Alternative pathways in the ruthenium catalysed hydrogenation of CO to alcohols†
Jan H.
Blank
a,
Robert
Hembre
b,
James
Ponasik
b and
David J.
Cole-Hamilton
*a
aEaStCHEM, School of Chemistry, University of St. Andrews, St. Andrews, Fife, KY16 9ST, Scotland, UK. E-mail: djc@st-and.ac.uk; Fax: +44 (0) 1334 463808; Tel: +44 (0) 1334 463805
bEastman Chemical Company, Kingsport, TN, USA
First published on 8th November 2013
Abstract
CO hydrogenation in [PBu4]Br in the presence of [Ru3(CO)12] gives predominantly methanol, ethanol and propanol with small amounts of 1,2-ethanediol. Using RuO2 as the catalyst precursor, the same products are formed along with higher alcohols (1-butanol –1-heptanol). Reactions carried out using added 13CH3OH or 13CO show that ethanol and propanol come from homologation reactions of methanol and ethanol respectively, but that the higher alcohols are not formed through the lower alcohols as intermediates.
Introduction
The hydrogenation of carbon monoxide using heterogeneous catalysts, the Fischer Tropsch (FT) process, has been well-developed and fine-tuned on a huge scale for the production of alkanes and alkenes from coal or natural gas.1–3 In these processes all of the oxygen is discarded as water so oxygen functionalised molecules are minor side-products. With the rising scarcity of oil, the production of oxygenates such as alcohols from coal, natural gas or CO24–7 (C1 feedstocks) has become more attractive, but despite this, following a flurry of activity in the 70's and 80's the conversion of CO into higher alcohols is still underdeveloped, Homogeneous CO hydrogenation may potentially give better selectivity and control over product formation and especially selectivity towards oxygenates.8Homogeneous carbon monoxide hydrogenation was pioneered by Gresham and Schweizer using cobalt based systems at high temperatures and pressures.9 Lower pressure operation was possible using rhodium10,11 or ruthenium, mainly [Ru3(CO)12],12–14 based catalysts with the major products being methanol and 1,2-ethanediol.
Improvements were obtained by adding halides to the ruthenium system and Knifton and co-workers introduced15–17 phosphonium bromides as the solvent as well as RuO2 as an alternative catalyst precursor.15 In these cases, ethanol also became a significant product.
We have recently reported18 studies on [Ru3(CO)12] catalysed CO hydrogenation in [Bu4P]Br, under milder conditions (250 bar, 200 °C), than those studied by Knifton in which we discovered that [Bu3PH]Br, sometimes present as an adventitious impurity in the solvent acts as a significant promoter of the reaction by releasing HBr into the system.
The main products were methanol, ethanol and propanol in declining amounts. Significant amounts of ethylene glycol were also formed. We now report that the product distribution obtained when using RuO2 as the ruthenium precursor is significantly different from that obtained when using [Ru3(CO)12] and use 13C labelling studies to examine the mechanisms of formation of the various products.
Experimental
Unless stated otherwise, all chemicals where obtained from Sigma Aldrich and used as received. Air-sensitive compounds where handled under N2 using standard Schlenk techniques. NMR spectra where recorded on Varian 300 NMR or Bruker AM 300/400 NMR spectrometers. The chemical shifts where referenced to the solvent, which were in turn referenced to a TMS standard. IR spectra were recorded by pressing a sample of the liquid product between two KBr plates in a holder. The samples were recorded on a Thermo Nicolet Avatar FTIR spectrometer with a nitrogen cooled MCT detector. For quantitation Gas Chromatography analysis was performed using a Supelcowax-10 capillary column (60 m × 0.32 mm × 1.0 μm film thickness) using an Agilent 6890 N Network GC system equipped with a flame ionisation detector. For identification a HP 6890 series GC system equipped with a HP 5973 mass selective detector was used. Both machines used the same column using 1 ml min−1 helium carrier gas flow and 250 °C injector and detector temperatures. The temperature programmes was as follows: 50 °C, hold 3 minutes, ramp 20 °C min−1, 150 °C, hold 5 min, ramp 20 °C min−1, 220 °C, hold 13 minutes. Split ratio: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100.
:100.
Full details of the methods used to analyse the positions and amounts of 13C incorporated into the products are provided in the ESI.†
Catalytic experiments
:H2 1:1) by repeatedly pressurising up to 10 barg and venting. The system was then tested for leaks at 170 bar before being heated up to 200 °C with the vessel sealed and under continuous stirring. At 200 °C the pressure was adjusted to 250 bar and the reaction mixture was allowed to react for 4 h at constant pressure, with CO/H2 (1:1) being fed from a ballast vessel. The heating was switched off and the reactor swiftly cooled to room temperature. The excess gas was vented and the liquid product was collected for analysis. When employing 13CO the reactor was not kept at constant pressure (no feed from the ballast vessel) in order to prevent unnecessary dilution of the 13C label.
For instance: in an unlabelled control experiment (used as a reference for GCMS analysis) [Ru3(CO)12] (0.5 g, 2.3 mmol Ru) and [PBu4]Br (15 g, 44.2 mmol) were added to the autoclave. The autoclave was screwed onto the holder and purged by pressuring to 11 bar and venting to ambient pressure 6 times using CO/H2 1:1 v/v before pressurising to over 170 bar. A leak test was performed and no pressure drop was observed overnight other than that resulting from cooling the autoclave. The following morning the heating jacket was mounted, and switched on, the stirrer was switched on. When the temperature reached 200 °C, the pressure was adjusted to 250 bar and the stirrer was set to a fixed power input. The temperature and pressure were held constant until the heating and stirring were switched off after 4 h. Taps to the autoclave were closed to prevent gas flowing into the reactor because of the cooling. When the autoclave temperature reached below 30 °C the autoclave was vented and opened. The product mixture was usually a red liquid of which a small sample was stored and was often analysed using NMR or IR. The remainder of the liquid was transferred to a flask and stripped of volatiles by vacuum distillation using temperatures up to 250 °C and a liquid N2 cold trap. The volatiles that where collected were diluted using acetonitrile/NMP stock solution (2 mL of 5% (v/v)) and analysed using GC. The total product amounts were calculated using the NMP and acetonitrile peaks as internal references.
:1 syngas and brought to 185 bar to test for leaks. When the system did not leak the autoclave was vented to ambient pressure. Then the pressure was raised to approximately 13 bar using 13C labelled CO and H2 was added to approximately 24 bar total pressure. Then the pressure was increased to 180 bar using regular CO/H2 (1:1). These pressures were measured using the Back Pressure Regulator (BPR) which is accurate at higher pressures but not very accurate at low pressures. The autoclave was closed and subsequently heated to 200 °C under constant stirring. The work-up and analysis were as described above. In this case the distillate was analysed by GC–MS to assess the compound individual masses and fragmentation patterns.
A similar reaction and work-up procedure was carried out using [PBu4]Br (15 g, 44.2 mmol), [Ru3(CO)12] (0.5 g, 2.34 mmol), 13CO (17 bar) and H2 (pressure to 33 bar) before pressurising to 180 bar.
:1, 180 bar). CO/H2 were fed continuously from a ballast vessel to maintain constant pressure and to make up for CO/H2 used in the reaction: [PBu4]Br (15 g (44.2 mmol)), RuO2 (0.3 g (1.6 mmol)), and methanol (99% 13C, 12% 18O, 0.5 ml, 12.35 mmol) were added to the autoclave. The system was purged using 1:1 syngas and then pressurised to 180 bar. The autoclave was closed, heated to 200 °C and stirred for 4 h. The autoclave was cooled to 30 °C and the work up was performed as described above.
A similar reaction and work-up procedure was carried out using [PBu4]Br (15 g, 44.2 mmol), [Ru3(CO)12] (0.5 g, 2.34 mmol) and methanol (99% 13C, 12% 18O, 0.5 ml).
Results
During the course of our studies on CO hydrogenation in melt systems, we discovered that the product distribution was different when using RuO2 as the catalyst precursor from that obtained with [Ru3(CO)12]. In both cases the major products were methanol, ethanol and propanol in declining amounts together with 1,2-ethandiol (Fig. 1). However, when using RuO2 as the catalyst precursor, significant amounts of higher alcohols (1-butanol –1-heptanol, with an almost Gaussian distribution peaking at 1-pentanol) were observed as products (Fig. 2). | ||
| Fig. 1 Product distribution from the homogeneous hydrogenation of CO in [PBu4]Br using [Ru3(CO)12] (grey rectangles) or RuO2 (black rectangles) as the catalyst precursor. CO/H2(1:1, 250 bar), 200 °C, 4 h. | ||
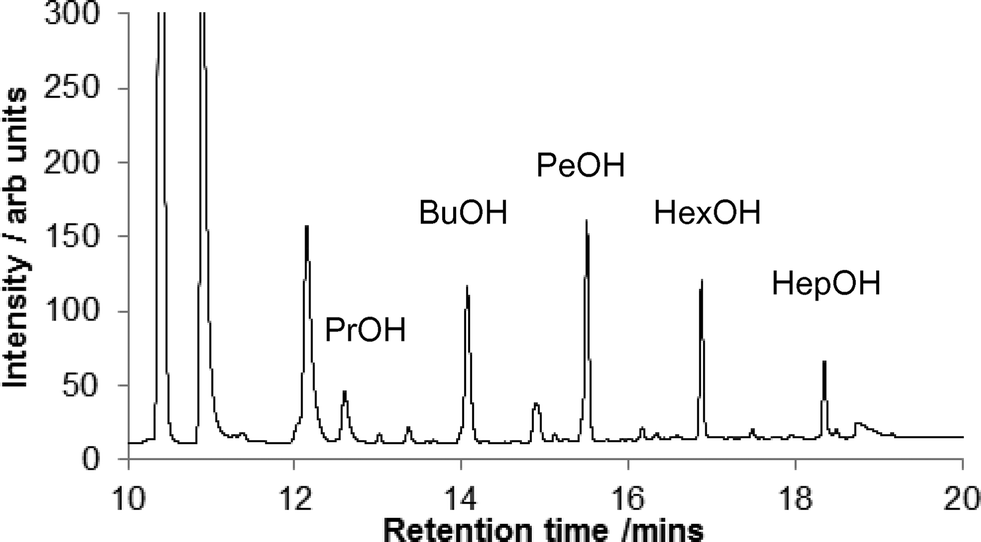 | ||
| Fig. 2 Vertically expanded GC trace from CO hydrogenation in the presence of RuO2, showing higher alcohol products; conditions as in Fig. 1. | ||
This unusual product distribution (the higher alcohols do not follow the expected Schulz–Flory distribution adopted by the lower alcohols) led us to believe that at least two different mechanisms may operate in the formation of C–C coupled products depending on the catalyst species present during the reaction. One possibility was that the tetrabutylphosphonium bromide solvent could act as a C4 source in the synthesis of the higher alcohols, via solvent degradation. We were also interested to discover whether ethanol and propanol in the [Ru3(CO)12] and/or RuO2·H2O catalysed reactions might be formed from intermediate methanol, as has previously been reported.19–23 Knifton has shown that, when 13CH3OH is added to a similar system ([Ru3(CO)12], [Bu4P]Br, 220 °C, CO/H2, 276 bar) the ethanol produced contains significant 13C in the methyl group,21 whilst Ono et al. have shown similar results when using [Ru3(CO)12] and [Co2(CO)8] in toluene containing [heptylPPh3]Br and PPh3,23 or when using [(Ph3P)2 N]Cl.20 Using heterogeneous catalysts and 13CH3OH, 13C can be incorporated into both the methylene and methyl groups of ethanol.22 When we added some methanol at the start of the reaction we found increased formation of ethanol.
Ethanol and propanol formation
To confirm that methanol homologation was occurring for the production of ethanol (and ethanol homologation for propanol), we carried out the CO hydrogenation with 13CH3OH added at the start if the reaction.When 99% 13C enriched methanol was added to reactions using either [Ru3(CO)12] or RuO2 catalyst precursors. The 1H NMR spectra of the product mixtures showed large 13C coupled satellites for the ethanol beta protons.‡ Interestingly, the protons from the α-position did not show increased 13C labelling in either case. Due to the crowded nature of the 1H NMR spectra only rough estimates of the isotopic abundances for some compounds found in the products can be made. Fortunately, the GC–MS data analysis (Table 1) showed that ethanol contained 40% and 34% of the 13C isotope for the reactions using RuO2 and [Ru3(CO)12], respectively. The GC–MS pattern once again indicated exclusive labelling at the β-position. Thus, in combination with the evidence from NMR studies, we conclude that methanol carbonylation is the major pathway to ethanol.
| Precursor | Methanol | Ethanol | Propanol | Butanol | Pentanol |
|---|---|---|---|---|---|
| a The isotopic enrichment is calculated as 13C/(12C + 13C) × 100% based on GC–MS isotopic patterns of the relevant peak groups. | |||||
| RuO2 | 22 | 40 | 33 | 2 | 0 |
| Ru3(CO)12 | 14 | 34 | 34 | 22 | 7 |
Interestingly, for propanol, the isotopic enrichment does not occur exclusively at the γ-position. Instead, the label is also found with almost equal abundance in the β-position but not in the α-position (see Scheme 1). This indicates that the ethyl moiety has time to switch carbons on the metal centre, probably via reversible β-hydride elimination and H addition steps before the CO insertion takes place.
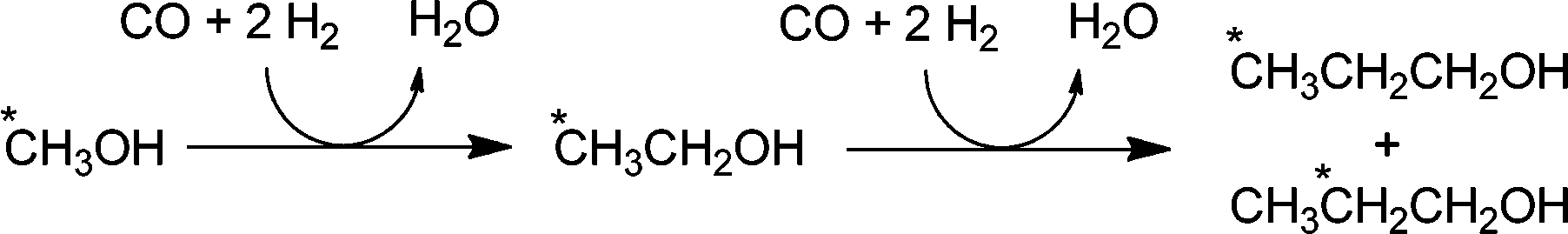 | ||
| Scheme 1 Ethanol and propanol synthesis via alcohol carbonylation. The * indicates the position of 13C when starting with 13CH3OH. | ||
Similar conclusions have been drawn by other workers when adding 13CH3OH to CO hydrogenation systems (see above) but the position of the label in propanol has not previously been discussed.
Higher alcohol formation
In order to understand the mechanisms of formation of the C4−6 alcohols in the two systems, we carried out studies using 13C-labelled CO with no added methanol.We considered four main mechanistic routes towards C–C coupled products:
1. Chain growth at the metal centre with irreversible release of the product alcohol
2. Chain growth through methanol and other alcohol carbonylation
3. C–C coupling via aldol condensation from free aldehyde intermediates
4. Alcohol synthesis via solvent degradation
These mechanisms and their expected outcomes when using RuO2 as the catalyst precursor and partially labelled CO or added 13CH3OH are shown in Table 2.
| Mechanism | Expected labelling pattern for butanol using RuO2 | |
|---|---|---|
| Using 13CO | Adding 13MeOH | |
| All C entirely from gas | ||
| CO + H2 → BuOH | Up to 4 13 C in butanol | No 13 C in butanol |
| Methanol reincorporation | ||
| MeOH + CO + H2 → → BuOH | Up to 4 13 C in butanol | A single 13 C in butanol |
| Acetaldehyde and aldol | ||
| MeOH + CO → CH3CHO → → BuOH | Up to 4 13 C in butanol | Up to two 13 CO in butanol in the β and δ positions |
| Solvent degradation | ||
| [Bu4P]Br → BuOH | No 13 C in butanol | No 13 C in butanol |
GC–MS analysis of the higher alcohols produced when using RuO2 as the catalyst and adding 13C labelled methanol shows that no label is present in the higher alcohol products. Hence, all the C atoms come from the gas phase and the mechanism of formation of these longer chain alcohols is different from that of the formation of ethanol and propanol, thus ruling out mechanism 2.
If the synthesis of higher alcohols occurs through the synthesis of methanol, which is then carbonylated to ethanal and undergoes subsequent aldol condensation we expect to find some product doubly labelled exclusively at the β and δ-position. Since there is no label in the higher products, mechanism 3 is also eliminated.
Using 13C enriched syngas and no added methanol we would expect that 13C incorporation should take place uniformly throughout the products, regardless of the mechanism except when product formation occurs through degradation of the solvent, [Bu4P]Br.
It is possible to work out the average 13C percentage incorporation into each C position of the products using mass spectrometry (see ESI†). If all the C atoms come from the CO, the average % incorporation normalised for chain length should be the same for all the products. If, however, the C4+-alcohols display a lower 13C abundance than the C1−3 alcohols this would indicate that the higher alcohols were made from degradation of the solvent, [Bu4P]Br.
Fig. 3 shows the % of 13CO incorporated into each product after reaction of 13C enriched syngas (H2:CO, 1:1) with [Ru3(CO)12] or RuO2 in tetrabutylphosphonium bromide at 200 °C and at a starting pressure of 260 bar (at 200 °C) in a closed batch system.
 | ||
| Fig. 3 The incorporation of 13CO into alcohol products during CO hydrogenation using 13CO enriched synthesis gas, based on the isotope pattern found in the GC-MS analysis for each product. The values given are 13C/12C + 13C × 100%, normalised for chain length. ▲[Ru3(CO)12] as catalyst precursor; ■ RuO2 as catalyst precursor. | ||
Fig. 3 clearly shows that a reaction using RuO2 in 13C enriched syngas yields isotopically enriched products. The average 13CO incorporation found for this reaction was 8.5%. More interestingly, the average 13C incorporation for all compounds is of the same order, indicating that solvent degradation does not contribute significantly to the formation of the longer chain alcohols.
For the reaction using [Ru3(CO)12] the average level of 13C in the products was higher because the initial partial pressure of 13CO used for the 13CO –12CO mixture was higher.§ Again, the GC–MS of the methanol, ethanol and propanol show patterns consistent with extensive labelling in the products. The found % incorporations for the first three alcohols are more or less the same at 11.5–12%. The amount of higher alcohols formed when using [Ru3(CO)12] is not sufficient to obtain meaningful information.
We are not aware of any other studies that report labelling experiments for the production of higher alcohols from homogeneous CO hydrogenation.
Discussion
It has been proposed14,24–26 that methanol is formed on ruthenium catalysts by a series of intermolecular hydride transfers and protonations (Scheme 2a). The initial hydride transfer is believed to occur from a species such as [Ru3H(CO)11]− to CO bound to another ruthenium centre, probably derived from [Ru(CO)3Br3]− in this system. The reaction is proposed to proceed via formyl and hydroxycarbene intermediates to a hydroxymethyl group which can be protonated to give methanol24 (see left side of Scheme 2a). Since we have shown that ethanol is formed by homolgation of methanol, the mechanism probably involves reaction of methanol with HBr (HBr is a promoter for this reaction) to give methyl bromide which is attacked by the ruthenium centre to give a coordinated methyl group. Methyl migration onto coordinated CO then provides the C–C bond forming step (right side of Scheme 2a). | ||
| Scheme 2 Possible mechanisms for the formation of a) methanol, ethanol and propanol; b), c) higher alcohols from the hydrogenation of CO using RuO2 as the catalyst precursor. | ||
For the higher alcohols, a different mechanism clearly operates and it must involve a different ruthenium centre since it is only observed when using RuO2 not when using [Ru3(CO)12] as the catalyst precursor. IR studies of post reaction mixtures do not show major differences between the two systems, so it is not possible to speculate on what the alternative ruthenium centre may be, although the production of water during the formation of active species from RuO2 may make the systems different.
One possible chain growth mechanism is that the hydroxymethyl intermediate in this alternative catalytic complex is reduced to methyl and that a sequence of migrations and reductions occurs (Scheme 2b). If this is the case, the methyl complex formed on this catalytic centre by reduction of the hydroxymethyl complex cannot be available from a reaction of the related ruthenium centre with MeBr derived from methanol as this would allow homologation. A further alternative mechanistic possibility is that the hydroxymethyl ligand migrates onto CO and the acyl is then reduced to hydroxyethyl (Scheme 2c). Repeating this sequence leads to chain growth. Interestingly, these two mechanisms leave the hydroxide on opposite ends of the growing chain in the final alcohol product, but our labelling studies cannot distinguish between them.
We do note, however, that, when adding 13CH3OH to either system, NMR and GCMS give no evidence for 13C incorporation into the 1,2-ethanediol formed, although 13CO is incorporated from the gas phase. The most logical initial steps in the formation of 1,2-ethanediol are those shown in Scheme 2c, with reductive elimination of 2-hydroxyethanal (glycolaldehyde) from the hydroxyacetyl complex, followed by aldehyde hydrogenation. It may be that hydrodeoxygenation of the hydroxyacetyl complex competes with reductive elimination and thus that chain growth occurs by the mechanism of Scheme 2c.
Both mechanisms 2b and 2c require the reduction of acyl complexes to alkyl complexes. This is proposed as a key step in the Pichler–Schulz mechanism for chain growth in Fischer–Tropsch reactions,27 but does not appear to have been demonstrated in model systems using molecular hydrogen as the reducing agent. Both silanes28 and boranes29–31 can affect the reduction so it is plausible that a ruthenium hydride donor similar to that involved in the initial formation of the formyl complex could be one component of the reducing system.
Conclusions
CO hydrogenation using ruthenium based catalyst precursors in [PBu4]Br under relatively mild conditions (250 bar, 200 °C) gives mainly methanol ethanol and propanol together with small amounts of ethylene glycol. Studies, in which 13CH3OH is added at the start of the reaction, show that ethanol and propanol are formed by homologation of methanol and ethanol respectively. When using RuO2 as the ruthenium source, higher alcohols (butanol–heptanol) are produced with a Gaussian distribution centred on pentanol. Studies using either added 13CH3OH or 13CO and no added methanol show that these higher alcohols are not formed by homologation but rather by a chain growth mechanism occurring at the Ru centre.Acknowledgements
We are greatly indebted to the Eastman Chemical Company for financial support and to Mr. Robert Cathcart and Mr. Peter Pogorzelec for invaluable technical support.Notes and references
- F. Fischer and H. Tropsch, Brennst.-Chem., 1926, 7, 97 CAS.
- S. Abello and D. Montane, ChemSusChem, 2011, 4, 1538 CrossRef CAS PubMed.
- D. A. Wood, C. Nwaoha and B. F. Towler, J. Nat. Gas Sci. Eng., 2012, 9, 196 CrossRef CAS PubMed.
- K. Tominaga, Y. Sasaki, M. Saito, K. Hagihara and T. Watanabe, J. Mol. Catal., 1994, 89, 51 CrossRef CAS.
- K. Tominaga, Y. Sasaki, T. Watanabe and M. Saito, Bull. Chem. Soc. Jpn., 1995, 68, 2837 CrossRef CAS.
- C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18122 CrossRef CAS PubMed.
- S. Wesselbaum, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499 CrossRef CAS PubMed.
- W. Keim, Pure Appl. Chem., 1986, 58, 825 CrossRef CAS.
- W. F. Gresham and C. E. Schweizer, US Patent, 2534018, 1950 Search PubMed.
- J. L. Vidal and Z. C. Mester, US 4115428, 1978 Search PubMed.
- J. L. Vidal and W. E. Walker, Inorg. Chem., 1980, 19, 896 CrossRef CAS.
- J. S. Bradley, J. Am. Chem. Soc., 1979, 101, 7419 CrossRef CAS.
- B. D. Dombek, J. Organomet. Chem., 1983, 250, 467 CrossRef CAS.
- B. D. Dombek, Organometallics, 1985, 4, 1707 CrossRef CAS.
- J. F. Knifton, J. Am. Chem. Soc., 1981, 103, 3959 CrossRef CAS.
- J. F. Knifton, J. Chem. Soc., Chem. Commun., 1983, 729 RSC.
- J. F. Knifton, J. J. Lin, D. A. Storm and S. F. Wong, Catal. Today, 1993, 18, 355 CrossRef.
- J. H. Blank, R. Hembre, J. Ponasik and D. J. Cole-Hamilton, ChemCatChem, 2013, 5, 1075 CrossRef CAS.
- B. K. Warren and B. D. Dombek, J. Catal., 1983, 79, 334 CrossRef CAS.
- H. Ono, K. Fujiwara, M. Hashimoto, E. Sugiyama and K. Yoshida, J. Mol. Catal., 1989, 57, 113 CrossRef CAS.
- J. F. Knifton, R. A. Grigsby and J. J. Lin, Organometallics, 1984, 3, 62 CrossRef CAS.
- J. G. Nunan, C. E. Bogdan, K. Klier, K. J. Smith, C. W. Young and R. G. Herman, J. Catal., 1988, 113, 410 CrossRef CAS.
- H. Ono, M. Hashimoto, K. Fujiwara, E. Sugiyama and K. Yoshida, J. Organomet. Chem., 1987, 331, 387 CrossRef CAS.
- D. S. Barratt and D. J. Cole-Hamilton, J. Organomet. Chem., 1986, 306, C41 CrossRef CAS.
- D. S. Barratt, C. Glidewell and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 1988, 1079 RSC.
- D. S. Barratt and D. J. Cole-Hamilton, J. Chem. Soc., Chem. Commun., 1985, 1559 RSC.
- W. A. Hermann, Angew. Chem., Int. Ed. Engl., 1982, 21, 117 CrossRef , and ref. therein.
- M. Akita, Appl. Catal., A, 2000, 200, 153 CrossRef CAS.
- J. A. Van Doorn, C. Masters and H. C. Volger, J. Organomet. Chem., 1976, 105, 245 CrossRef CAS.
- S. L. Brown and S. G. Davies, J. Chem. Soc., Chem. Commun., 1986, 84 RSC.
- R. E. Stimson and D. F. Shriver, Organometallics, 1982, 1, 787 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cy00423f |
| ‡ Unlabelled ethanol is also produced, presumably from methanol synthesised during the reaction from CO and H2. Unlabelled methanol is also present (86 and 78% from [Ru3(CO)12] and RuO2·2H2O respectively). |
| § Different pressures were used because the cylinder containing the 13CO depleted between the reactions. |
| This journal is © The Royal Society of Chemistry 2014 |