced oxygen mobility and/or the increased thermodynamic factor in the thinner LSC film.
In order to check which condition caused enhanced Dchem in the thinner LSC film, we first track change in oxygen vacancy concentration (equivalent to oxygen non-stoichiometry, δ) of the LSC film. It has been known that the oxygen non-stoichiometry (δ) of dense LSC films which have been estimated from impedance spectroscopy was smaller than that of the bulk.19 Recently, however, la O' et al.6 found also from impedance spectroscopy that the oxygen non-stoichiometry (δ) of epitaxial LSC films were higher than that of the bulk. These conflicting results in the extent of non-stoichiometry (δ) of LSC thin film and its relation to temperature and equilibrium oxygen partial pressure are not clearly explained yet.
In general, the valence state of cobalt ion varies between 2+ and 4+, while those of La(3+), Sr(2+), and O(2−) are fixed. Therefore, when a portion, x, of La3+ in LaCoO3 is replaced by Sr2+ to form La1−xSrxCoO3−δ, electroneutrality is maintained by both a decrease in the oxygen content and an increase in mean cobalt valence.20 Hence the oxygen non-stoichiometry, δ of LSC films can be inferred from the mean cobalt valence, n, as follows;
| δ = 1/2((3 + x) − n) = 1/2(3.4 − n), (x = 0.4) | (2) |
In order to evaluate the extent of oxygen non-stoichiometry, δ, from eqn (2), the depth distribution of Co oxidation states in the LSC thin films were analyzed by Electron Energy Loss Spectroscopy (EELS) depth profiling. According to previous reports,21 intensity ratios of L3 to L2 peaks of the L2, 3 edges in EEL spectra of Co cation are known to carry information on the oxidation state of Co. Hence, from the traced depth profile of the Co oxidation state along the film thickness, the relative distribution of oxygen vacancies from the film surface to the LSC–LAO interface can be quantified, because the abundance of Co cations with lower oxidation states reveals the existence of more oxygen vacancies for satisfying the charge neutrality criterion.20
Fig. 4a and b shows the EELS depth profiles of LSC films with different thicknesses of 60 and 243 nm. As shown in Fig. 4a and b, each EEL spectrum kept a nearly equivalent intensity profile throughout the whole film thickness which indicated no variation in oxygen vacancy concentration with respect to the position along the film thickness even though some anisotropic lattice distortion was gradually given across the vertical direction from the interface. Furthermore, as shown in Fig. 4c, both 60 and 243 nm thick films showed no difference in Co L2 and L3 intensities and also showed nearly the same intensity profile as that of bulk LSC, indicating that the oxygen vacancy concentration of thin films is not much different from that of bulk and has also no thickness dependence. Hence if we are only concerned with oxygen vacancy concentration, the higher Dchem of the thinner film than thicker or bulk LSC cannot be explained. Actually these results are contradictory to our initial expectation that the oxygen vacancy concentration of LSC films would be different from that of bulk as explained by Z. Cai et al.22
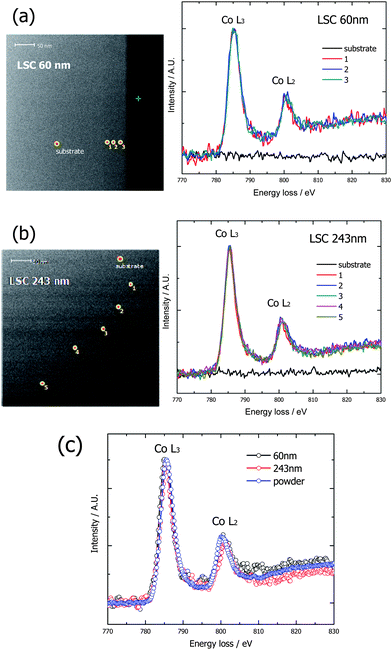 | ||
| Fig. 4 Depth profiling of L3 and L2 peaks of the L2, 3 edges in EEL spectra of the Co cation in (a) 60 nm thick LSC films and (b) 243 nm thick LSC films. (c) Comparison between thin films and bulk. | ||
According to their first principles-based calculation, oxygen vacancy formation was facilitated by the tensile strain given to the LSC film by the lattice mismatch with the substrate. In our case, the LSC film was compressed along the lateral directions due to constraint given by the LAO substrate, which has a smaller lattice constant than LSC, indicating that vacancy formation cannot be easier in a thin film than bulk. Nonetheless, since our EELS analysis showed the oxygen vacancy formation in LSC thin films was not affected by the constrained stress, we had to consider the possible change in thermodynamic factor with respect to the strained state of the thin film.
Oxygen mobility, which can also be affected by the lattice strain, is another factor to be considered in explaining the difference in Dchem values between the 60 nm and 243 nm thick films.16,23,24 The different Dchem activation energies in each case can be circumstantial evidence for the change in oxygen mobility with respect to different thin film strain states. The strain effect on mobility, however, is still in a controversial state that is beyond the scope of the present investigation. This topic will be addressed in a further study in a forthcoming paper. Nevertheless, it is worth mentioning that the method used for measuring Dchem in the thin film cathode is a very unique and effective tool, which has never been used before. Furthermore, the distinctly observed enhanced Dchem of the thin film cathode is very encouraging, providing clear insight into the development of thin film cathodes for high-performance μ-SOFCs.
In summary, chemical diffusion coefficients (Dchem) of the LSC cathode thin film are measured by the modified conductivity relaxation technique, which has been uniquely designed to secure a longer diffusion length by avoiding a situation in which the entire film surface is directly exposed to the atmosphere, thereby limiting the diffusion length to the thickness of the film. According to the measurement, the Dchem of epitaxial LSC thin films was found to be higher than that of the bulk LSC by up to two orders of magnitude, and was further enhanced as the film became thinner. To the best of our knowledge, this is the first proposed interpretation of enhanced oxygen diffusion kinetics in thin film cathodes by applying the viable measurement method. This remarkable discovery based on the enhanced oxygen transport property in the thin film cathode affords great potential for the proper architecturing of a highly efficient electrode for μ-SOFCs and also for other electrochemical devices, such as electrolysis cells, membrane reactors, and gas sensors.
Acknowledgements
This work was supported by the Fundamental R&D Program for Core Technology of Materials funded by the Ministry of Knowledge Economy, Republic of Korea and Institutional Research Program of Korea Institute of Science and Technology (KIST)Notes and references
- C.-W. Kwon, J.-W. Son, J.-H. Lee, H.-M. Kim, H.-W. Lee and K.-B. Kim, Adv. Funct. Mater., 2011, 21, 1154 Search PubMed; P.-C. Su, C.-C. Chao, J. H. Shim, R. Fasching and F. B. Prinz, Nano Lett., 2008, 8, 2289 CrossRef CAS; A. Evans, A. Bieberle-Hutter, J. L. M. Rupp and L. J. Gauckler, J. Power Sources, 2009, 194, 119 CrossRef CAS.
- S. Adler, Chem. Rev., 2004, 104, 4791 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345 CrossRef CAS; N. Q. Minh, J. Am. Ceram. Soc., 1993, 76, 563 CAS; N. Q. Minh, C. Singhal and M. Williams, ECS Trans., 2009, 17, 211 Search PubMed; E. Fabbri, L. Bi, D. Pergolesi and E. Traversa, Adv. Mater., 2012, 24, 195 CrossRef CAS.
- M. Sase, F. Hermes, K. Yashiro, K. Sato, J. Mizusaki, T. Kawada, N. Sakai and H. Yokokawa, J. Electrochem. Soc., 2008, 155, B793 CrossRef CAS; Y. M. L. Yang, A. J. Jacobson, C. L. Chen, G. P. Luo, K. D. Ross and C. W. Chu, Appl. Phys. Lett., 2001, 79, 776 Search PubMed.
- E. Mutoro, E. J. Crumlin, M. D. Biegalski, H. M. Christen and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 3689 RSC.
- G. J. la O', S.-J. Ahn, E. Crumlin, Y. Orikasa, M. D. Biegalski, H. M. Christen and Y. Shao-Horn, Angew. Chem., Int. Ed., 2010, 49, 5344 CrossRef CAS.
- M. Burriel, G. Garcia, J. Santiso, J. A. Kilner, J. C. C. Richard and S. J. Skinner, J. Mater. Chem., 2008, 18, 416 RSC.
- L. Wang, R. Merkle, J. Maier, T. Acarturk and U. Starke, Appl. Phys. Lett., 2009, 94, 071908 CrossRef.
- M. Prestat, A. Infortuna, S. Korrodi, S. Rey-Mermet, P. Muralt and L. J. Gauckler, J. Electroceram., 2007, 18, 111 CrossRef CAS; D. Beckel, U. P. Muecke, T. Gyger, G. Florey, A. Infortuna and L. J. Gauckler, Solid State Ionics, 2007, 178, 407 CrossRef CAS.
- R. H. E. van Doorn and A. J. Burggraaf, Solid State Ionics, 2000, 128, 65 CrossRef CAS.
- S. Geller and V. B. Bala, Acta Crystallogr., 1956, 9, 1019 CrossRef CAS.
- D. O. Klenov, W. Donner, B. Foran and S. Stemmer, Appl. Phys. Lett., 2003, 82, 3427 Search PubMed.
- X. Chen, S. Wang, Y. L. Yang, L. Smith, N. J. Wu, B. I. Kim, S. S. Perry, A. J. Jacobson and A. Ignatiev, Solid State Ionics, 2002, 146, 405 CrossRef CAS; G. Kim, S. Wang, A. J. Jacobson and C. L. Chen, Solid State Ionics, 2006, 177, 1461 CrossRef CAS; A. Karthikeyan and S. Ramanathan, Appl. Phys. Lett., 2008, 92, 243109 CrossRef.
- H.-R. Kim, J.-C. Kim, K.-R. Lee, H.-I. Ji, H.-W. Lee, J.-H. Lee and J.-W. Son, Phys. Chem. Chem. Phys., 2011, 13, 6133 RSC.
- C.-R. Song and H.-I. Yoo, Solid State Ionics, 1999, 120, 141 CrossRef CAS.
- C.-R. Song and H.-I. Yoo, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3975 CrossRef CAS.
- S. Wang, A. Verma, Y. L. Yang, A. J. Jacobson and B. Abeles, Solid State Ionics, 2001, 140, 125 CrossRef CAS.
- M. van der Haar, M. W. den Otter, M. Morskate, H. J. M. Bouwmeester and H. Verweij, J. Electrochem. Soc., 2002, 149, J41 CrossRef CAS.
- T. Kawada, J. Suzuki, M. Sase, A. Kaimai, K. Yashiro, Y. Nigara, J. Mizusaki, K. Kawamura and H. Yugami, J. Electrochem. Soc., 2002, 149(7), E252 CrossRef CAS.
- J. Mizusaki, Y. Mima, S. Yamauchi, K. Fueki and H. Tagawa, J. Solid State Chem., 1989, 80, 102 CrossRef CAS.
- Y.-M. Kim, J. He, M. D. Biegalski, H. Ambaye, V. Lauter, H. M. Christen, S. T. Pantelides, S. J. Pennycook, S. V. Kalinin and A. Y. Borisevich, Nat. Mater., 2012, 11(10), 888 Search PubMed.
- Z. H. Cai, Y. Kuru, J. W. Han, Y. Chen and B. Yildiz, J. Am. Chem. Soc., 2011, 133, 17696 CrossRef CAS.
- J. W. Han and B. Yildiz, J. Mater. Chem., 2011, 21, 18983 RSC.
- R. A. De Souza, A. Ramadan and S. Horner, Energy Environ. Sci., 2012, 5, 5545 Search PubMed; C. Korte, A. Peters, J. Janek, D. Hesse and N. Zakharov, Phys. Chem. Chem. Phys., 2008, 10, 4623 RSC; N. Schichtel, C. Korte, D. Hesse and J. Janek, Phys. Chem. Chem. Phys., 2009, 11, 3043 RSC; N. Schichtel, C. Korte, D. Hesse, N. Zakharov, B. Buz, D. Gerthsen and J. Janek, Phys. Chem. Chem. Phys., 2010, 12, 14596 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and detailed results. See DOI: 10.1039/c2ee21647g |
| This journal is © The Royal Society of Chemistry 2013 |