User-friendly synthesis of highly selective and recyclable mesoporous titanium-silicate catalysts for the clean production of substituted p-benzoquinones†
Irina D.
Ivanchikova
,
Mikhail K.
Kovalev
,
Maxim S.
Mel'gunov
,
Alexander N.
Shmakov
and
Oxana A.
Kholdeeva
*
Boreskov Institute of Catalysis, Lavrentieva 5, Novosibirsk 630090, Russia. E-mail: khold@catalysis.ru; Fax: +7 383 330 95 73; Tel: +7 383 326 94 33
First published on 7th October 2013
Abstract
Mesoporous titanium-silicates have been prepared following the evaporation-induced self-assembly (EISA) methodology and characterized by elemental analysis, XRD, N2 adsorption, SEM, DRS UV–Vis and Raman techniques. The use of acetylacetone during synthesis allowed the formation of highly dispersed dimeric and/or small oligomeric Ti species, within the mesostructured silica network, to be realized. The materials catalyse oxidation of alkylsubstituted phenols to corresponding p-benzoquinones with 100% selectivity using the green oxidant – 30% aqueous hydrogen peroxide. The titanium-silicates prepared by the convenient and versatile EISA-based procedure reveal the true heterogeneous nature of the catalysis and do not suffer from titanium leaching. They show advantages over other types of mesoporous Ti,Si-catalysts, such as TiO2–SiO2 mixed oxides and grafted Ti/SiO2, in terms of the catalyst stability and reusability.
Introduction
Oxidative transformations of substituted phenols offer efficient access to quinones which are the building blocks for a wide variety of bioactive compounds, e.g., vitamins E and K.1 Microporous titanium-silicates TS-1 and TS-2 are effective catalysts for the hydroxylation of phenol to hydroquinone and cathechol2 but they cannot be employed for oxidation of bulky substituted phenols due to their pore size limitation. On the contrary, H2O2-based oxidations of substituted phenols readily occur over mesoporous titanium-silicates.3–7 Pinnavaia and co-workers first reported oxidation of 2,6-di-tert-butylphenol (DTBP) into a mixture of benzoquinone (BQ) and diphenoquinone (DPQ) using Ti-HMS and Ti-MCM-41.3 Two groups independently reported the oxidation of 2,3,6-trimethylphenol (TMP) to 2,3,5-trimethyl-1,4-benzoquinone (TMBQ, vitamin E precursor) with a 77–82% yield over Ti-MMM4 and Ti-MCM-415 catalysts prepared by hydrothermal templated synthesis, under basic conditions. The major by-product of this reaction was 2,2′,3,3′,5,5′-hexamethyl-4,4′-biphenol (BP).A hydrothermally stable mesostructured catalyst Ti-MMM-2 synthesized under weak acidic conditions showed a similar TMBQ yield but revealed better resistance with respect to deactivation with aqueous H2O2.8 An almost quantitative yield of TMBQ was attained using TiO2–SiO2 mixed oxides prepared by a sol–gel technique6 and catalysts obtained by grafting Ti(IV) precursors onto SBA-159 or commercial mesoporous silica.10 It was found that the key point to achieve high TMBQ selectivity is the presence of Ti(IV) dimers or small oligomers on the surface of mesoporous silica.10c Such species are manifested by the presence of a broad band centred at 250–290 nm in DR UV–Vis spectra. The presence of two (several) adjacent Ti atoms is required to ensure fast oxidation of the key intermediates, phenoxyl radicals, thus preventing the formation of dimeric by-products.10b,c The proper state of Ti centers in the grafted catalysts can be achieved either by controlling the surface concentration of Ti (the optimum value lies within the range of 0.7–1.0 Ti nm−2)10b,c or by using di(poly)nuclear Ti(IV) precursors.9,10c Although the grafted catalysts produce TMBQ with excellent yields, they require the use of concentrated (70%) H2O2 for a stable recycling behavior.10c Therefore, the development of a simple, user-friendly procedure for the synthesis of mesoporous titanium-silicate catalysts, which would combine excellent selectivity with stable recycling performance for the oxidation of TMP and other alkylphenols, with the environmentally friendly oxidant – 30% aqueous H2O2 still remains a demanding task.
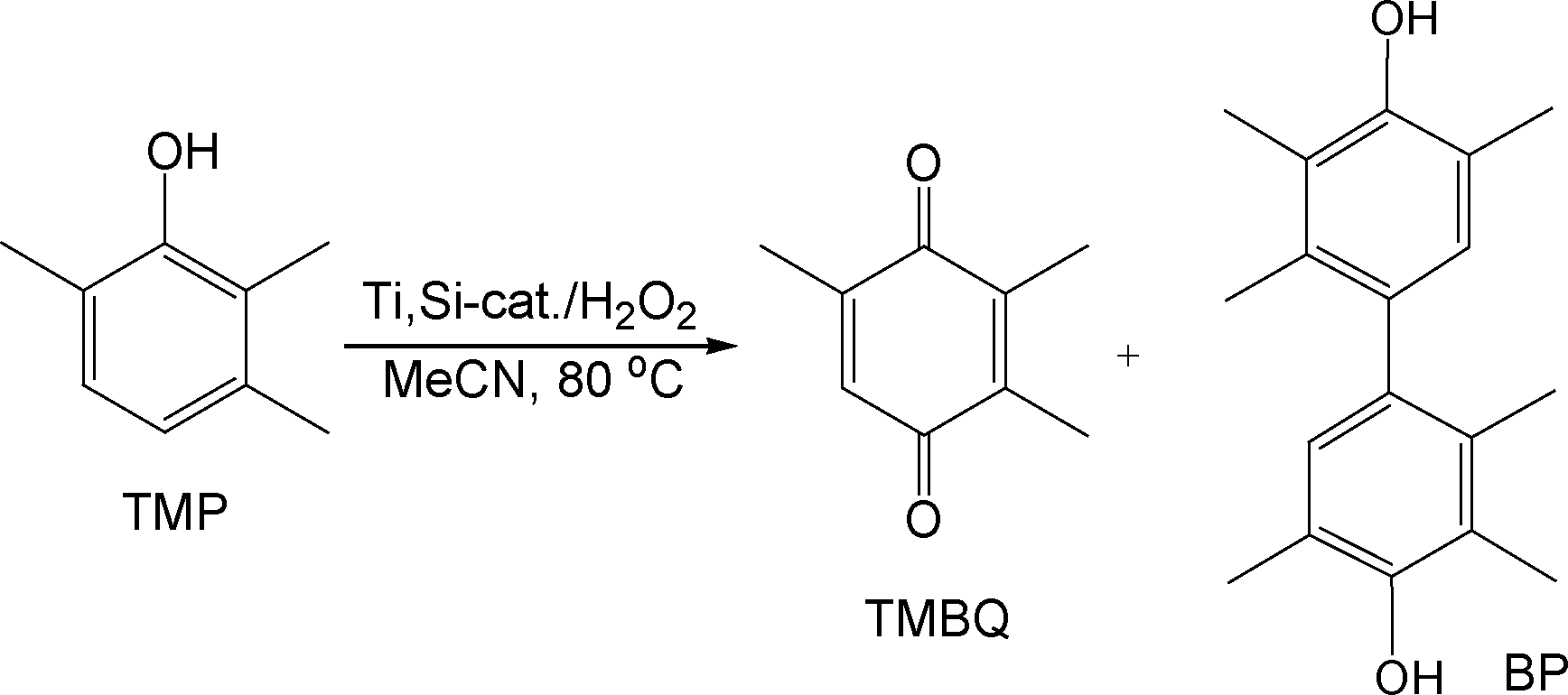
Evaporation-induced self-assembly (EISA) is a simple technique which enables rapid production of ordered thin silica films11–18 and can be applied for the preparation of mesoporous materials in the form of fibres or powders.19 The surfactant concentration progressively increases during solvent evaporation and triggers the self-assembly of silica-surfactant micelles and their further organization into mesophases. The EISA method is widely used in the synthesis of ordered mesoporous metal oxides.20 A few studies demonstrated the utility of the EISA method for the synthesis of Ti-containing ordered silica films.21–23 Ogawa et al.21 reported the preparation of films with a Si/Ti ratio of 50 and demonstrated their photocatalytic reactivity. Thin films with Si/Ti = 30–80 containing mostly site-isolated Ti(IV) species and with a DRS UV–Vis signal at 215 nm were prepared using acetylacetone (acac) to retard the hydrolysis rate of Ti(IV) isopropoxide.22 Hüsing et al.23 achieved titanium loading of up to a Si/Ti ratio of 5 in silica films, using the EISA technique. Oligo(ethylene oxide) alkyl ether surfactant Brij56 was used as both a template for mesostructure formation and a complexing agent for Ti(IV). DRS UV–Vis studies of the calcined films showed only a single intense band at 205 nm, characteristic of isolated Ti(IV) atoms in tetrahedral coordination.24
In the present work, we extended the EISA approach to the preparation of mesostructured titanium-silicate catalysts. We report a simple and versatile procedure for the synthesis of highly efficient and reusable catalysts, with well-dispersed di(oligo)nuclear active Ti centers, which allow for the oxidation of alkylphenols to the target p-benzoquinones with 100% selectivity using 30% H2O2.
Experimental
Materials
Cetyltrimethylammonium bromide (CTAB, 99+%) and tetraethylorthosilicate (TEOS, 98+%) were purchased from Aldrich. Tetraethoxy titanium (IV) (TEOT, technical grade) and acetylacetone (99%), 2,6-di-tert-butylphenol (DTBP, 99%), 2,6-dimethylphenol (DMP, 99%) and 2,6-di-tert-butyl-1,4-benzoquinone (DTBQ, 98%) were used as received from Aldrich. 2,3,6-Trimethylphenol (TMP, 97+%) was obtained from Fluka. Acetonitrile (Fluka) was dried and stored over activated 4 A molecular sieves. Ethanol (95%) and other reactants were obtained commercially and used without additional purification. The concentration of H2O2 (ca. 30 wt% in water) was determined iodometrically prior to use. Deionized water (EASY pure, RF, Barnsted) was used for the preparation of catalysts.Synthesis of catalysts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O:acac were 1:2:1 (sample A) or 1:2:2 (sample B). The resulting solutions were stirred for ~1 h at room temperature before use in the next step.
:25/C2H5OH:6.7/H2O:0.0012/HCl:0.15/CTAB:0.018/Ti. In a standard procedure, 5.5 g of surfactant, CTAB, was dissolved in 115 g of ethanol (95%) for 2 h at room temperature. Then 20.8 g of TEOS, 12.0 g of 0.01 M HCl and 1.52 g of the Ti(acac)x precursor solution were added dropwise under vigorous stirring. The resulting clear sol was left in an open Petri dish under ambient conditions until the solvent had fully evaporated. The resulting solids were calcined at 550 °C for 5 h in air with a temperature ramp of 1 °C min−1 to remove the organic species. The freshly calcined samples were characterized by elemental analysis, N2 adsorption, XRD, DRS UV–Vis and Raman spectroscopy and used for catalytic tests.
:H2O:acac were 1:2:1 (sample A) or 1:2:2 (sample B). The resulting solutions were stirred for ~1 h at room temperature before use in the next step.
:25/C2H5OH:6.7/H2O:0.0012/HCl:0.15/CTAB:0.018/Ti. In a standard procedure, 5.5 g of surfactant, CTAB, was dissolved in 115 g of ethanol (95%) for 2 h at room temperature. Then 20.8 g of TEOS, 12.0 g of 0.01 M HCl and 1.52 g of the Ti(acac)x precursor solution were added dropwise under vigorous stirring. The resulting clear sol was left in an open Petri dish under ambient conditions until the solvent had fully evaporated. The resulting solids were calcined at 550 °C for 5 h in air with a temperature ramp of 1 °C min−1 to remove the organic species. The freshly calcined samples were characterized by elemental analysis, N2 adsorption, XRD, DRS UV–Vis and Raman spectroscopy and used for catalytic tests.
To estimate the hydrothermal stability and stability toward aqueous H2O2, sample A was treated either with boiled water for 6 h or with aqueous 30% H2O2 for 1 h (15 mg of catalyst, 0.11 M H2O2, 3 mL MeCN, 25 °C), dried in air and calcined at 550 °C before physicochemical measurements.
Catalytic oxidations
Catalytic oxidations were performed under vigorous stirring (600 rpm) in thermostated glass vessels. Typical reaction conditions were as follows: phenolic substrate, 0.1 M; H2O2, 0.35 M; catalyst 20 mg (Ti 0.006 mmol); MeCN, 1 mL, 80 °C, 20 min. The reaction was started by the addition of H2O2. Samples of the reaction mixture were withdrawn periodically during the reaction course by a syringe. The oxidation products were identified by GS-MS and 1H NMR spectroscopic techniques. The yield of quinone and conversion of alkylphenol were quantified by GC using an internal standard, biphenyl. Turnover frequency (TOF) values were determined from the initial rates of phenol consumption. Each experiment was reproduced 3–5 times.Catalyst reusability was examined in 4–6 time scaled experiments (total reaction mixture volume 15–20 mL). Following the reaction, the catalyst was filtered off and washed with hot acetonitrile and acetone. Before use in the next catalytic run, the catalyst was dried in air at room temperature overnight and calcined in air at 250 °C for 2 h and then at 550 °C for 4 h. The nature of the catalysis was verified by hot filtration tests.
Instrumentation
GC analyses were performed using a gas chromatograph Tsvet-500 equipped with a flame ionization detector and a quartz capillary column (30 × 0.25) filled with Supelco MDN-5S. GC-MS analyses were carried out using an Agilent 7000B system with the triple-quadrupole mass-selective detector Agilent 7000 and GC Agilent 7890B (quartz capillary column 30 m × 0.25 mm/HP-5ms). 1H NMR spectra were recorded on a Bruker Avance-400 spectrometer (400.13 MHz).XRD measurements were performed on a high precision X-ray diffractometer mounted on a beamline No. 2 of VEPP-3 storage ring at the Siberian Synchrotron Radiation Center (SSRC). The radiation wavelength was λ = 0.15393 nm. High natural collimation of the synchrotron radiation beam, flat perfect crystal analyzer and parallel Soller slit on the diffracted beam, limited its azimuthal divergence and provided extremely high instrumental resolution of the diffractometer in a small angle region of 2θ = 0.5 ÷ 10° and higher.
Nitrogen adsorption measurements were carried out at 77 K using an ASAP-2400 instrument (Micromeritics) within the partial pressure range 10−4–1.0. The catalysts were degassed at 90 °C for 24 h before the measurements. Textural characteristics were calculated using a comparative method reported elsewhere.25 Pore size distributions were calculated from the adsorption branches of the nitrogen isotherms by means of the regularization procedure, using reference local isotherms calculated in a cylindrical silica pore model in the framework of the density functional theory (DFT) approach. Special software provided by Quantachrome Corp. was used for this purpose. Mean pore diameters were calculated as mathematical expectation values from these distributions.
The state of titanium in the catalysts was probed by DR UV–Vis spectroscopy under ambient conditions using a Shimadzu UV–VIS 2501PC spectrophotometer. FT-Raman spectra (3600–100 cm−1, 300 scans, resolution 4 cm−1, 180° geometry) were recorded using a RFS 100/S spectrometer (Bruker). Excitation of the 1064 nm line was provided by a Nd-YAG laser (100 mW power output). Scanning electron microscopy images were acquired using a JEOL JSM-6460 LV microscope. Titanium content in the filtrate was determined by ICP-AES using a Thermo Scientific iCAP-6500 instrument.
Results and discussion
Catalyst synthesis and characterization
A sol–gel technique enables molecular-scale control, intimate mixing of reactants and promotes Si–O–Ti bond formation in titania–silica mixed oxides.26 However, the reactivities of silicon and transition metal alkoxides in hydrolysis/condensation reactions differ significantly and special measures are required to avoid segregation of the active metal into a separate metal oxide phase. Different strategies have been applied to adjust the relative precursor reactivities, including pre-hydrolysis of TEOS, the use of a more reactive silicon precursor, viz. tetramethyl orthosilicate (TMOS) and/or modification of metal alkoxides with complexing agents, such as peroxides, alkoxyalcohols, aminoalcohols, β-diketones, β-ketoesters, carboxylic and phosphonic acids.26,27 Acetylacetone has been widely used as a hydrolysis-retarding agent in the sol–gel synthesis of amorphous titania–silica6,26,27 and the templated synthesis of ordered mesoporous titanium-silicates.22,28 On the other hand, acac is known to favour the formation of Ti dimers,27 the presence of which is crucial for the selective oxidation of bulky phenols to p-benzoquinones.10b,cKeeping all these facts in mind, we employed acac for the stabilization of the Ti precursor (see Experimental for details). The molar ratio of Ti/acac was chosen as 1:1 and 1:2 (samples A and B, respectively). When no acac was used in the synthesis, immediate precipitation of titanium dioxide occurred. Furthermore, the use of acac allowed us not only to avoid the TiO2 precipitation but also to generate a proper, di(oligo)meric state of Ti centers in the catalysts (vide infra).
The twenty-fold excess of the alcohol solvent (relative to the concentration of CTAB at which the formation of micelles normally starts) prevented the formation of any precipitate, and the synthetic solution remained transparent after mixing of all the reactants. Upon evaporation of the alcohol, the critical micelle concentration was achieved and self-assembly of a silica–Ti-surfactant mesophase occurred, resulting in the formation of transparent monoliths with a characteristic planar size of ca. 1 cm and height of 3–5 mm without visible internal defects (Fig. 1a). After calcination was used to remove the organic template, the monoliths cracked to yield a solid with particles of 0.1–2 mm (Fig. 1b). According to the EISA methodology, the composition of starting mixtures is directly reflected in the composition of the products, so that the introduction of titanium into the final material was nearly quantitative, as confirmed by the elemental analysis data (Table 1).
 | ||
| Fig. 1 Sample A after evaporation under ambient conditions (a) and after calcination (b). | ||
| Sample | Ti:acaca |
Ti,b wt% | S BET, m2 g−1 | V,c cm3 g−1 | D,d nm |
|---|---|---|---|---|---|
| a Molar ratio used in the synthesis. b Elemental analysis data for calcined samples. c Mesopore volume. d Mean pore diameter. e After 1 run of TMP oxidation with H2O2. f After 7 runs of TMP oxidation; the reaction conditions as in Table 2 (entry 3). | |||||
| A | 1:1 |
1.43 | 1288 | 0.70 | 3.1 |
| B | 1:2 |
1.36 | 1105 | 0.70 | 3.4 |
| A-1e | 1:1 |
1.43 | 1165 | 0.64 | 3.0 |
| A-7f | 1:1 |
1.43 | 1000 | 0.48 | 2.7 |
After calcination, the solids were ground to give a fine powder. SEM images of ground sample A are shown in Fig. 2 (top). One can observe relatively sharp particles with a size of 100–500 μm. The particles have internal cracks and rows of “bubbles”, which could be the reason for their further breaking, as confirmed by SEM images of sample A after the first catalytic run (Fig. 2, bottom) and after 7 reuses in TMP oxidation (Fig. S1 in ESI†). Indeed, the size of the particles reduced to 50–200 μm after the catalytic runs, and the distribution of particle sizes seems to be narrower relative to the starting material.
 | ||
| Fig. 2 SEM images of calcined sample A: (top) before catalysis and (bottom) after the 1st catalytic run (A-1). | ||
The small angle XRD pattern of sample A is shown in Fig. 3. The broad diffraction peak that can be indexed as (10), in a two-dimensional hexagonal lattice with d10 ≈ 4.5 nm and a corresponding lattice constant of a ≈ 5.2 nm, indicates a long-range structural order in an array of regularly arranged cylindrical mesopores with a uniform pore size. Treatments with a MeCN solution of H2O2 or boiling water produced no changes in the lattice constant. At the same time, strong broadening of the (10) reflection showed a high degree of structural imperfection. Therefore, the material prepared certainly represents a mesophase however it has a rather poor organization.
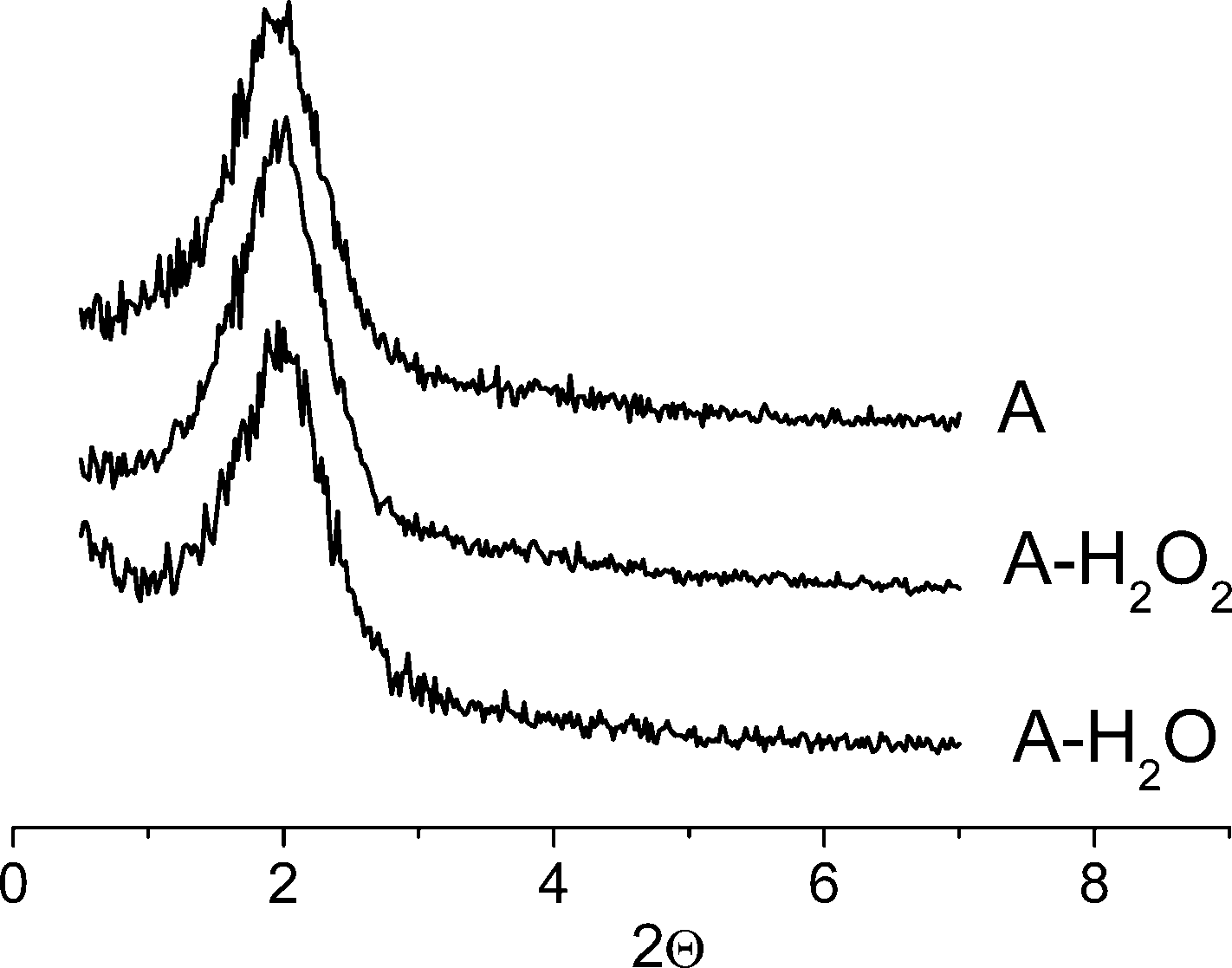 | ||
| Fig. 3 XRD patterns of calcined sample A: initial, after treatment with aqueous H2O2 in MeCN (25 °C, 1 h) and after treatment with boiled water for 6 h. | ||
The nitrogen adsorption–desorption isotherms for samples A and B are presented in Fig. 4. Textural properties of the materials (specific BET surface areas, mesopore volumes and average mesopore diameters) acquired from the adsorption data are given in Table 1. Importantly, the solids had no micropores. A rough estimation of the silica wall thickness as a difference between the lattice constant a and average mesopore diameter D gives a value of ca. 2 nm.
 | ||
| Fig. 4 Nitrogen adsorption–desorption isotherms for calcined samples A and B (filled and empty symbols correspond to adsorption and desorption, respectively). | ||
Variation in the amount of acac used during synthesis had two consequences for the final material. First, the increase of the acac/Ti molar ratio from 1:1 to 2:1 led to a slightly increased mesopore diameter (3.4 nm versus 3.1 nm). Secondly, the acac/Ti molar ratio affected the state of Ti centers.
It is well-known that the nature of Ti species in titanium-silicate catalysts has a strong impact on their catalytic performance.2,24 As was mentioned in the Introduction, well-dispersed dimeric (small oligomeric) Ti centers are required for the selective formation of p-benzoquinones in the oxidation of alkylated phenols.9,10c DR UV–Vis spectroscopy is a useful technique for characterization of the local geometry and coordination environment of the titanium ions in solids. DR UV–Vis spectra of samples A and B are shown in Fig. 5. Both samples reveal broad absorption bands centred at 235 (A) and 255 nm (B) although the Ti content was rather low (the formal surface density of Ti calculated based on the Ti content and surface area would be ca. 0.14 atom nm−2). Such broad absorptions centred at 235–260 nm are attributed to six-coordinated TiIV dimers and small oligomers.29 The long-wave shift observed for sample B relative to sample A indicates a higher degree of Ti oligomerization in the former sample. The absence of a characteristic absorption at 330 nm typical of anatase microcrystallites is consistent with the absence of the band at 145 cm−1 in the Raman spectra of the samples.
 | ||
| Fig. 5 DR UV–Vis spectra of calcined samples A, B and sample A after treatments with aqueous H2O2 in MeCN (25 °C, 1 h) and with boiled water for 6 h. | ||
Hydrothermal stability and stability toward H2O2
Hydrothermal stability is crucial for catalysts that are supposed to be applied for oxidations with aqueous H2O2. Earlier, some of us reported the synthesis of the hydrothermally stable mesostructured titanium-silicate Ti-MMM-2 that demonstrated a fairly good resistance towards boiling water and dilute solutions of H2O2 in MeCN.8 Ti-MMM-2 showed high activity in the H2O2-based oxidation of TMP, but the selectivity for TMBQ never exceeded 82% as this catalyst contains mostly isolated Ti atoms (DRS UV maximum at 208 nm). Interestingly, the use of acac in the synthesis of Ti-MMM-2 did not allow us to obtain samples with predominantly di(oligo)meric active Ti sites.The hydrothermal stability of sample A was examined by treatments with boiling water for 6 h and with 0.1 M solution of H2O2 in MeCN for 1 h at room temperature (the same tests were previously employed for Ti-MMM-2).8Fig. 3 shows XRD patterns of sample A after the treatments in comparison with the initial catalyst. Both the position and width of the peaks are very close, which confirms the good hydrothermal stability of the solids synthesized by the EISA technique. DR UV–Vis spectra of sample A after the treatments are given in Fig. 5. One can see that the state of Ti centers changed insignificantly after the mild treatment with H2O2 while the boiling procedure caused a long-wave shift of the absorption, thus indicating some further agglomeration of TiO2 oligomers.
Catalytic study
The catalytic performance of the titanium-silicates prepared by the EISA methodology has been explored in the oxidation of TMP with 30% hydrogen peroxide in MeCN. The main results are presented in Table 2. Under the reaction conditions that had been previously determined as optimal for the production of TMBQ over Ti,Si-catalysts,10b both sample A and sample B revealed 100% selectivity toward the target quinone (entries 1 and 2). The substrate conversion attained 95 and 93% for samples A and B, respectively, when a 1.75-fold excess of the oxidant was employed (the reaction stoichiometry for the oxidation of TMP with H2O2 to produce TMBQ is 1:2). The efficiency of the oxidant utilization was, therefore, 53–54%. The conversion of TMP could be increased up to 100% by increasing the amount of hydrogen peroxide (entry 3). In turn, the use of only a stoichiometric amount of H2O2 led to a significant decrease in both TMP conversion and TMBQ selectivity (entry 6). Attempts to reduce the amount of catalyst also resulted in decreasing substrate conversion, although the product selectivity still remained high (entries 7 and 8).
| Entry | Substrate | [H2O2], M | Sample | Substrate conversion, % | Product selectivity, % | TOF,b min−1 |
|---|---|---|---|---|---|---|
| a Reaction conditions: substrate, 0.1 M; catalyst, 0.006 mmol of Ti; CH3CN 1 mL; 80 °C; 20–40 min. b TOF = (moles of TMP consumed)/(moles of Ti × time), determined by GC from the initial rates of TMP consumption. c 0.003 mmol of Ti. d 0.0015 mmol of Ti. | ||||||
| 1 | TMP | 0.35 | A | 95 | 100 | 2.2 |
| 2 | TMP | 0.35 | B | 93 | 100 | 2.2 |
| 3 | TMP | 0.44 | A | 100 | 100 | 2.3 |
| 4 | DTBP | 0.44 | A | 50 | 100 | 0.5 |
| 5 | DMP | 0.44 | A | 90 | 100 | 0.9 |
| 6 | TMP | 0.2 | A | 48 | 75 | 1.5 |
| 7 | TMP | 0.44 | A | 82c | 98 | 2.3 |
| 8 | TMP | 0.44 | A | 65d | 95 | 2.4 |
The catalytic activity expressed by a TOF value was the same for samples A and B (2.2 min−1) and was a little higher than the TOF values previously found for the grafted Ti/SiO2 catalysts (1.4–1.8 min−1). Taking into account that the EISA samples had smaller or comparable mesopores (3.1–3.4 nm) than the grafted Ti catalysts (e.g., 3.3 nm for Ti/SiO2 Nippon-Kasei or 15.4 nm for Ti/SiO2 Davicat), we may suggest that no internal diffusion limitation occurs in the TMP oxidation over samples A and B.
To evaluate the scope of the catalytic material, two other representative phenols, DTBP and DMP, were oxidized with H2O2 over sample A under standard reaction conditions. The corresponding quinones, DTBQ and DMQ, formed with 100% selectivity (Table 2, entries 4 and 5, respectively). The lower conversion of DTBP as compared to TMP and DMP was most likely due to its steric bulk.
Catalyst recyclability
The recycling behaviour of sample A has been studied in seven consecutive operation cycles of the TMP oxidation (conditions of entry 3, Table 2; total TON = 120). As one can judge from Fig. 6, the excellent (99–100%) selectivity towards TMBQ remained unchanged while TMP conversion slightly decreased (from 100 to 90%). A small decrease in reaction rate (TOF) was observed, mostly in the first cycles, but starting from the forth cycle, this parameter has been stabilized. In fact, the decrease in TOF results in an increase in reaction time from 20 to 50 min, which is not crucial. More importantly, no drop in TMBQ selectivity occurs after the catalyst recycling. This is a clear advantage of the Ti,Si-catalysts prepared by the EISA method as compared to the grafted Ti/SiO2 catalysts, for which only the use of 70% H2O2 allowed the selectivity to be kept quasi constant while with 35% H2O2 the selectivity decreased to 85% by the third reuse.10c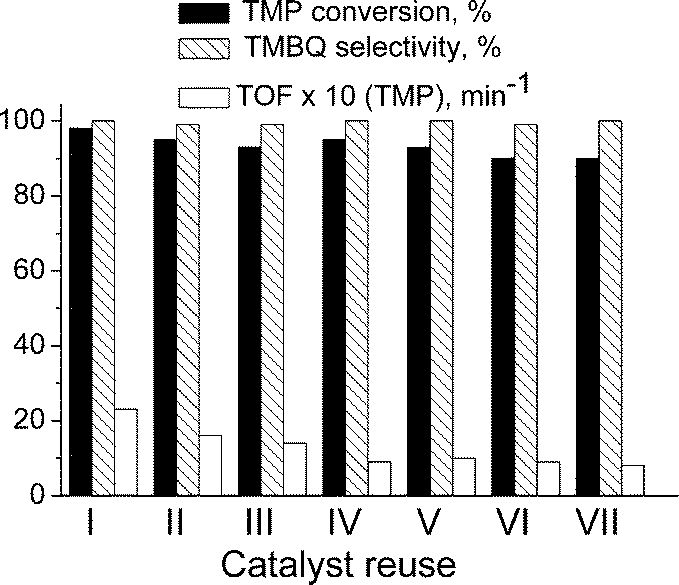 | ||
| Fig. 6 Catalyst recycling (sample A) in TMP oxidation with H2O2. Reaction conditions as in Table 2 (entry 3). | ||
Although there is no direct spectroscopic technique that would allow us to give an unambiguous answer as to whether the Ti di(oligo)meric species are truly incorporated within the silica network or not, the higher stability of the EISA catalysts toward deactivation with 30% H2O2 might suggest that the new procedure reported in this work favours insertion of the Ti species into the silica mesostructure. Relatively thick silica walls may be another factor responsible for the higher stability to deactivation.
Catalyst stability under reaction conditions
The hot catalyst filtration test revealed neither TMP conversion nor TMBQ formation in the filtrate after removal of the catalyst (Fig. 7). This proves the true heterogeneous nature of the catalysis in the catalytic system studied.30 In addition, the amount of titanium in the filtrate determined by ICP-AES was below 0.1 ppm. Accordingly, the content of titanium in the catalysts remained the same after seven reuses (see Table 1). | ||
| Fig. 7 Hot catalyst filtration test for TMP oxidation with H2O2 over sample A (filled and empty symbols correspond to substrate consumption and product yield, respectively). Reaction conditions as in Table 2 (entry 1). | ||
Meanwhile, after the catalyst recycling, a progressive decrease in the surface area and mesopore volume was detected (Table 1). After the first run, the average mesopore diameter remained practically unchanged but it reduced from 3.1 to 2.7 nm after seven catalytic runs (Table 1; the corresponding N2 adsorption isotherms and PSDs are given in Fig. S2 in ESI†). This decrease is most likely due to shrinkage of the material subjected to the turnover conditions and several calcinations during the recycling. However, we should note that the reduction of TOF values was more pronounced after the first use (Fig. 6); therefore, it can hardly be attributed to the deterioration of the textural properties. On the other hand, studies by DR UV–Vis spectroscopy showed that the state of Ti in sample A changed more drastically namely after the first catalyst use, while very little difference was observed between the fifth and seventh uses (Fig. 8). This agrees favourably with stabilization of the catalytic activity after the fourth cycle (Fig. 6).
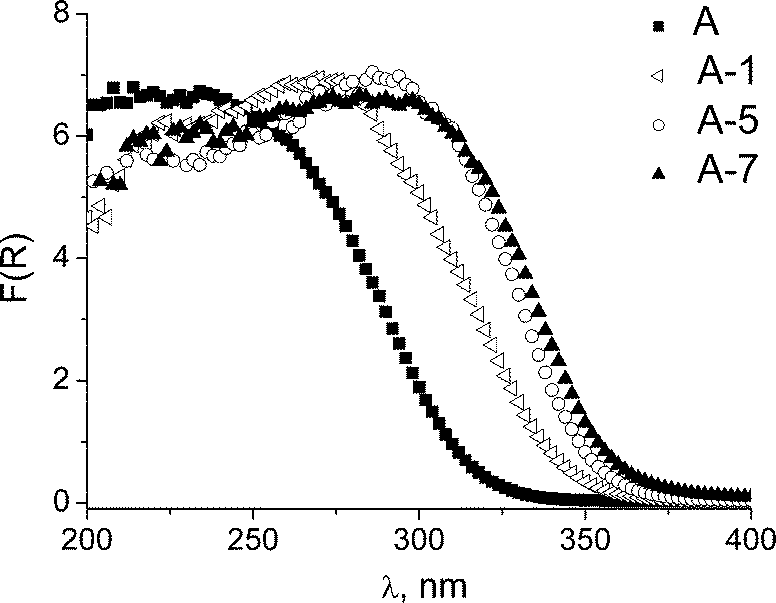 | ||
| Fig. 8 DR UV–Vis spectra of calcined sample A: initial (A) and after 1st (A-1), 5th (A-5) and 7th (A-7) runs of TMP oxidation with H2O2. Reaction conditions as in Table 2 (entry 3). | ||
The observed long-wave shift in the DR UV–Vis spectra is a manifestation of progressive oligomerization of titanium on the catalyst surface.10c,29 The growth of TiO2 oligomers initially caused some decrease in TOFs however, more importantly, it produced no negative effect on the reaction selectivity which remained close to 100% during seven reuses (Fig. 6). The Raman spectroscopic technique, which enables detection of early stages in the emergence of anatase microcrystallites by the presence of an intense band at 140–145 cm−1,24,31 identified just a very weak signal at ca. 150 cm−1 (Fig. S3 in ESI†). A rough estimation made from a comparison of the intensity of this peak with the corresponding band of anatase (Fig. S3,† inset) showed that, even after the seventh cycle, the amount of anatase in the catalyst was below 1% relative to the total Ti content. Earlier, it was demonstrated that the presence of TiO2 microcrystallites is detrimental for the selective formation of p-benzoquinones.32 Therefore, the lack of anatase and isolated Ti centres in the mesoporous titanium-silicates, prepared by the EISA method, ensures the high selectivity towards substituted p-benzoquinones.
Conclusions
In summary, an original and convenient procedure for the preparation of mesoporous titanium-silicate catalysts has been developed using the EISA approach. This procedure does not require either rigorously controlled reaction conditions or special handling, which makes it safe, cheap and sustainable. Coupled with the use of acac as a Ti modificator, the EISA methodology enables control of the Ti active center state to favour the formation of Ti dimers or small oligomers on the catalyst surface. The presence of such centers ensures highly selective formation of p-benzoquinones during the H2O2-based oxidation of alkylphenols. The materials prepared by the EISA technique behave as true heterogeneous catalysts, do not suffer from titanium leaching and can be easily recovered and reused without loss of their catalytic properties. Although some agglomeration of TiO2 clusters occurs on the surface due to the negative action of aqueous H2O2, this process is significantly slower for the EISA-based materials than for Ti,Si-catalysts prepared by other methods, e.g., sol–gel TiO2–SiO2 mixed oxides, grafted Ti/SiO2 or Ti-MCM-41 like mesostructures. Further studies are in progress to understand the reasons behind the higher stability of the catalysts prepared by the EISA methodology.Acknowledgements
The authors thank Dr. Y. A. Chesalov and A. N. Serkova for the Raman and SEM measurements, respectively. This work was partially supported by the Russian Foundation for Basic Research (grant no. 13-03-92699).Notes and references
- (a) P. Schudel, H. Mayer and O. Isler, in The Vitamins, ed. W. H. Sebrell and R. S. Harris, Academic, New York, 1972, vol. 5, pp. 168–317 Search PubMed; (b) W. Bonrath and T. Netscher, Appl. Catal., A, 2005, 280, 55 CrossRef CAS PubMed; (c) O. A. Kholdeeva, O. V. Zalomaeva, A. B. Sorokin, I. D. Ivanchikova, C. Della Pina and M. Rossi, Catal. Today, 2007, 121, 58 CrossRef CAS PubMed; (d) M. Eggersdorfer, D. Laudert, U. Letinois, T. McClymont, J. Medlock, T. Netscher and W. Bonrath, Angew. Chem., Int. Ed., 2012, 51, 12960 CrossRef CAS PubMed.
- (a) M. G. Clerici, in Fine Chemicals through Heterogeneous Catalysis, ed. R. A. Sheldon and H. van Bekkum, Wiley, Weinheim, 2001, pp. 538–551 Search PubMed; (b) U. Romano and M. Ricci, in Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications, ed. M. G. Clerici and O. A. Kholdeeva, Wiley, New Jersy, 2013, pp. 451–462 Search PubMed.
- (a) P. T. Tanev, M. Chibwe and T. J. Pinnavaia, Nature, 1994, 368, 321 CrossRef CAS PubMed; (b) W. Zhang, M. Fröba, J. Wang, P. T. Tanev, J. Wong and T. J. Pinnavaia, J. Am. Chem. Soc., 1996, 118, 9164 CrossRef CAS.
- N. N. Trukhan, V. N. Romannikov, E. A. Paukshtis, A. N. Shmakov and O. A. Kholdeeva, J. Catal., 2001, 202, 110 CrossRef CAS.
- A. Sorokin and A. Tuel, Catal. Today, 2000, 57, 45 CrossRef CAS.
- (a) O. A. Kholdeeva, N. N. Trukhan, M. P. Vanina, V. N. Romannikov, V. N. Parmon, J. Mrowiec-Bialon and A. B. Jarzebski, Catal. Today, 2002, 75, 203 CrossRef CAS; (b) J. Mrowiec-Białoń, A. B. Jarzębski, O. A. Kholdeeva, N. N. Trukhan, V. I. Zaikovskii, V. V. Kriventsov and Z. Olejniczak, Appl. Catal., A, 2004, 273, 47 CrossRef PubMed.
- O. A. Kholdeeva, O. V. Zalomaeva, A. N. Shmakov, M. S. Melgunov and A. B. Sorokin, J. Catal., 2005, 236, 62 CrossRef CAS PubMed.
- O. A. Kholdeeva, M. S. Mel'gunov, A. N. Shmakov, N. N. Trukhan, V. V. Kriventsov, V. I. Zaikovskii, M. E. Malyshev and V. N. Romannikov, Catal. Today, 2004, 91–92, 205 CrossRef CAS PubMed.
- A. Tuel and L. G. Hubert-Pfalzgraf, J. Catal., 2003, 217, 343 CAS.
- (a) O. A. Kholdeeva, I. D. Ivanchikova, M. Guidotti and N. Ravasio, Green Chem., 2007, 9, 731 RSC; (b) O. A. Kholdeeva, I. D. Ivanchikova, M. Guidotti, N. Ravasio, M. Sgobba and M. V. Barmatova, Catal. Today, 2009, 141, 330 CrossRef CAS PubMed; (c) O. A. Kholdeeva, I. D. Ivanchikova, M. Guidotti, C. Pirovano, N. Ravasio, M. V. Barmatova and Y. A. Chesalov, Adv. Synth. Catal., 2009, 351, 1877 CrossRef CAS; (d) C. Pirovano, M. Guidotti, V. Dal Santo, R. Psaro, O. A. Kholdeeva and I. D. Ivanchikova, Catal. Today, 2012, 197, 170 CrossRef CAS PubMed.
- (a) M. Ogawa, J. Am. Chem. Soc., 1994, 116, 7941 CrossRef CAS; (b) M. Ogawa, Chem. Commun., 1996, 1149 RSC.
- (a) Y. Lu, R. Ganguli, C. A. Drewien, M. T. Anderson, C. J. Brinker, W. Gong, Y. X. Guo, H. Soyez, B. Dunn, M. H. Huang and J. I. Zink, Nature, 1997, 389, 364 CrossRef CAS PubMed; (b) K. Yu, A. J. Hurd, A. Eisenberg and C. J. Brinker, Langmuir, 2001, 17, 7961 CrossRef CAS.
- P. J. Bruinsma, A. Y. Kim, J. Liu and S. Baskaran, Chem. Mater., 1997, 9, 2507 CrossRef CAS.
- H. Yang, N. Coombs and G. A. Ozin, J. Mater. Chem., 1998, 8, 1205 RSC.
- M. Ogawa and N. Masukawa, Microporous Mesoporous Mater., 2000, 38, 35 CrossRef CAS.
- D. Zhao, P. Yang, B. F. Chmelka and G. D. Stucky, Chem. Mater., 1999, 11, 1174 CrossRef CAS.
- S. Besson, T. Gacoin, C. Jacquiod, C. Ricolleau, D. Babonneau and J.-P. Boilot, J. Mater. Chem., 2000, 10, 1331 RSC.
- P. Innocenzi, L. Malfatti, T. Kidchob and P. Falcaro, Chem. Mater., 2009, 21, 2555 CrossRef CAS.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579 CrossRef CAS.
- (a) P. Yang, D. Zhao, D. I. Margolese, B. F. Schmelka and G. D. Stucky, Nature, 1998, 396, 152 CrossRef CAS PubMed; (b) P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Chem. Mater., 1999, 11, 2813 CrossRef CAS; (c) L. Chen, B. Yao, Y. Cao and K. Fan, J. Phys. Chem. C, 2007, 111, 11849 CrossRef CAS.
- M. Ogawa, K. Ikeue and M. Anpo, Chem. Mater., 2001, 13, 2900 CrossRef CAS.
- Z.-L. Hua, J.-L. Shi, W.-H. Zhang and W.-M. Huang, Mater. Lett., 2002, 53, 299 CrossRef CAS.
- N. Hüsing, B. Launay, D. Doshi and G. Kickelbick, Chem. Mater., 2002, 14, 2429 CrossRef.
- (a) B. Notari, Adv. Catal., 1996, 41, 253 CrossRef CAS; (b) P. Ratnasamy, D. Srinivas and H. Knozinger, Adv. Catal., 2004, 48, 1 CrossRef CAS.
- V. B. Fenelonov, V. N. Romannikov and A. Y. Derevyankin, Microporous Mesoporous Mater., 1999, 28, 57 CrossRef CAS.
- (a) M. V. Landau, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2008, pp. 119–160 Search PubMed; (b) R. Hutter, T. Mallat, D. Dutoit and A. Baiker, Top. Catal., 1996, 3, 421 CrossRef CAS; (c) R. J. Davis and Z. Liu, Chem. Mater., 1997, 9, 2311 CrossRef CAS; (d) M. Schneider and A. Baiker, Catal. Today, 1997, 35, 339 CrossRef CAS.
- U. Schubert, J. Mater. Chem., 2005, 15, 3701 RSC.
- (a) S. A. Bagshaw, F. D. Renzo and F. Fajula, Chem. Commun., 1996, 2209 RSC; (b) W.-H. Zhang, J. Lu, B. Han, M. Li, J. Xiu, P. Ying and C. Li, Chem. Mater., 2002, 14, 3413 CrossRef CAS.
- (a) L. Marchese, T. Maschmeyer, E. Gianotti, S. Coluccia and J. M. Thomas, J. Phys. Chem. B, 1997, 101, 8836 CrossRef CAS; (b) A. Carati, C. Flego, E. Previde Massara, R. Millini, L. Carluccio, W. O. Parker Jr. and G. Bellussi, Microporous Mesoporous Mater., 1999, 30, 137 CrossRef CAS; (c) E. Gianotti, A. Frache, S. Coluccia, J. M. Thomas, T. Maschmeyer and L. Marchese, J. Mol. Catal. A: Chem., 2003, 204–205, 483 CrossRef CAS.
- R. A. Sheldon, M. Wallau, I. W. C. E. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485 CrossRef CAS.
- (a) X. Gao and I. E. Wachs, Catal. Today, 1999, 51, 233 CrossRef CAS; (b) A. Corma, Chem. Rev., 1997, 97, 2373 CrossRef CAS PubMed.
- N. N. Trukhan, V. N. Romannikov, A. N. Shmakov, M. P. Vanina, E. A. Paukshtis, V. I. Bukhtiyarov, V. V. Kriventsov, I. Y. Danilov and O. A. Kholdeeva, Microporous Mesoporous Mater., 2003, 59, 73 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM, N2 adsorption and Raman data for the reused catalyst. See DOI: 10.1039/c3cy00615h |
| This journal is © The Royal Society of Chemistry 2014 |