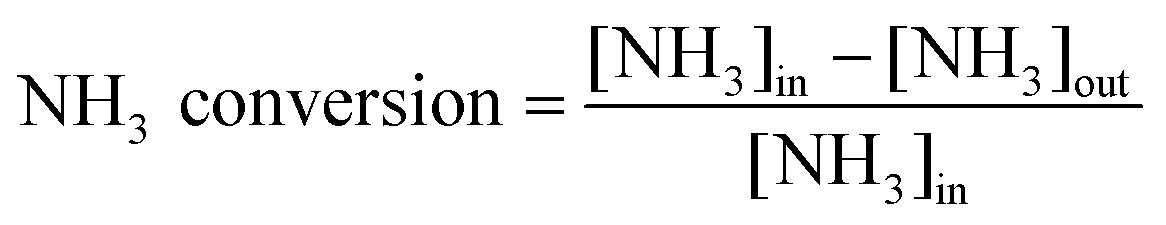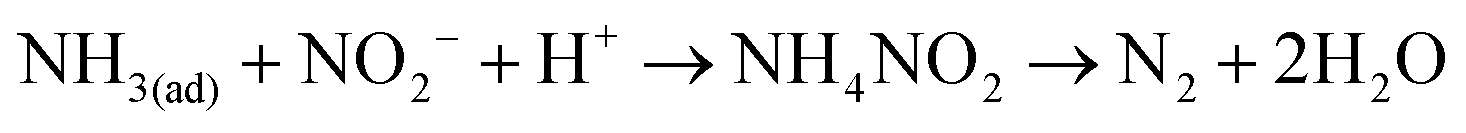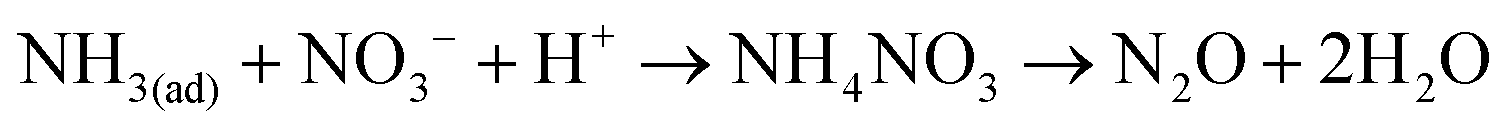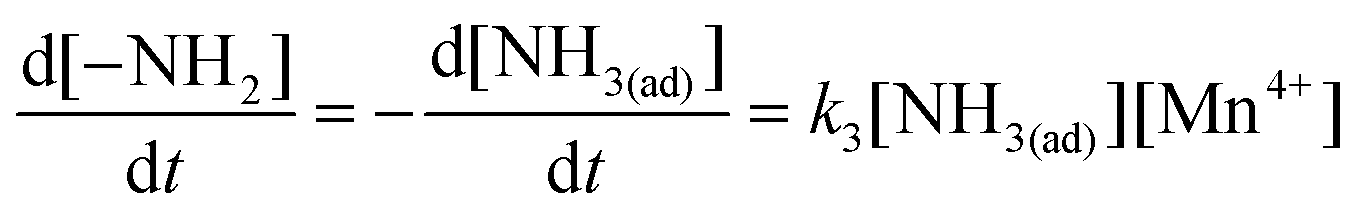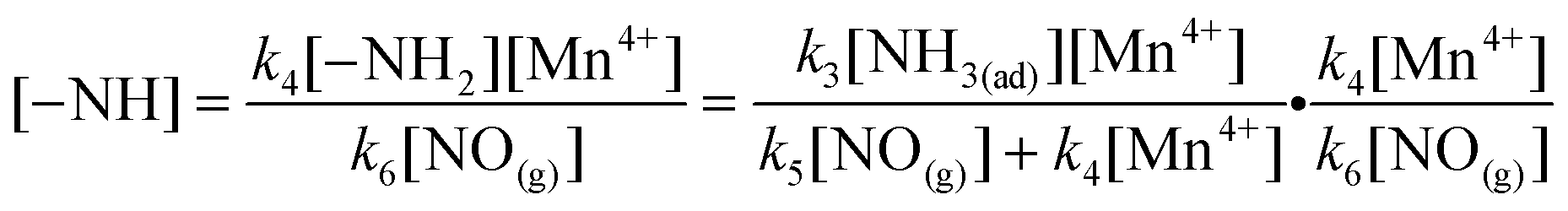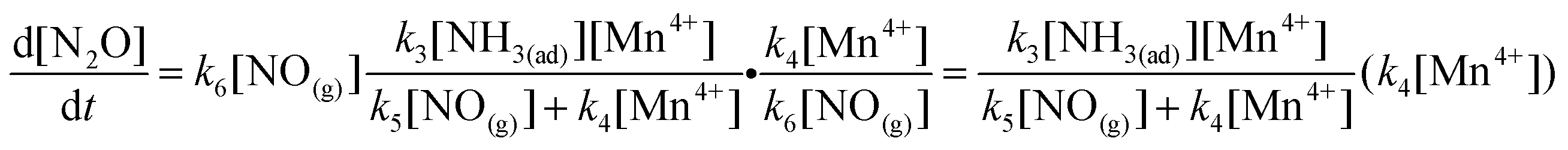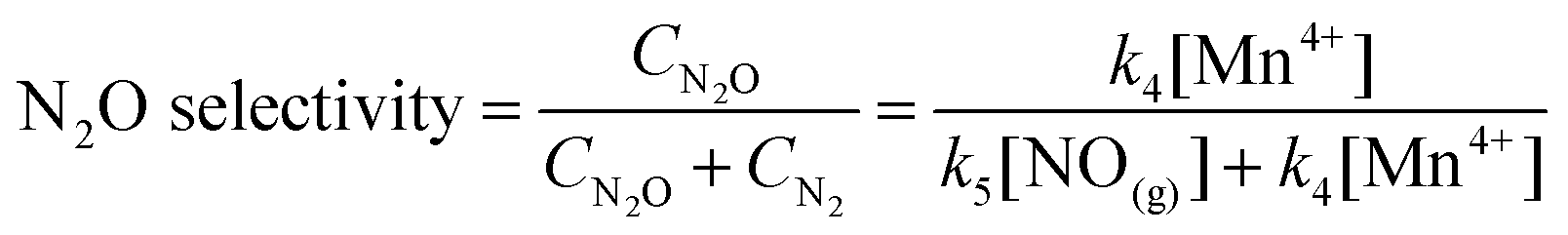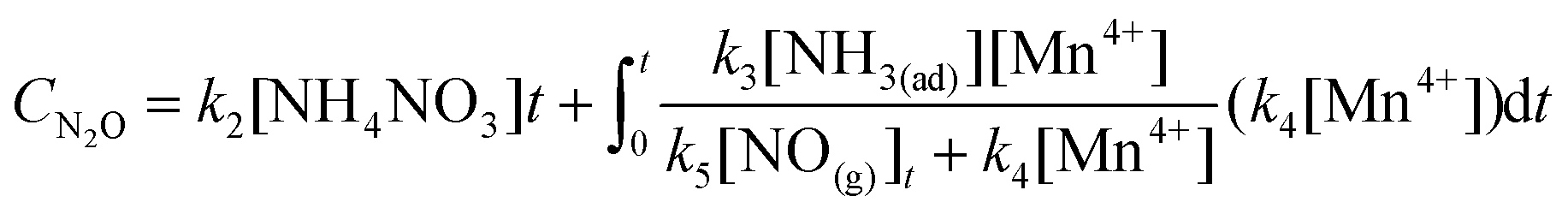Competition of selective catalytic reduction and non selective catalytic reduction over MnOx/TiO2 for NO removal: the relationship between gaseous NO concentration and N2O selectivity†
Received
30th August 2013
, Accepted 8th October 2013
First published on 9th October 2013
Abstract
In this work, a novel phenomenon was discovered that N2O selectivity of NO reduction over MnOx/TiO2 was related to the concentration of gaseous NO and that lower concentration of gaseous NO would cause higher N2O selectivity. In situ DRIFTS and transient reaction studies demonstrated that both the Eley–Rideal mechanism (the reaction of over-activated NH3 with gaseous NO) and the Langmuir–Hinshelwood mechanism (the reaction of adsorbed NO3− with adsorbed NH3 on the adjacent sites) could contribute to the formation of N2O. Kinetic study demonstrated that N2O selectivity would be independent of gaseous NO concentration if NO reduction over MnOx/TiO2 mainly followed the Langmuir–Hinshelwood mechanism. If NO reduction over MnOx/TiO2 mainly followed the Eley–Rideal mechanism, there was competition between the selective catalytic reduction (SCR) reaction and non selective catalytic reduction (NSCR) reaction. As gaseous NO concentration increased, more –NH2 was used to reduce gaseous NO to form N2 and the further oxidization of –NH2 to –NH was restrained, resulting in an obvious decrease of N2O selectivity. The Eley–Rideal mechanism played an important role in NO reduction over MnOx/TiO2, especially at higher temperatures. Therefore, N2O selectivity of the low temperature SCR reaction over MnOx/TiO2 decreased especially at higher temperatures after the increase of gaseous NO concentration.
1. Introduction
Nitrogen oxides (NO and NO2), emitted from automobiles and stationary sources, greatly contribute to the formation of smog, acid rain and ozone.1 Selective catalytic reduction (SCR) of NO with NH3 has been an efficient technique for the control of NOx emission from coal fired power plants and automobiles.2 The standard SCR process is based on the following reaction between NH3 and NO:3| | | 4NO + 4NH3 + O2 → 4N2 + 6H2O | (1) |
V2O5–WO3(MoO3)/TiO2 has been widely used as a SCR catalyst to control the emission of NO from stationary coal fired power plants for several decades.2 The temperature window of V2O5–WO3(MoO3)/TiO2 is about 300–400 °C, so the SCR unit is located upstream of the desulfurizer and electrostatic precipitator in order to avoid reheating of the flue gas.4 However, retrofitting the SCR devices into existing systems is difficult because the space and access in many power plants are extremely limited.5 Therefore, there has been strong demand in developing highly active SCR catalysts at low temperatures, which will be placed downstream of the electrostatic precipitator and desulfurizer.6 Mn based catalysts, for example MnOx–CeO2,7,8 MnO2/TiO2,9–13 MnOx–CeO2/TiO214 and Fe2O3–MnO2/TiO2,15 show excellent low temperature SCR activity among the first row transition metal based catalysts.16–20 However, some N2O would form during the low temperature SCR reaction over Mn based catalysts.6,7,21 The non selective catalytic reduction (NSCR) reaction is based on the following reaction between NH3 and NO:22
| | | 4NH3 + 4NO + 3O2 → 4N2O + 6H2O | (2) |
N2O is now considered as a pollutant due to its greenhouse effect and its depletion of the ozone layer.22–25 However, only a little work focused on the mechanism of N2O formation during the low temperature SCR over Mn based catalyst.21 Some groups have demonstrated that one of the two N atoms in N2O originates from NH3 and the other from NO under the SCR conditions.3,22 However, there is no agreement on the mechanism of N2O formation: by (1) reaction of gaseous NO with over-activated NH3 (–NH) to N2O (i.e. the Eley–Rideal mechanism), or (2) adsorption of NO3− on the adjacent sites of adsorbed NH3, followed by reaction to an activated transition state (i.e. NH4NO3) and then decomposition to N2O (i.e. the Langmuir–Hinshelwood mechanism).3 Recently, a novel phenomenon has been found that N2O selectivity of the low temperature SCR reaction over MnOx/TiO2 was related to the concentration of gaseous NO. N2O selectivity obviously decreased after the increase of gaseous NO concentration in the inlet. Herein, the mechanism of N2O formation during the low temperature SCR reaction over MnOx/TiO2 was studied, and the effect of NO concentration on N2O selectivity was investigated.
2. Experimental
2.1 Catalyst preparation
MnOx/TiO2 (Mn loading was 5 wt%) was prepared by the impregnation method using Degussa TiO2 P25 as support and manganese nitrate as precursor. The sample was dried at 110 °C for 12 h, and it was then calcined at 500 °C under air atmosphere for 3 h.
2.2 Catalytic test
The reduction of NO was performed on a fixed-bed quartz tube reactor (6 mm of internal diameter). The catalyst with 40–60 mesh was placed on the quartz wool held in the reactor, which was heated by a vertical electrical furnace. The total flow rate was 200 mL min−1 (room temperature), and the mass of catalyst was 200 mg. The corresponding gas hourly space velocity (GHSV) was 6 × 104 cm3 g−1 h−1 (i.e. 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1). The feed contained 500 or 1000 ppm of NO, 500 or 1000 ppm of NH3, 2% of O2, and balance of N2. The concentrations of NO, NO2, NH3 and N2O in the outlet were continually monitored by an FTIR spectrometer (MKS Instruments). The ratios of NOx and NH3 conversion, the amount of N2 formed and N2O selectivity were calculated using the following equations:
000 h−1). The feed contained 500 or 1000 ppm of NO, 500 or 1000 ppm of NH3, 2% of O2, and balance of N2. The concentrations of NO, NO2, NH3 and N2O in the outlet were continually monitored by an FTIR spectrometer (MKS Instruments). The ratios of NOx and NH3 conversion, the amount of N2 formed and N2O selectivity were calculated using the following equations:| |  | (3) |
| |  | (4) |
| |  | (5) |
| |  | (6) |
where, [NH3]in and [NOx]in were the concentrations of NH3 and NOx (including NO and NO2) in the inlet, and [NH3]out, [NOx]out and [N2O]out were the concentrations of NH3, NOx (including NO and NO2) and N2O in the outlet.
2.3
In situ DRIFTS study
In situ DRIFT spectra were recorded on a Fourier transform infrared spectrometer (FTIR, Nicolet NEXUS 870) equipped with a smart collector and an MCT detector cooled by liquid N2.26 The catalyst was finely ground and placed in a ceramic crucible and manually pressed. The FTIR spectra were recorded by accumulating 100 scans with a resolution of 4 cm−1.
3. Results
3.1 SCR performance of MnOx/TiO2 for the low temperature SCR reaction
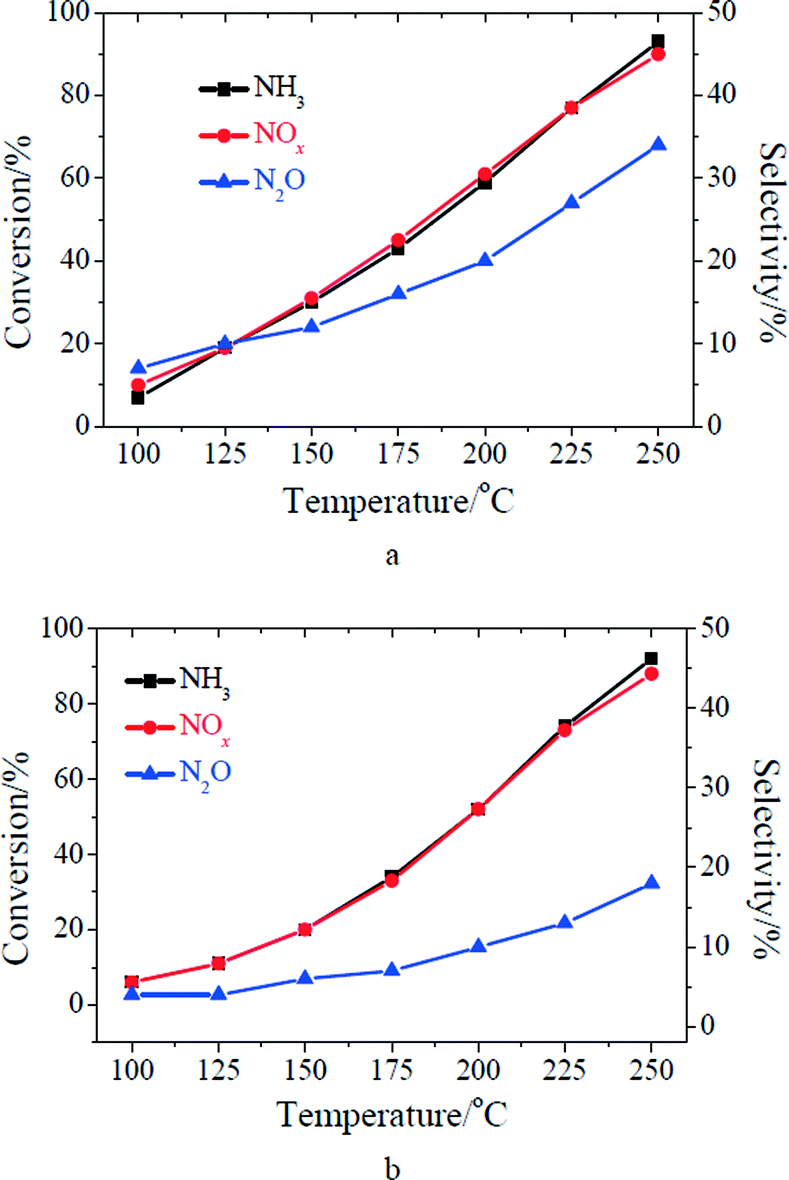
The performance of MnOx/TiO2 for the low temperature SCR reaction at 100–250 °C is shown in Fig. 1. Both the ratio of NO conversion and N2O selectivity increased with the increase of reaction temperature, which is consistent with previous research on the low temperature SCR reaction over MnOx/TiO2.13,27,28 As the concentrations of NO and NH3 in the inlet increased from 500 to 1000 ppm, the ratios of NO and NH3 conversion over MnOx/TiO2 slightly decreased. A similar result once happened on Mn–Fe spinel.6 However, N2O selectivity obviously decreased after the increase of the concentrations of gaseous NO and NH3 especially at higher temperatures (shown in Fig. 1). It suggests that N2O selectivity of the low temperature SCR reaction over MnOx/TiO2 was related to the concentrations of gaseous NO and NH3 in the inlet. This phenomenon was seldom previously reported in the literature.
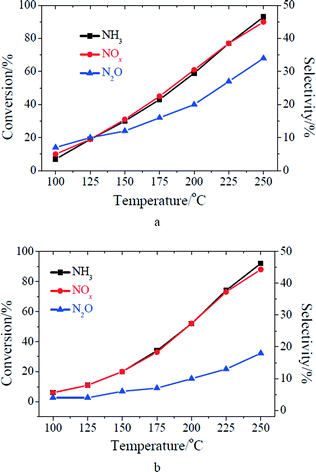 |
| | Fig. 1 SCR performance of MnOx/TiO2: (a) [NO] = [NH3] = 500 ppm; (b) [NO] = [NH3] = 1000 ppm. Reaction conditions: [O2] = 2%, catalyst mass = 200 mg, total flow rate = 200 mL min−1, GHSV = 60000 cm3 g−1 h−1. | |
3.2 Effect of gaseous NO concentration on N2O selectivity
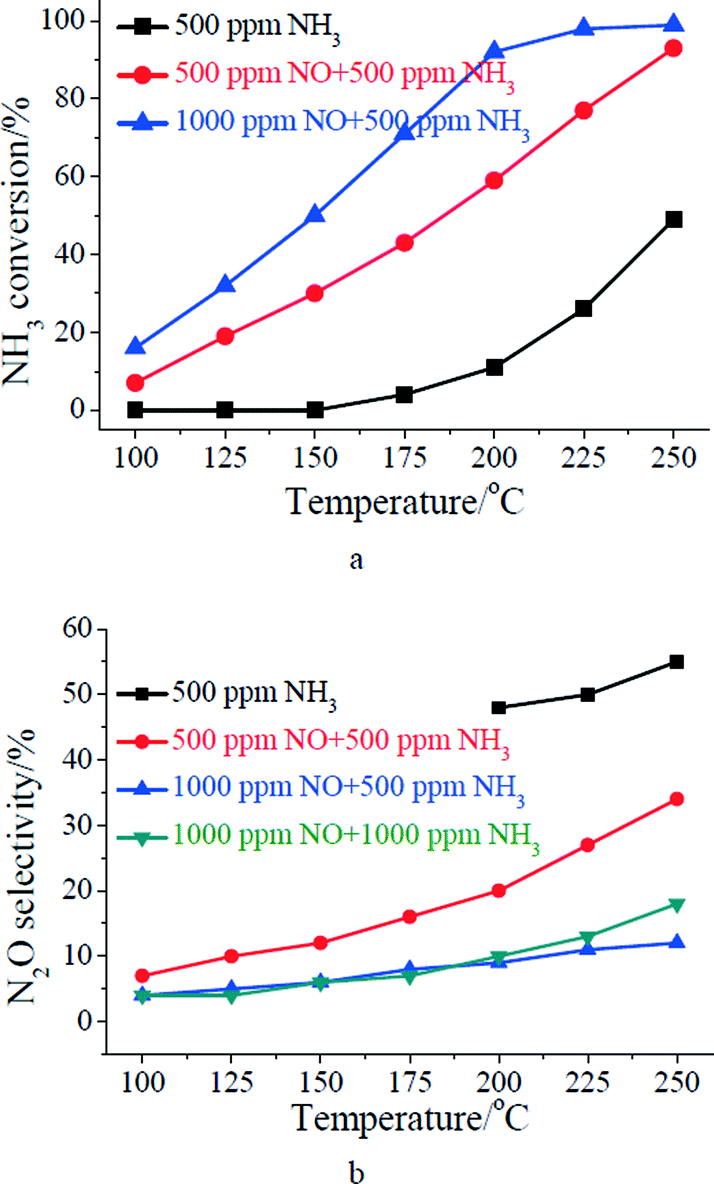
Fig. 2 shows the effect of gaseous NO concentration on NH3 conversion and N2O formation. As shown in Fig. 2a, little NH3 can be oxidized over MnOx/TiO2 below 175 °C in the absence of NO. With the increase of reaction temperature from 175 to 250 °C, NH3 oxidation was promoted (shown in Fig. 2a). However, more than 50% of NH3 was oxidized to N2O at 200–250 °C (shown in Fig. 2b).
 |
| | Fig. 2 Effect of NO concentration on: (a) NH3 conversion; (b) N2O selectivity. Reaction conditions: [O2] = 2%, catalyst mass = 200 mg, total flow rate = 200 mL min−1, GHSV = 60000 cm3 g−1 h−1. | |
As 500 ppm of NO was introduced, NH3 conversion over MnOx/TiO2 was promoted (shown in Fig. 2a). Meanwhile, N2O selectivity of MnOx/TiO2 for the SCR reaction was much less than that for NH3 oxidation at 200–250 °C. As the concentration of gaseous NO increased from 500 to 1000 ppm, NH3 conversion was further promoted (shown in Fig. 2a). Meanwhile, the formation of N2O over MnOx/TiO2 was further restrained (shown in Fig. 2b). It suggests that N2O selectivity over MnOx/TiO2 was related to the concentration of gaseous NO.
3.3
In situ DRIFTS study
3.3.1 Transient reaction at 150 °C.
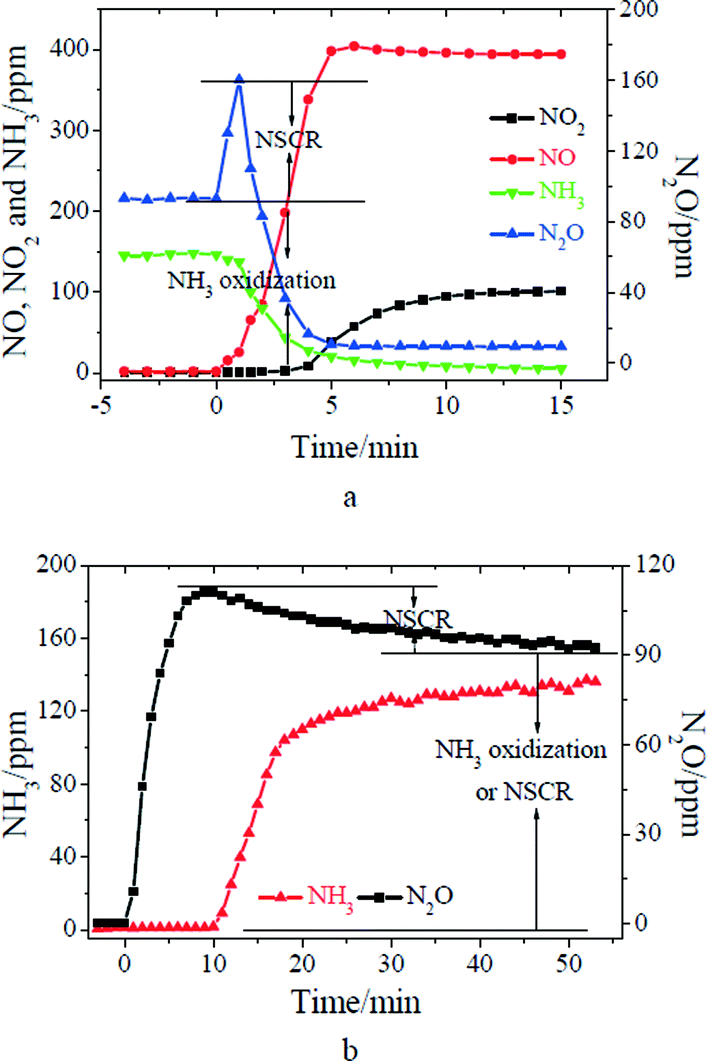
MnOx/TiO2 was first treated with 500 ppm of NH3 at 150 °C for 30 min followed by N2 purged for 5 min. 500 ppm of NO and 2% of O2 were then introduced into the IR cell (shown in Fig. 3a). After the adsorption of NH3, five characteristic vibrations at 1680, 1600, 1437, 1206 and 1165 cm−1 appeared on MnOx/TiO2. The bands at 1600 and 1206 cm−1 were assigned to coordinated NH3 bound to the Lewis acid sites, and the bands at 1680 and 1437 cm−1 were attributed to ionic NH4+ bound to the Brønsted acid sites.8,10 The band at 1165 cm−1 could be attributed to –NH2, which resulted from the activation of coordinated NH3 bound to the Lewis acid sites.29 After NO + O2 passed over NH3 pretreated MnOx/TiO2, the bands at 1680, 1600, 1437, 1206 and 1165 cm−1 corresponding to adsorbed ammonia species gradually diminished. Meanwhile, three characteristic vibrations at 1609, 1529 and 1280 cm−1 appeared. The band at 1609 cm−1 was assigned to monodentate nitrite, and the bands at 1529 and 1280 cm−1 were attributed to bidentate nitrate.30 Moreover, adsorbed H2O, which is the product of the SCR reaction, appeared at 1630 cm−1. These bands suggest that adsorbed NH3 can react with gaseous NO (i.e. the Eley–Rideal mechanism). The concentrations of N2O and NO in the outlet during the transient reaction were simultaneously recorded (shown in Fig. 4a). After NO + O2 passed over NH3 pretreated MnOx/TiO2, NO concentration gradually increased to about 480 ppm. Meanwhile, the concentration of N2O in the outlet rapidly increased to about 30 ppm, and it then gradually decreased to the background of N2O in NO (9 ppm) (shown in Fig. 4a). It suggests that the reaction between adsorbed NH3 and gaseous NO (i.e. the Eley–Rideal mechanism) at 150 °C can produce N2O.
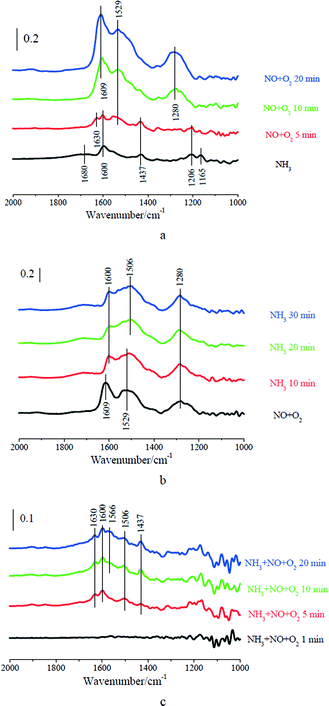 |
| | Fig. 3 (a) DRIFT spectra taken at 150 °C upon passing NO + O2 over NH3 presorbed MnO2/TiO2; (b) DRIFT spectra taken at 150 °C upon passing NH3 over NO + O2 presorbed MnO2/TiO2; (c) DRIFT spectra taken at 150 °C upon passing NH3 + NO + O2 over MnO2/TiO2. | |
 |
| | Fig. 4 (a) Transient reaction taken at 150 °C upon passing NO + O2 over NH3 presorbed MnO2/TiO2; (b) Transient reaction taken at 150 °C upon passing NH3 over NO + O2 presorbed MnO2/TiO2. | |
Then, the reactants were introduced to MnOx/TiO2 in the reverse order. MnOx/TiO2 was first treated with 500 ppm of NO and 2% of O2 for 30 min at 150 °C followed by N2 purged for 5 min. 500 ppm of NH3 was then introduced into the IR cell (shown in Fig. 3b). After the adsorption of NO + O2 at 150 °C, MnOx/TiO2 was mainly covered by monodentate nitrite (1609 cm−1) and bidentate nitrate (1529 and 1280 cm−1). After NH3 was introduced into the cell, the bands corresponding to monodentate nitrite (at 1609 cm−1) and bidentate nitrate (1529 cm−1) firstly shifted to 1600 and 1506 cm−1. Then, the intensities of the two bands gradually decreased (shown in Fig. 3b). They suggest that adsorbed NOx can react with adsorbed NH3 (i.e. the Langmuir–Hinshelwood mechanism). The concentrations of N2O and NH3 in the outlet during the transient reaction were simultaneously recorded (shown in Fig. 4b). After NH3 was introduced to NO + O2 pretreated MnOx/TiO2 for 5 min, little NH3 was observed. Meanwhile, the concentration of N2O in the outlet rapidly increased to about 13 ppm after the introduction of NH3, and it then gradually decreased to about 5 ppm in 60 min. It suggests that the reaction between adsorbed NH3 and adsorbed NOx (i.e. the Langmuir–Hinshelwood mechanism) can also produce N2O. Fig. 3b shows that the decrease of the band at 1600 cm−1 corresponding to adsorbed NH4NO2 was much faster than that at 1506 cm−1 corresponding to adsorbed NH4NO3. It suggests that the reaction through the nitrite route was faster than that through the nitrate route, which was consistent with the result of Mn–Fe spinel.6 Previous research demonstrated that the product of the nitrite route was N2, while that of nitrate route was N2O.22
Finally, the IR spectra during the SCR reaction (i.e. 500 ppm of NH3, 500 ppm of NO and 2% of O2 were simultaneously introduced) at 150 °C were recorded. As shown in Fig. 3c, adsorbed H2O (at 1630 cm−1), coordinated NH3 or adsorbed NH4NO2 (at 1600 cm−1), adsorbed NH4NO3 (at 1566 and 1506 cm−1) and ionic NH4+ (at 1437 cm−1) were all observed. It suggests that both the Langmuir–Hinshelwood mechanism and the Eley–Rideal mechanism could happen at 150 °C.
3.3.2 Transient reaction at 250 °C.
MnOx/TiO2 was first treated with 500 ppm of NH3 for 30 min at 250 °C, and 500 ppm of NO and 2% of O2 were then introduced into the IR cell (shown in Fig. 5a). After the adsorption of NH3, MnOx/TiO2 was mainly covered by coordinated NH3 bound to the Lewis acid sites (at 1602 cm−1). After NO + O2 passed over NH3 pretreated MnOx/TiO2, coordinated NH3 rapidly diminished, and adsorbed H2O (at 1620 cm−1) appeared. Then, MnOx/TiO2 was mainly covered by monodentate nitrite (at 1607 cm−1) and monodentate nitrate (1559 cm−1).30 The concentrations of NH3, N2O, NO and NO2 during the transient reaction were simultaneously recorded (shown in Fig. 6a). As NH3 was introduced to MnOx/TiO2, about 90 ppm of N2O was observed, which resulted from the oxidation of NH3 by the lattice oxygen of MnOx/TiO2. After NO + O2 passed over NH3 pretreated MnOx/TiO2 at 250 °C, N2O concentration rapidly increased from 90 to 160 ppm, and it then decreased to the background of N2O in NO (9 ppm). The concentration of N2O from the transient reaction at 250 °C was much higher than that at 150 °C. It suggests that the formation of N2O from the Eley–Rideal mechanism was obviously promoted with the increase of reaction temperature.
 |
| | Fig. 5 (a) DRIFT spectra taken at 250 °C upon passing NO + O2 over NH3 presorbed MnO2/TiO2; (b) DRIFT spectra taken at 250 °C upon passing NH3 over NO + O2 presorbed MnO2/TiO2; (c) DRIFT spectra taken at 250 °C upon passing NH3 + NO + O2 over MnO2/TiO2. | |
 |
| | Fig. 6 (a) Transient reaction taken at 250 °C upon passing NO + O2 over NH3 presorbed MnO2/TiO2; (b) Transient reaction taken at 250 °C upon passing NH3 over NO + O2 presorbed MnO2/TiO2. | |
Then, the reactants were introduced to MnOx/TiO2 in the reverse order. MnOx/TiO2 was first treated with 500 ppm of NO and 2% of O2 for 30 min followed by N2 purged for 5 min at 250 °C. 500 ppm of NH3 was then introduced into the IR cell (shown in Fig. 5b). After the adsorption of NO + O2 at 250 °C, MnOx/TiO2 was mainly covered by monodentate nitrite (1607 cm−1) and monodentate nitrate (1559 cm−1). After NH3 was introduced into the cell, the band at 1607 cm−1 corresponding to monodentate nitrite (at 1607 cm−1) rapidly diminished. However, monodentate nitrate (1559 cm−1) firstly shifted to 1534 cm−1. Then, it gradually diminished. Finally, MnOx/TiO2 was mainly covered by coordinated NH3 (at 1602 cm−1). The concentrations of N2O and NH3 during the transient reaction were simultaneously recorded (shown in Fig. 6b). After NH3 was introduced to NO + O2 pretreated MnOx/TiO2 for 10 min, NH3 in the outlet was observed. Meanwhile, the concentration of N2O in the outlet rapidly increased to about 110 ppm after the introduction of NH3, and it then gradually decreased to about 90 ppm in 50 min (shown in Fig. 6b). In this case, the origination of N2O from NH3 oxidation or the Langmuir–Hinshelwood mechanism was difficult to be differentiated. However, the concentration of N2O in the first 5 min was slightly higher than the concentration of N2O from the oxidation of 500 ppm of NH3. It suggests that the Langmuir–Hinshelwood mechanism can also contribute to N2O formation at 250 °C.
Finally, the IR spectra during the SCR reaction (i.e. 500 ppm of NH3, 500 ppm of NO and 2% of O2 were simultaneously introduced) at 250 °C were recorded. As shown in Fig. 5c, only adsorbed H2O (at 1620 cm−1) and coordinated NH3 or adsorbed NH4NO2 (at 1602 cm−1) can be clearly detected. However, the band at 1534 cm−1 corresponding to adsorbed NH4NO3 can not be observed. Fig. 5a shows that the disappearance of NH4NO3 was much slower than that of NH4NO2. It suggests that NH4NO3 did not form during NO reduction over MnOx/TiO2 at 250 °C. Therefore, the contribution of NH4NO3 decomposition to N2O formation can be neglected and N2O formation at 250 °C mainly resulted from the Eley–Rideal mechanism.
4. Discussion
4.1 Mechanism of N2O formation
4.1.1 N2O formation from the Langmuir–Hinshelwood mechanism.
The reduction of NO (including the SCR reaction and the NSCR reaction) through the Langmuir–Hinshelwood mechanism can be approximately described as:3,6,22| |  | (9) |
| |  | (10) |
| |  | (11) |
| |  | (12) |
| |  | (13) |
Reaction 7 is the adsorption of gaseous ammonia on the acid sites (i.e. Brønsted acid sites and Lewis acid sites) to form adsorbed ammonia species including ionic NH4+ and coordinated NH3. Reaction 8 is the physical adsorption of gaseous NO on MnOx/TiO2. Then, adsorbed NO is oxidized by Mn4+ on MnOx/TiO2 to form adsorbed NO2− (i.e.reaction 9). Reaction 10 is the oxidation of adsorbed NO by Mn4+ on MnOx/TiO2 to NO3−. Subsequently, adsorbed NO2− and NO3− react with adsorbed NH3 species on the adjacent sites to form NH4NO2 and NH4NO3 (i.e.reactions 11 and 12), respectively. Finally, NH4NO2 and NH4NO3 are decomposed to N2 and N2O, respectively. Reaction 13 is the regeneration of Mn4+ on MnOx/TiO2.
The kinetic equations of the formation of N2 and N2O over MnOx/TiO2 through the Langmuir–Hinshelwood mechanism can be approximately described as:
| | 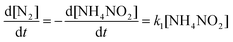 | (14) |
| |  | (15) |
where,
k1,
k2, [NH
4NO
2] and [NH
4NO
3] are the decomposition rate constants of NH
4NO
2 and NH
4NO
3, and the concentrations of NH
4NO
2 and NH
4NO
3 on MnO
x/TiO
2, respectively.
[NH4NO2] and [NH4NO3] are mainly related to the concentrations of NO adsorbed ([NO(ad)]) and Mn4+ on MnOx/TiO2 (the deduction is shown in the ESI†). The GHSV used in this work was quite high and there were generally large amounts of NOx and NH3 in the outlet (shown in Fig. 7a and b), so MnOx/TiO2 was almost saturated with the adsorption of NO and NH3. Furthermore, there is generally agreement that the SCR reaction starts with the adsorption of NH3, which is very strong compared to the adsorption of NO + O2 and the products.3 Thus, the increase of gaseous NO concentration from 500 to 1000 ppm could not break the adsorption equilibrium of NH3 and NO. It suggests that [NO(ad)] and [NH3(ad)] would not vary after increasing the concentrations of gaseous NO and NH3. As a result, the concentrations of NH4NO2 and NH4NO3 on MnOx/TiO2 were independent of the concentrations of gaseous NO and NH3.
 |
| | Fig. 7 Concentrations of NOx (a), NH3 (b), N2 formed (c) and N2O (d) in the outlet of the reactor. Reaction conditions: [O2] = 2%, catalyst mass = 200 mg, total flow rate = 200 mL min−1, GHSV= 60000 cm3 g−1 h−1. | |
4.1.2 N2O formation from the Eley–Rideal mechanism.
The reduction of NO (including the SCR reaction and the NSCR reaction) through the Eley–Rideal mechanism can be approximately described as:3,6,22| | | NH3(ad) + ≡Mn4+ → –NH2 + ≡Mn3+ + H+ | (16) |
| | | –NH2 + ≡Mn4+ → –NH + ≡Mn3+ + H+ | (17) |
| | | –NH2 + NO(g) → N2 + H2O | (18) |
| | | –NH + NO(g) → N2O + H+ | (19) |
| |  | (13) |
Reaction 16 is the activation of adsorbed ammonia species by Mn4+ on MnOx/TiO2 to form amide species (–NH2). –NH2 on MnOx/TiO2 can be further oxidized to –NH (i.e.reaction 17). Then, gaseous NO was reduced by –NH2 and –NH on the surface to form N2 and N2O (i.e.reactions 18 and 19), respectively.
The kinetic equation of reaction 16 can be described as:
| |  | (20) |
where
k3 and [–NH
2] are the kinetic constant of
reaction 16 and the concentration of –NH
2 on MnO
x/TiO
2, respectively.
The kinetic equation of reaction 17 can be described as:
| |  | (21) |
where
k4 and [–NH] are the kinetic constant of
reaction 17 and the concentration of –NH on MnO
x/TiO
2.
The kinetic equations of reactions 18 and 19 can be described as:
| |  | (22) |
| |  | (23) |
where,
k5,
k6 and [NO
(g)] are the kinetic constants of
reactions 18 and 19, and the concentration of gaseous NO, respectively.
According to eqn (20)–(22), the variation of –NH2 concentration on MnOx/TiO2 can be described as:
| |  | (24) |
As the reaction reached the steady state, –NH2 concentration on MnOx/TiO2 would not vary. Therefore,
| |  | (25) |
Thus,
| |  | (26) |
Then, the formation of N2 (eqn (22)) can be transformed as:
| | 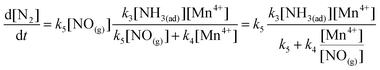 | (27) |
According to eqn (21) and (23), the variation of –NH concentration on MnOx/TiO2 can be described as:
| |  | (28) |
As the reaction reached the steady state, –NH concentration on MnOx/TiO2 would not vary. Therefore,
| |  | (29) |
Thus,
| |  | (30) |
Then, the formation of N2O (eqn (23)) can be transformed as:
| |  | (31) |
Taking account of the contributions of both the Langmuir–Hinshelwood mechanism and the Eley–Rideal mechanism, the formation of N2 and N2O can be approximately described as follows:
| |  | (32) |
| |  | (33) |
The concentrations of NH4NO2 and NH4NO3 on MnOx/TiO2 were independent of gaseous NO and NH3, so the contribution of the Langmuir–Hinshelwood mechanism to the formation of N2 and N2O would not vary after the increase of gaseous NO concentration. However, the contribution of the Eley–Rideal mechanism to N2 formation would increase after the increase of gaseous NO concentration (hinted by eqn (32)). Meanwhile, the contribution of the Eley–Rideal mechanism to N2O formation would decrease after the increase of gaseous NO concentration (hinted by eqn (33)).
If the reduction of NO mainly followed the Langmuir–Hinshelwood mechanism, N2O selectivity can be described as:
| |  | (34) |
The concentrations of NH4NO2 and NH4NO3 did not change after the increase of gaseous NO and NH3, so N2O selectivity was independent of gaseous NO concentration.
If the reduction of NO mainly followed the Eley–Rideal mechanism, N2O selectivity can be described as:
| |  | (35) |
Eqn (35) suggests that N2O selectivity would decrease after the increase of gaseous NO concentration.
There is generally agreement that the Langmuir–Hinshelwood mechanism plays an important role on the SCR reaction at lower temperatures.3,6 Meanwhile, the Eley–Rideal mechanism can also contribute to NO reduction at lower temperatures. The Eley–Rideal mechanism was obviously promoted with the increase of reaction temperature and it predominated over the SCR reaction at higher temperatures.3,6
4.2 Effect of gaseous NO concentration in the inlet on N2O selectivity
If the concentrations of gaseous NO and NH3 were sufficiently high, the whole catalyst bed was saturated with the adsorption of gaseous NO and NH3. Thus, [NH3(ad)], [NH4NO2] and [NH4NO2] can be regarded as constants on the whole catalyst bed. However, the concentration of gaseous NO at the bottom of the catalyst bed was much less than that at the top of the catalyst bed due to the reduction of NO. Therefore, the amounts of N2 and N2O formed over the whole catalyst bed should be described as follows:| |  | (36) |
| |  | (37) |
where t is the time how long gaseous NO passed through the catalyst column, which is inversely proportional to the GHSV.
Fig. 7a shows that the concentrations of gaseous NO in the outlet of the reaction with 1000 ppm of NO and 500 ppm of NH3 were much higher than that with 500 ppm of NO and 500 ppm of NH3. Meanwhile, the concentration of gaseous NO in the inlet of the reaction with 1000 ppm of NO and 500 ppm of NH3 were twice that with 500 ppm of NO and 500 ppm of NH3. They suggest that the concentrations of gaseous NO at each section of the catalyst bed during the reaction with 1000 ppm of NO and 500 ppm of NH3 were all higher than those with 500 ppm of NO and 500 ppm of NH3. Hinted by eqn (36), the amount of N2 formed during the reaction with 1000 ppm of NO and 500 ppm of NH3 was higher than that with 500 ppm of NO and 500 ppm of NH3, which was demonstrated in Fig. 7c. Hinted by eqn (37), the amount of N2O formed during the reaction with 1000 ppm of NO and 500 ppm of NH3 was less than that with 500 ppm of NO and 500 ppm of NH3, which was demonstrated in Fig. 7d. As a result, N2O selectivity of the reaction with 1000 ppm of NO and 500 ppm of NH3 was less than that with 500 ppm of NO and 500 ppm of NH3 (shown in Fig. 2b).
The concentrations of gaseous NO in the outlet and in the inlet during the reaction with 1000 ppm of NO and 1000 ppm of NH3 were both close to those with 1000 ppm of NO and 500 ppm of NH3 below 200 °C (shown in Fig. 7a). It suggests that the concentrations of gaseous NO at each section of the catalyst bed during the reaction with 1000 ppm of NO and 1000 ppm of NH3 were all close to those with 1000 ppm of NO and 500 ppm of NH3 below 200 °C. Therefore, the amounts of N2 and N2O formed during the reaction with 1000 ppm of NO and 1000 ppm of NH3 were close to those with 1000 ppm of NO and 500 ppm of NH3 below 200 °C, which is demonstrated in Fig. 7c and d. As a result, N2O selectivity of the reaction with 1000 ppm of NO and 1000 ppm of NH3 was close to that with 1000 ppm of NO and 500 ppm of NH3 below 200 °C (shown in Fig. 2b). However, little NH3 can be observed in the outlet of the reaction with 1000 ppm of NO and 500 ppm of NH3 above 200 °C (shown in Fig. 7b). It suggests that some catalyst in the bottom of the catalyst bed did not take part in the reaction because gaseous NH3 had been completely consumed. As the concentration of gaseous NH3 increased from 500 to 1000 ppm, a large amount of NH3 can be observed in the outlet above 200 °C (shown in Fig. 7b). Therefore, most of the catalyst took part in the reaction with 1000 ppm of NO and 1000 ppm of NH3 above 200 °C. They suggest that t of the reaction with 1000 ppm of NO and 1000 ppm of NH3 was higher than that with 1000 ppm of NO and 500 ppm of NH3 above 200 °C. Hinted by eqn (36) and (37), the amounts of N2 and N2O formed during the reaction with 1000 ppm of NO and 1000 ppm of NH3 above 200 °C were both much higher than that with 1000 ppm of NO and 500 ppm of NH3 (shown in Fig. 7c and d). Gaseous NO concentration at the top of the catalyst bed was much higher than that at the bottom of the catalyst bed due to NO reduction. It suggests that N2O selectivity at the top of the catalyst bed was much less than that at the bottom of the catalyst bed. As a result, N2O selectivity of the reaction with 1000 ppm of NO and 1000 ppm of NH3 above 200 °C were higher than that with 1000 ppm of NO and 500 ppm of NH3, which was demonstrated in Fig. 2b.
Fig. 7a shows that the concentrations of gaseous NO at each section of the catalyst column during the reaction with 1000 ppm of NO and 1000 ppm of NH3 were all higher than those with 500 ppm of NO and 500 ppm of NH3. Hinted by eqn (35), N2O selectivity of the reaction with 1000 ppm of NO and 1000 ppm of NH3 was less than that with 500 ppm of NO and 500 ppm of NH3 (shown in Fig. 2b).
5. Conclusion
N2O selectivity of the low temperature SCR reaction over MnOx/TiO2 was related to gaseous NO concentration in the flue gas. The lower concentration of gaseous NO in the flue gas would cause the higher N2O selectivity. If the concentration of gaseous NO in the flue gas is very low, low temperature SCR of NO with MnOx/TiO2 as the catalyst could not be the right choice for the control of NO emission due to the lower N2 selectivity. Furthermore, N2O selectivity at the bottom of the catalyst bed was much higher than that at the top of the catalyst column due to the lower gaseous NO concentration. Therefore, the decrease of GHSV to excessively pursue the removal efficiency of NO will cause lower N2 selectivity.
Acknowledgements
This study was financially supported by the National Natural Science Fund of China (Grant No. 21207067 and 41372044), Environmental scientific research of Jiangsu Province (2012026), the Fundamental Research Funds for the Central Universities (grant no. 30920130111023), the Zijin Intelligent Program, Nanjing University of Science and Technology (grant no. 2013–0106), and special fund of State Key Joint Laboratory of Environment Simulation and Pollution Control.
Notes and references
- G. S. Qi and R. T. Yang, J. Catal., 2003, 217, 434–441 CAS.
- N. Y. Topsoe, Science, 1994, 265, 1217–1219 CAS.
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS.
- S. J. Yang, J. H. Li, C. Z. Wang, J. H. Chen, L. Ma, H. Z. Chang, L. Chen, Y. Peng and N. Q. Yan, Appl. Catal., B, 2012, 117, 73–80 CrossRef PubMed.
- Y. Liu, T. T. Gu, X. L. Weng, Y. Wang, Z. B. Wu and H. Q. Wang, J. Phys. Chem. C, 2012, 116, 16582–16592 CAS.
- S. J. Yang, C. Z. Wang, J. H. Li, N. Q. Yan, L. Ma and H. Z. Chang, Appl. Catal., B, 2011, 110, 71–80 CrossRef CAS PubMed.
- G. S. Qi, R. T. Yang and R. Chang, Appl. Catal., B, 2004, 51, 93–106 CrossRef CAS PubMed.
- G. S. Qi and R. T. Yang, J. Phys. Chem. B, 2004, 108, 15738–15747 CrossRef CAS.
- P. G. Smirniotis, P. M. Sreekanth, D. A. Pena and R. G. Jenkins, Ind. Eng. Chem. Res., 2006, 45, 6436–6443 CrossRef CAS.
- Z. B. Wu, B. Q. Jiang, Y. Liu, H. Q. Wang and R. B. Jin, Environ. Sci. Technol., 2007, 41, 5812–5817 CrossRef CAS.
- B. Q. Jiang, Y. Liu and Z. B. Wu, J. Hazard. Mater., 2009, 162, 1249–1254 CrossRef CAS PubMed.
- Y. J. Kim, H. J. Kwon, I. S. Nam, J. W. Choung, J. K. Kil, H. J. Kim, M. S. Cha and G. K. Yeo, Catal. Today, 2011, 151, 244–250 CrossRef PubMed.
- S. M. Lee, K. H. Park, S. S. Kim, D. Kwon and S. C. Hong, J. Air Waste Manage. Assoc., 2012, 62, 1085–1092 CAS.
- Z. B. Wu, R. B. Jin, Y. Liu and H. Q. Wang, Catal. Commun., 2008, 9, 2217–2220 CrossRef CAS PubMed.
- G. S. Qi and R. T. Yang, Appl. Catal., B, 2003, 44, 217–225 CrossRef CAS.
- P. G. Smirniotis, D. A. Pena and B. S. Uphade, Angew. Chem., Int. Ed., 2001, 40, 2479–2481 CrossRef CAS.
- M. Wallin, S. Forser, P. Thormahlen and M. Skoglundh, Ind. Eng. Chem. Res., 2004, 43, 7723–7731 CrossRef CAS.
- D. A. Pena, B. S. Uphade and P. G. Smirniotis, J. Catal., 2004, 221, 421–431 CrossRef CAS PubMed.
- D. A. Pena, B. S. Uphade, E. P. Reddy and P. G. Smirniotis, J. Phys. Chem. B, 2004, 108, 9927–9936 CrossRef CAS.
- S. Roy, B. Viswanath, M. S. Hegde and G. Madras, J. Phys. Chem. C, 2008, 112, 6002–6012 CAS.
- X. F. Tang, J. H. Li, L. A. Sun and J. M. Hao, Appl. Catal., B, 2010, 99, 156–162 CrossRef CAS PubMed.
- G. Madia, M. Koebel, M. Elsener and A. Wokaun, Ind. Eng. Chem. Res., 2002, 41, 4008–4015 CrossRef CAS.
- G. Delahay, B. Coq, S. Kieger and B. Neveu, Catal. Today, 1999, 54, 431–438 CrossRef CAS.
- M. H. Kim and S. W. Ham, Top. Catal., 2010, 53, 597–607 CrossRef CAS PubMed.
- S. Suarez, J. A. Martin, M. Yates, R. Avila and J. Blanco, J. Catal., 2005, 229, 227–236 CrossRef CAS PubMed.
- L. A. Chen, J. H. Li and M. F. Ge, Environ. Sci. Technol., 2010, 44, 9590–9596 CrossRef CAS PubMed.
- P. R. Ettireddy, N. Ettireddy, S. Mamedov, P. Boolchand and P. G. Smirniotis, Appl. Catal., B, 2007, 76, 123–134 CrossRef CAS PubMed.
- K. Zhuang, J. Qiu, F. S. Tang, B. L. Xu and Y. N. Fan, Phys. Chem. Chem. Phys., 2011, 13, 4463–4469 RSC.
- F. D. Liu, K. Asakura, H. He, W. P. Shan, X. Y. Shi and C. B. Zhang, Appl. Catal., B, 2011, 103, 369–377 CrossRef CAS PubMed.
- K. I. Hadjiivanov, Catal. Rev., 2000, 42, 71–144 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cy00648d |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.