Manganese complexes with non-porphyrin N4 ligands as recyclable catalyst for the asymmetric epoxidation of olefins†
Nabin Ch.
Maity
a,
Prasanta
Kumar Bera
a,
Debashis
Ghosh
ab,
Sayed H. R.
Abdi
*ab,
Rukhsana I.
Kureshy
ab,
Noor-ul H.
Khan
ab,
Hari C.
Bajaj
ab and
E.
Suresh
bc
aDiscipline of Inorganic Materials and Catalysis, Central Salt and Marine Chemicals Research Institute (CSMCRI), Council of Scientific & Industrial Research (CSIR), Bhavnagar 364 0002, Gujarat, India
bAcademy of Scientific and Innovative Research (AcSIR), Central Salt and Marine Chemicals Research Institute (CSMCRI), Council of Scientific & Industrial Research (CSIR), Bhavnagar 364 0002, Gujarat, India. E-mail: shrabdi@csmcri.org; Fax: +91 0278 2566970; Tel: +91 278 2567760
cAnalytical Discipline and Centralized Instrument Facility, Central Salt and Marine Chemicals Research Institute (CSMCRI), Council of Scientific & Industrial Research (CSIR), Bhavnagar 364 0002, Gujarat, India
First published on 4th October 2013
Abstract
New chiral manganese complexes of N4 ligands derived from 2-acetylpyridine were prepared and used as catalysts in the enantioselective epoxidation of olefins, using H2O2 as an oxidant to give epoxides, with excellent conversions (up to 99%) and enantiomeric excess (up to 88%) within 1 h at 0 °C. A detailed mechanistic study was undertaken based on the information obtained by single crystal X-ray, optical rotation, UV-Vis, CD spectra and kinetic studies, and revealed that the reaction is first order with respect to the concentration of catalyst and oxidant and independent of substrate concentration. The complex (0.1 mol%) was successfully subjected to recyclability experiments over 3 cycles in the epoxidation of styrene with H2O2 as an oxidant and acetic acid as an additive at 0 °C with retention of performance.
Introduction
Catalytic asymmetric epoxidation is an important reaction1,2 as the resulting enantiomerically pure epoxides are highly useful intermediates and building blocks.1–8 A host of work has been carried out on catalyst design with prime focus on achieving the targeted epoxide with high enantioselectivity and yield.1 The use of Mn–salen catalysts,9–12 Shi's ketone based system13,14 and porphyrin-inspired catalysts15,16 are excellent methods to epoxidize a wide range of olefins with high enantioselectivity (>99% enantiomeric excess (ee)), yet substantial room for improvement remains in terms of catalyst loading, enantioselectivity and use of ecofriendly oxidants with high atom efficiency.1 While numerous variants of salen ligands have been reported, only a handful of bio-inspired non-heme N4 ligands3,4,7,8,17–25 with Mn have been studied. Nevertheless, these N4 ligands have shown very interesting results in asymmetric epoxidation of olefins with ee up to 96% with selected substrates, but mostly at temperatures of −20 °C or below. Moreover, the synthetic protocols of these ligands are tedious, multi-step and often require expensive chiral starting materials. With this backdrop, we have come up with a simple methodology to synthesize chiral N4 ligands with readily available chiral cyclohexanediamine and 2-acetylpyridine as starting materials in three convenient steps (Scheme 1). We then used their manganese complexes in the asymmetric epoxidation of olefins with low catalyst loading (0.1 mol%) to get the product epoxide in ee 44–88% with a high turnover number (1000, which reached up to ~2500 after 3 catalytic cycles) in a short reaction time (45–60 min) for the epoxidation of olefins, α,β-unsaturated ketones26 and chromenes, with H2O2 as an oxidant and acetic acid as an additive at 0 °C.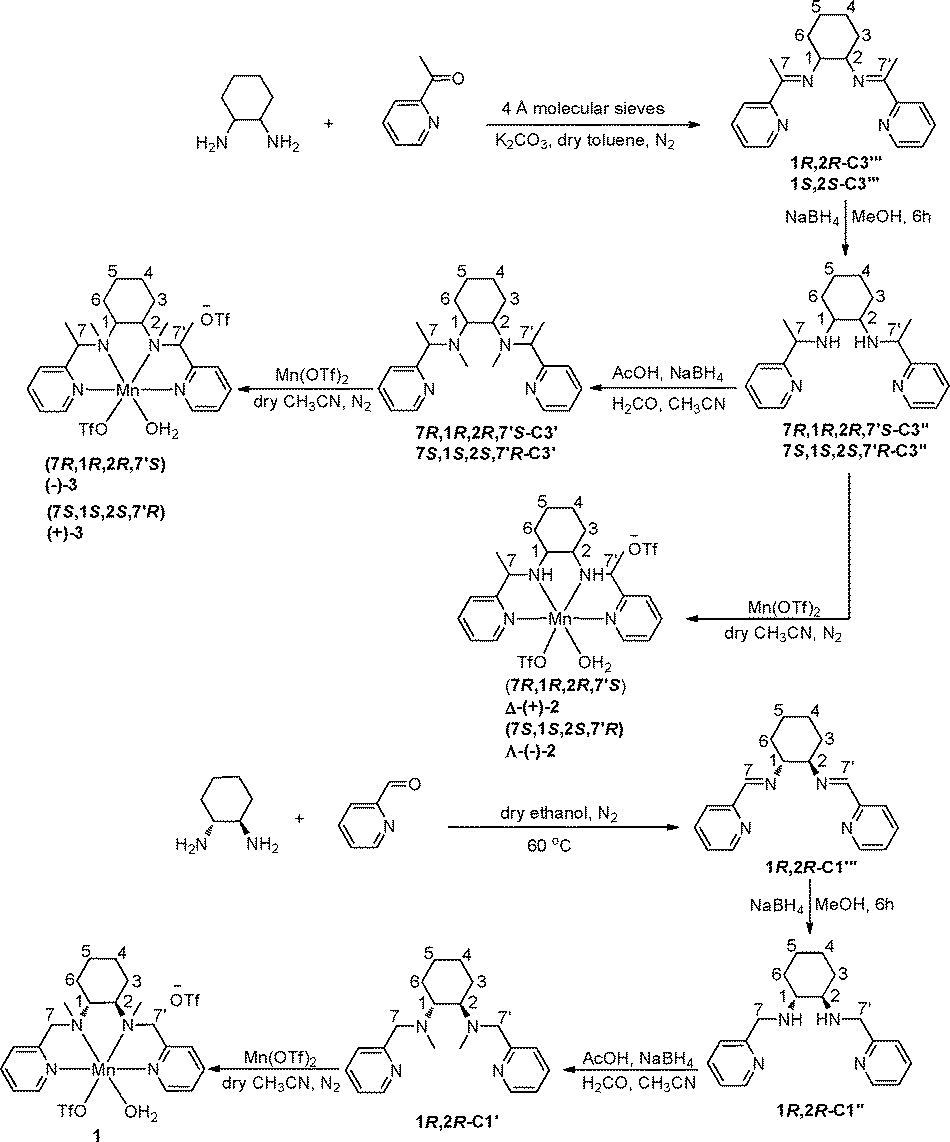 | ||
| Scheme 1 Scheme for synthesis of the complexes. | ||
Results and discussion
The protocol used in the present study to synthesize bio-inspired novel N4 ligands is very simple and requires no chromatographic purification. The starting materials, 2-acetylpyridine, pyridine 2-carboxaldehyde and optically pure 1,2-diaminocyclohexane, are inexpensive and readily available. These tetradentate ligands display C2-symmetry with two pyridine and two sp3 nitrogen donor atoms. The corresponding manganese complexes 1, 2 and 3 from these ligands, respectively, were readily obtained by the reaction of equimolar quantities of ligand and Mn(OTf)2 in CH3CN under argon atmosphere (Fig. 1). Generally this type of N4 ligand co-ordinates with manganese in a cis-α-topology,27 which is evident from the single crystal structures28 of the complexes Δ-(+)-2 and Λ-(−)-2 (Fig. 2) grown by diffusion of dry ether into saturated CH3CN solutions of the respective complexes. The complexes Δ-(+)-2 and Λ-(−)-2 are non-superimposable mirror images (Fig. S1, see in ESI†) of each other with chirality at the metal center. The complexes Δ-(+)-2 and Λ-(−)-2 were crystalline and in pure phase, as evident from their optical rotation and CD spectra (Fig. 3 and 4). It is interesting to note that during hydrogenation of the imine bond of the ligand precursor by NaBH4, the configuration of positions 7C and 7′C is guided by the configuration of the 1,2-cyclohexanediamine collar. Although these complexes were not so promising in epoxidation of model olefin (ee, 50%), their structures gave important structural information (by analogy) for the active catalysts (−)-3 and (+)-3 (Fig. 5). Even after repeated attempts we were not able to get single crystals of the active catalysts (−)-3 and (+)-3.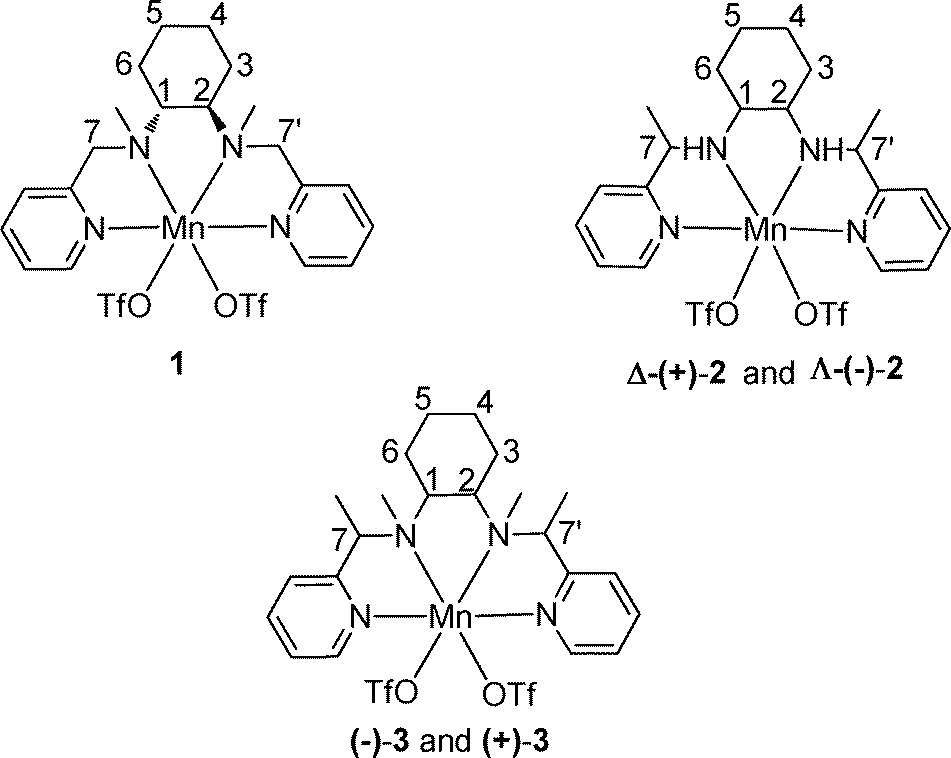 | ||
| Fig. 1 Schematic representation of the complexes used in this study. | ||
 | ||
| Fig. 2 Mercury diagram depicting the cationic catalysts Δ-(+)-2 and Λ-(−)-2 with an atom numbering scheme (hydrogen atoms are omitted for clarity). | ||
 | ||
| Fig. 3 CD spectra with 1.1 × 10−6 M solutions of complexes Λ-(−)-2 and Δ-(+)-2 in CH3CN. | ||
 | ||
| Fig. 4 CD spectra with 1.4 × 10−6 M solutions of complexes (+)-3 and (−)-3 in CH3CN. | ||
 | ||
| Fig. 5 Stereochemistry of the metal complexes. | ||
Catalysts 1–3 (0.1 mol%; this catalyst loading was found to be optimum based on experiments with catalyst loadings ranging from 0.5–0.025 mol%; Table S1, see in ESI†) were screened for their activity in the epoxidation of chalcone and styrene as model olefins using hydrogen peroxide–acetic acid as an active oxidant25 in acetonitrile at 0 °C. Individually, if we compare catalysts 1 and 2, the N-methylated catalyst 1 fared better than 2. In fact, for a reason not known at this point in time, in the case of catalyst 2 (NH-group containing) the epoxidation reaction failed to proceed beyond 40–66% conversion (Table 1, entries 1–4).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Substrate | Time [min] | Conversionb [%] | ee [%] | Config. |
| a Reaction conditions: catalyst (0.1 mol%, 1 mL CH3CN), substrate (1.4 mmol), 1.2 equiv. of 50% H2O2, AcOH (3 equiv.) at 0 °C. b Determined by 1H NMR. c Determined by HPLC using a Daicel IC column. d Determined on a chiral capillary column of GTA-type. | ||||||
| 1 | 1 | Chalcone | 60 | 92 | 78c | 2S,3R |
| 2 | 1 | Styrene | 45 | 80 | 27d | R |
| 3 | Δ-(+)-2 | Chalcone | 60 | 40 | 50c | 2S,3R |
| 4 | Δ-(+)-2 | Styrene | 45 | 66 | 13d | R |
| 5 | (−)-3 | Chalcone | 60 | 95 | 86c | 2S,3R |
| 6 | (−)-3 | Styrene | 45 | >99 | 43d | R |
| 7 | (+)-3 | Chalcone | 60 | >93 | 84c | 2R,3S |
| 8 | Nil | Styrene | 60 | 3 | — | — |
| 9 | Nil | Chalcone | 60 | 2 | — | — |
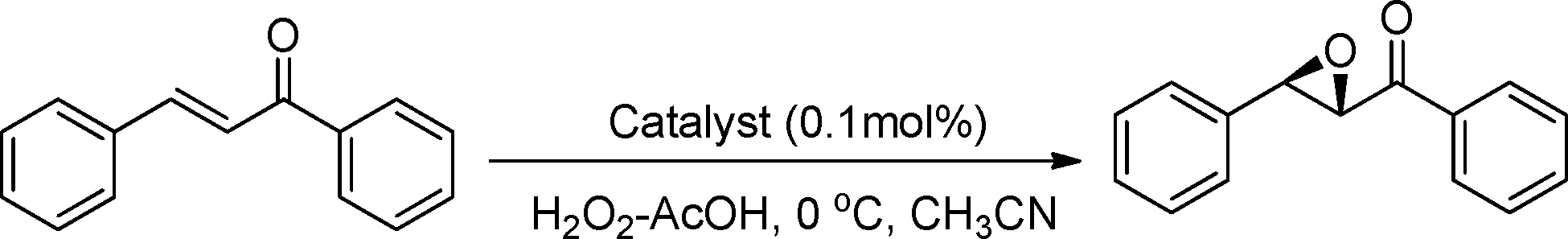
However, when we have a methyl group at both the N and the 7C and 7′C positions, the performance of the catalyst improves dramatically, particularly the enantioselectivity (entries 1, 2 and 5, 6), possibly due to the improved electronic and steric features, which restrict the number of possible transition states at the oxygen atom transfer stage from the oxo intermediate to the olefin. It is to be noted that in the absence of the catalyst, keeping other reaction parameters constant, only traces of the epoxide product were detected after 60 min (entries 8, 9). This clearly shows the involvement of catalyst in the epoxidation reaction, with negligible or very little background reaction. Change of the chiral diamine collar from (1R,2R) to (1S,2S) only changes the configuration of the product and as such, the chiral center developed at the 7,7′ positions during the course of the reaction does not have that much of a role in the product stereochemistry, which was also found out by Sun et al.3 But the steric crowding generated by the methyl group at the 7,7′ positions greatly improves both the enantioselectivity, as well as the yield of the epoxide product.
The above screening of the catalysts clearly indicates that the catalyst (−)-3 performed better than the rest of the complexes synthesized in the present study, hence this catalyst was subjected to reaction parameter optimization for further improvement in the product yield and ee. Since the oxidant used in the present study is a combination of H2O2 (50% aqueous) and acetic acid, it was prudent to find the optimum amount of acetic acid (AcOH) required for higher product yield and ee. This is also important because higher acid concentration often triggers an epoxide ring opening reaction, particularly for acid sensitive epoxides. Accordingly, we varied the equivalents of AcOH from 1 to 5, while maintaining the amount of H2O2 as 1.2 equivalents with respect to the substrate throughout (Table S1,† entries 5, 6 and 7). When this was done, a H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOH ratio of 1.2:3 was found to be best. In the absence of AcOH, very low conversion (10%) and enantioselectivity (ee 20%) were found (entry 8). Stack et al. have deliberated on a pH window of 2–4 at which Mn complexes with similar ligand systems worked well,29 and the use of strong acid caused decomposition of the catalyst itself. We attempted the use of dilute trifluoroacetic acid (2.0 × 10−7 M trifluoroacetic acid in CH3CN, pH ~3.3) and benzoic acid (0.087 M benzoic acid in CH3CN, pH ~3.4) in place of acetic acid, but the results were not promising both in terms of conversion (4 and 33%) and ee (8 and 71%) of the product epoxide (entries 9, 10). So the role of acetic acid may not only be to maintain pH, but its nucleophilicity may also play an important role as it coordinates to the Mn center to form a catalytically active intermediate in the catalytic cycle (Scheme 2). Further, 30% aqueous H2O2 was found to be less active, due to the low concentration of peroxide compared to 50% aqueous H2O2 (Table 2, entries 1 and 2). Other oxidants including m-CPBA, cumine hydroperoxide (CHP, technical grade, 80%), tert-butyl hydroperoxide (TBHP, 70 wt% in H2O) and dry urea–H2O2 were also evaluated for their efficacy in the epoxidation of chalcone as a model compound, with the above optimized reaction conditions. Incidentally, dry urea–H2O2 fared very poorly both in terms of epoxide conversion (30%) and ee (29%), whereas the other oxidants gave the product in moderate to good conversion (40–84%) and ee (57–75%) (Table 2, entries 3–6).
:AcOH ratio of 1.2:3 was found to be best. In the absence of AcOH, very low conversion (10%) and enantioselectivity (ee 20%) were found (entry 8). Stack et al. have deliberated on a pH window of 2–4 at which Mn complexes with similar ligand systems worked well,29 and the use of strong acid caused decomposition of the catalyst itself. We attempted the use of dilute trifluoroacetic acid (2.0 × 10−7 M trifluoroacetic acid in CH3CN, pH ~3.3) and benzoic acid (0.087 M benzoic acid in CH3CN, pH ~3.4) in place of acetic acid, but the results were not promising both in terms of conversion (4 and 33%) and ee (8 and 71%) of the product epoxide (entries 9, 10). So the role of acetic acid may not only be to maintain pH, but its nucleophilicity may also play an important role as it coordinates to the Mn center to form a catalytically active intermediate in the catalytic cycle (Scheme 2). Further, 30% aqueous H2O2 was found to be less active, due to the low concentration of peroxide compared to 50% aqueous H2O2 (Table 2, entries 1 and 2). Other oxidants including m-CPBA, cumine hydroperoxide (CHP, technical grade, 80%), tert-butyl hydroperoxide (TBHP, 70 wt% in H2O) and dry urea–H2O2 were also evaluated for their efficacy in the epoxidation of chalcone as a model compound, with the above optimized reaction conditions. Incidentally, dry urea–H2O2 fared very poorly both in terms of epoxide conversion (30%) and ee (29%), whereas the other oxidants gave the product in moderate to good conversion (40–84%) and ee (57–75%) (Table 2, entries 3–6).
 | ||
| Scheme 2 Proposed mechanism. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidant | Time [min] | Conversionb [%] | eec [%] | Config. |
| a Reaction conditions: catalyst (0.1 mol%, 1 mL CH3CN), chalcone (1.4 mmol), oxidant (1.2 equiv). b Determined by 1H NMR. c Determined by HPLC using a Daicel IC column. | |||||
| 1 | 50% H2O2–AcOH | 60 | 94 | 86 | 2R,3S |
| 2 | 30% H2O2–AcOH | 60 | 84 | 81 | 2R,3S |
| 3 | m-CPBA | 60 | 84 | 75 | 2R,3S |
| 4 | CHP | 60 | 50 | 57 | 2R,3S |
| 5 | TBHP | 60 | 40 | 65 | 2R,3S |
| 6 | Urea–H2O2 | 60 | 30 | 29 | 2R,3S |
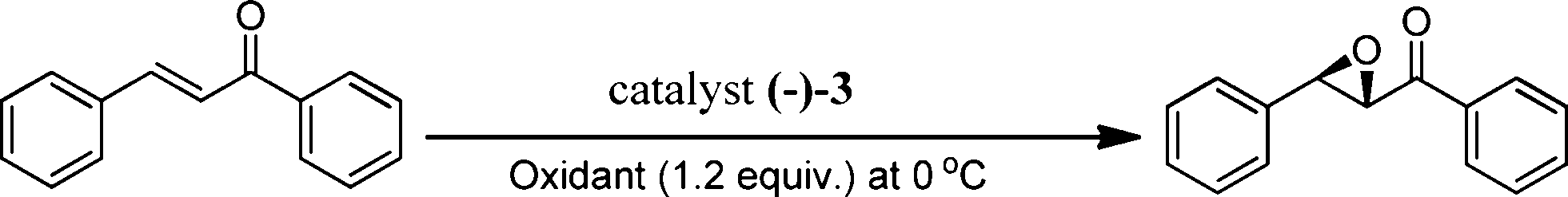
Further, temperature is a very important aspect in asymmetric epoxidation; therefore, the epoxidation of chalcone was carried out with the preferred catalyst, (−)-3 (0.1 mol%), using the above optimized conditions over a range of temperatures varied from 10 °C to −10 °C (Table S2; entries 1–4, see in ESI†). As such, the reaction is very fast at 10 °C (conversion >99%) in 60 min, but the ee (76%) (entry 1) was lower than that obtained at 0 °C (entry 2; ee 86%). Reducing the reaction temperature further caused a reduction in both the conversion and ee (entries 3 and 4) due to the solubility issue of the substrates at lower temperatures.
Therefore, next we screened the effect of solvent variation on the efficiency of the epoxidation protocol using the above optimized conditions, with chalcone as a model substrate (Table 3). Catalyst (−)-3 was found to be insoluble in dichloromethane (DCM) and toluene (both water immiscible), which adversely affected conversion (20–30%) and product ee (15–40%) (Table 3; entries 1 and 2). The reaction in green solvents like dimethyl carbonate (DMC) and propylene carbonate, however, gave moderate conversions (80–84%) and ee (70–75%), as in these solvents the catalyst solubility is fairly good and their affinity for water is also better than solvents like DCM and toluene. Using a mixed solvent system (e.g., DMC and CH3CN, 1:1) was not of much help as we got only 70% conversion and 69% ee (entry 5). Therefore, CH3CN was taken as the preferred solvent for the present epoxidation protocol (entry 6). Notwithstanding the suitability of CH3CN as the solvent, the amount of CH3CN is also very critical. On increasing the solvent amount (entry 7) the reaction slowed down (conversion 84%) with some loss in ee (78%) of the product. On the other hand decreasing the amount of solvent led to an increase in reaction rate (conversion 99%) but with a significant decrease in ee (70%), possibly due to background reactions (entry 8).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Time [min] | Conversionb [%] | eec [%] | Config. |
| a Reaction conditions: catalyst (0.1 mol%, 1 mL solvent), chalcone (1.4 mmol), 50% H2O2 (1.2 equiv.), AcOH (3 equiv.). b Determined by 1H NMR. c Determined by HPLC using a Daicel IC column. d Reaction conditions: catalyst (0.1 mol%, 3 mL solvent), chalcone (1.1 mmol), 50% H2O2 (1.2 equiv.), AcOH (3 equiv.). e Reaction conditions: catalyst (0.1 mol%, 0.5 mL solvent), chalcone (1.1 mmol), 50% H2O2 (1.2 equiv.), AcOH (3 equiv.) | |||||
| 1 | Toluene | 60 | 20 | 15 | 2R,3S |
| 2 | DCM | 60 | 30 | 40 | 2R,3S |
| 3 | Dimethyl carbonate | 60 | 80 | 75 | 2R,3S |
| 4 | Propylene carbonate | 60 | 84 | 70 | 2R,3S |
| 5 | DMC:CH3CN |
60 | 70 | 69 | 2R,3S |
| 6 | CH3CN | 60 | 96 | 86 | 2R,3S |
| 7 | CH3CNd | 60 | 84 | 78 | 2R,3S |
| 8 | CH3CNe | 60 | 99 | 70 | 2R,3S |
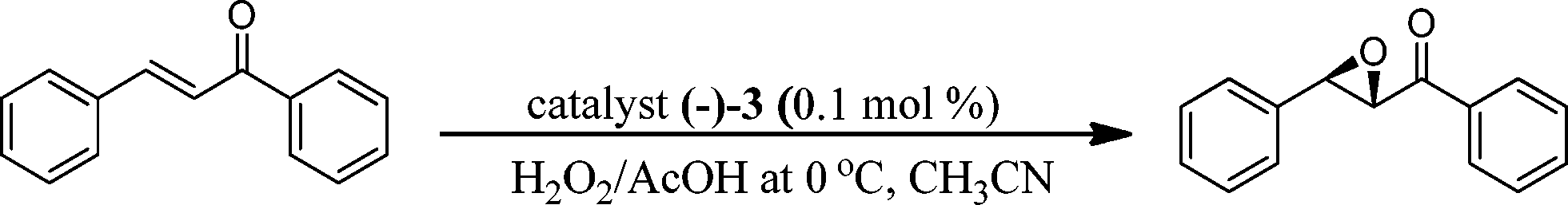
Catalyst (−)-3 (0.1 mol%) in the above optimized conditions was subsequently explored for its applicability in the epoxidation of a variety of olefins with 1.2 equiv. of H2O2 and 3 equiv. of AcOH as the oxidant mixture at 0 °C within ~60 min (Table 4). The present catalyst loading (0.1 mol%) is one of the lowest for Mn–N4 ligand systems to show high conversions (80–97%) of product, but with moderate ee (84–88%), particularly for substrates like chalcone, chromenes and indene under mild reaction parameters (entries 1–5). During the epoxidation of cis-β-methyl styrene, the cis product was preferentially formed (E:Z = 8:92) with no side products formed, however the formation of a small amount of trans product suggests that the oxygen atom transfer to the olefin is not entirely through ionic mechanisms (entry 6).30 In the case of the substrates styrene and 1-hexene (entries 7 and 8), though conversions were excellent (>99% for styrene) the enantioselectivity was moderate (~44%). Notwithstanding the moderate enantioselectivities, the synthetic protocol for the present N4 ligand system is very simple, forms epoxide products at low catalyst loadings with excellent conversions and has scope for further variation to improve enantioselectivity.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Time [min] | Conversion [%] | ee [%] | Config. | TOF × s−1g |
|
a Reaction conditions: catalyst (0.1 mol%, 1 mL CH3CN), substrate (1.4 mmol), 50% H2O2 (1.2 equiv.), AcOH (3 equiv.).
b Conversion determined by NMR.
c Determine by GC.
d Determined by HPLC using a Daicel IC, OD, OB or OD-H column.
e Determined on a chiral capillary column of GTA-type.
f Ratio of E:Z is 8:92.
g Turn over frequency (TOF) is calculated by the expression [product]/[catalyst] × time (s−1).
|
||||||
| 1 |
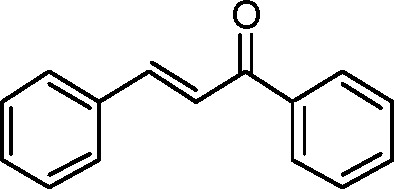
|
60 | 95b | 86d | 2R,3S | 0.26 |
| 2 |
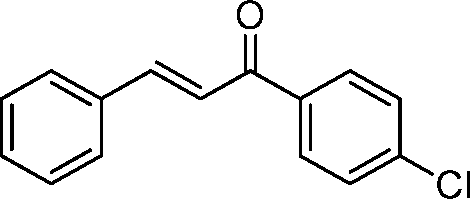
|
60 | 93b | 88d | 2R,3S | 0.26 |
| 3 |

|
60 | 90c | 86d | 3R,4R | 0.25 |
| 4 |
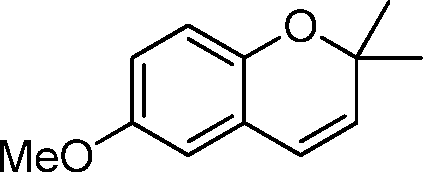
|
60 | 80c | 86d | 3R,4R | 0.24 |
| 5 |

|
45 | 97c | 84d | 1S,2R | 0.35 |
| 6 |

|
45 | >99c | 71ef | 1S,2R | 0.37 |
| 7 |

|
45 | 80c | 44e | R | 0.30 |
| 8 |
|
45 | >99c | 43e | R | 0.37 |

Kinetic study
To understand the mechanism of the epoxidation reaction in CH3CN medium, the kinetics of the epoxidation of styrene as a representative substrate were investigated using catalyst (−)-3 in the presence of 1.2 equiv. of 50% aqueous H2O2 and 3 equiv. of AcOH, as a function of the concentrations of the catalyst, styrene and oxidant, at −10 °C (Table 5). In all the kinetic runs, the plot for the formation of styrene epoxide with time was linear in the initial stage of the reaction, and attained saturation near completion (Fig. 6). Based on these results, the initial rate constants (kobs) (for the linear portion of the graph) were determined by estimating the amount of epoxide formed with time.| [Catalyst] × 104 M | [Substrate] × 10 M | [Oxidant] × 10 M | k obs × 104 M min−1 |
|---|---|---|---|
| Effect of substrate concentration | |||
| 3.625 | 3.08 | 4.35 | 5.1 |
| 3.5 | 5.04 | ||
| 3.92 | 4.98 | ||
| Effect of oxidant concentration | |||
| 3.625 | 3.625 | 2.18 | 2.3 |
| 3.62 | 4.0 | ||
| 4.71 | 5.2 | ||
| 5.80 | 6.2 | ||
| Effect of catalyst concentration | |||
| 1.5 | 3.625 | 4.35 | 2.1 |
| 2.17 | 2.9 | ||
| 3.7 | 5.1 | ||
| 5.08 | 6.9 | ||
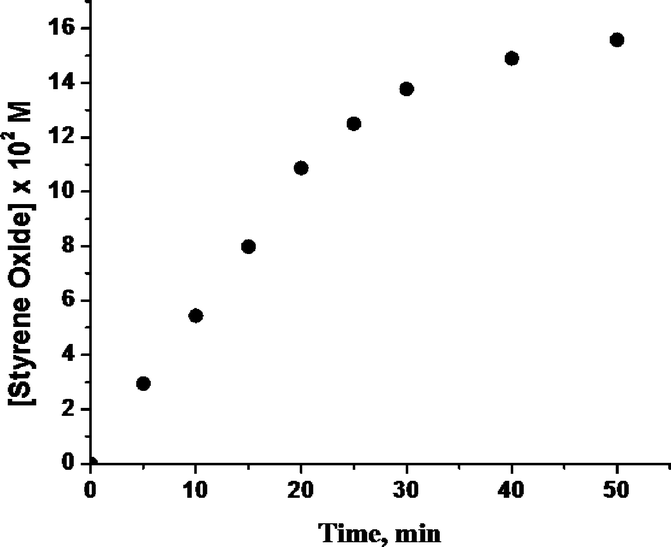 | ||
| Fig. 6 Time dependent plot of the formation of styrene oxide at −10 °C with catalyst (−)-3 in 1 mL CH3CN; [styrene] = 0.3625 M, [catalyst] = 3.625 × 10−4 M, [oxidant] = 0.435 M. | ||
The epoxidation of styrene with various concentrations of catalyst (−)-3 with a constant concentration of styrene and 1.2 equiv. of H2O2 and 3 equiv. of AcOH at −10 °C gave a straight line for the plot of rate constant (kobs) versus catalyst concentration with unit slope (d log kobs/d log[catalyst] = ~1, Fig. 7). This suggests that the epoxidation of styrene is first order with respect to the concentration of the catalyst.
 | ||
| Fig. 7 Plot of kobsversus [catalyst (−)-3] using styrene as the substrate at −10 °C; [oxidant] = 4.35 × 10−1 M, [substrate] = 3.625 × 10−1 M, 1 mL CH3CN. | ||
The epoxidation of styrene with various concentrations of H2O2 with constant concentrations of styrene and catalyst at −10 °C gave a straight line for the plot of (kobs) versus [H2O2] with unit slope (d log kobs/d log[H2O2] = ~1, Fig. 8), suggesting that the reaction is first order with respect to the oxidant concentration.
 | ||
| Fig. 8 Plot of kobsversus [oxidant] with catalyst (−)-3 using styrene as the substrate at −10 °C; [catalyst] = 3.625 × 10−4 M, [substrate] = 3.625 × 10−1 M, 1 mL CH3CN. | ||
Substrate concentration was varied at a constant concentration of catalyst (−)-3 and 1.2 equiv. of H2O2 at −10 °C. Zero-order dependence was observed for the initial concentration of styrene (74.00–110 × 10−2 M). In a nutshell, the rate of the reaction is dependent on the concentration of the catalyst and the oxidant; and was second order overall.
UV-Vis studies
UV-Vis studies with catalyst (−)-3 clearly showed the absorbance band for ligand to metal charge transfer (LMCT) at around ~305 nm when the solution was dilute, and at high concentration (when absorbance <400 nm is too high) a band at ~655 nm corresponding to MnV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species arrived during the course of the reaction (Fig. 9),19,31–34 and gets diminished when the substrate was added. This is due to the transfer of oxygen from Mn to the substrate. Visibly the color of the reaction mixture changed from colorless to light brown upon addition of H2O2. Upon addition of 1-hexene (1-hexene was chosen over other olefins as 1-hexene does not show any absorbance in the UV region, which makes it easier to highlight the interaction) to the (−)-3, the solution changed color from colorless (as a CH3CN solution of the MnII complex is colorless) to light red and there is also a red shift in the UV region. This UV change suggests that there exists a sufficiently strong interaction between the catalyst and the substrate (Fig. 10).
O species arrived during the course of the reaction (Fig. 9),19,31–34 and gets diminished when the substrate was added. This is due to the transfer of oxygen from Mn to the substrate. Visibly the color of the reaction mixture changed from colorless to light brown upon addition of H2O2. Upon addition of 1-hexene (1-hexene was chosen over other olefins as 1-hexene does not show any absorbance in the UV region, which makes it easier to highlight the interaction) to the (−)-3, the solution changed color from colorless (as a CH3CN solution of the MnII complex is colorless) to light red and there is also a red shift in the UV region. This UV change suggests that there exists a sufficiently strong interaction between the catalyst and the substrate (Fig. 10).
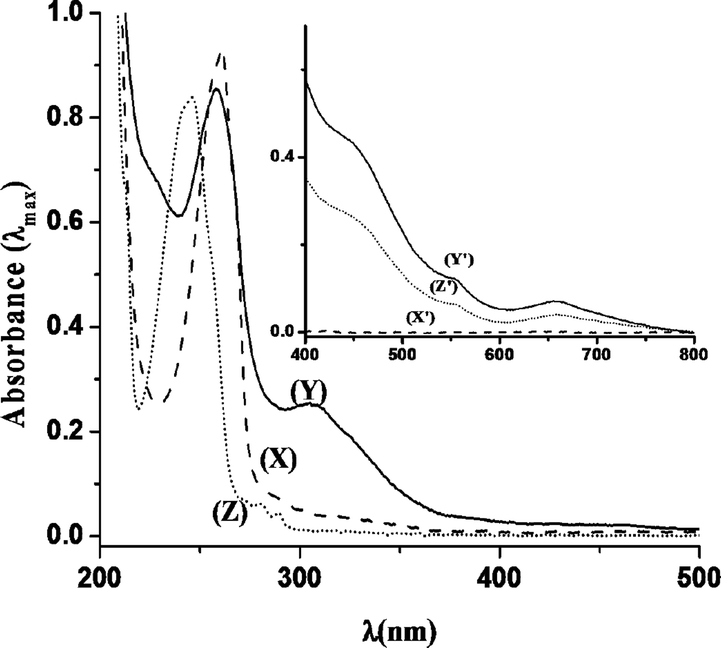 | ||
| Fig. 9 UV-Vis spectra of a 14.2 × 10−3 M solution of catalyst (−)-3 in CH3CN (X), with oxidant H2O2–AcOH (Y), on addition of substrate [styrene] (Z) and UV-Vis spectra of a 7.1 × 10−1 M solution of catalyst (−)-3 in CH3CN (X′), with oxidant H2O2–AcOH (Y′), on addition of substrate [styrene] (Z′). | ||
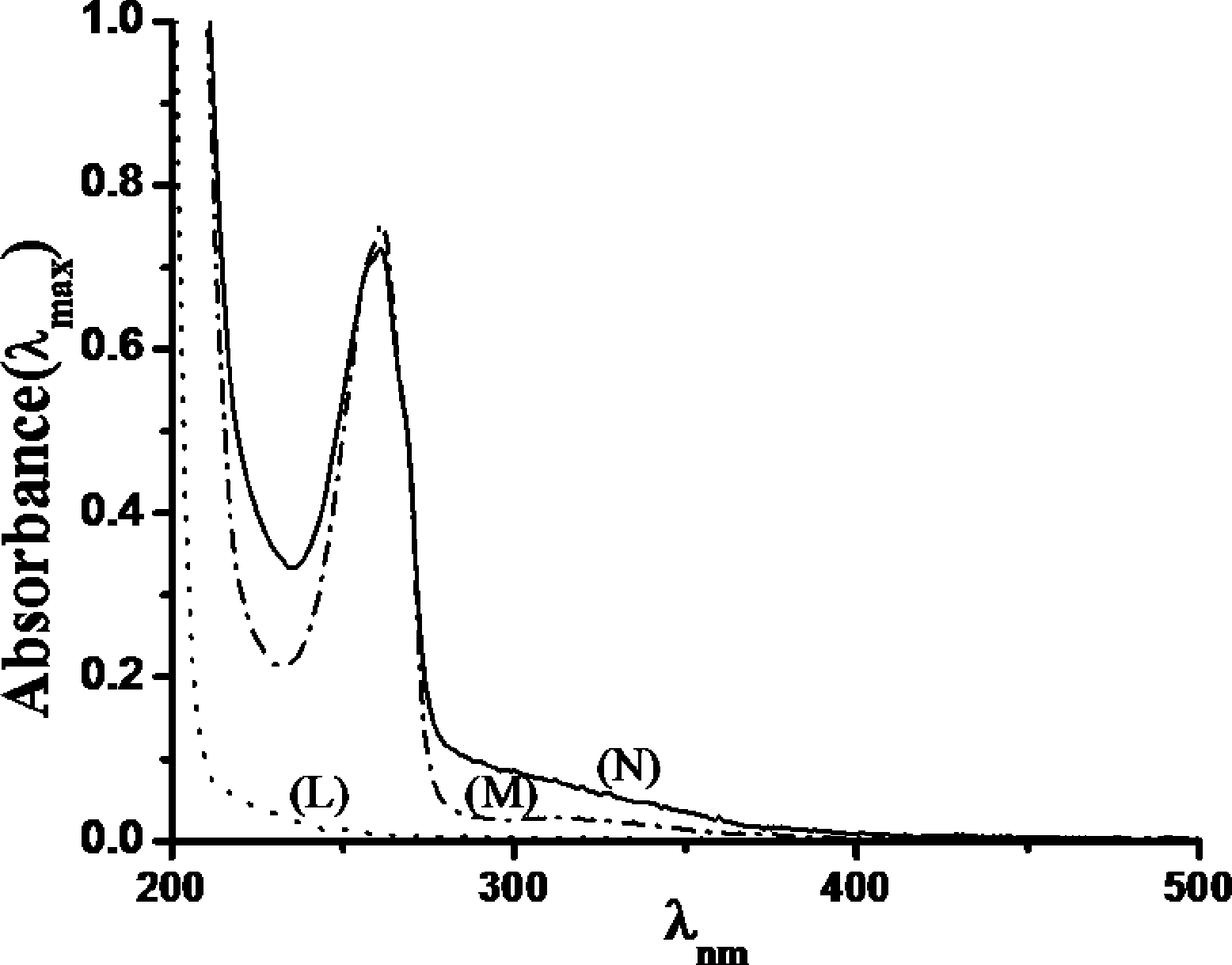 | ||
| Fig. 10 UV-Vis spectra of a 0.298 × 10−4 M solution of catalyst (−)-3 in CH3CN (M), on addition of 1-hexene (N), and only 1-hexene in CH3CN (L). | ||
Based on the kinetic and UV-Vis studies, by considering the role of AcOH in the catalytic reaction, a probable reaction mechanism has been proposed, though existence of other plausible alternatives cannot be ruled out (Scheme 2).25 The reaction is first order with respect to H2O2, where the starting MnII complex is converted to the hydroxoperoxo-MnIII complex [(L)MnIII-OOH]+2; such a species has been previously reported.35 The hydroperoxo complex [(L)MnIII-OOH(S)]+2 is unlikely to directly take part in the epoxidation reaction25 and can exchange its sixth position with acetic acid to form a MnO35 or Mn-peroxo species, whose presence can be seen in the UV-Vis spectrum with a band at ~655 nm. Costas and co-workers34,36 indeed have trapped the active oxo-iron(V) species in a related catalyst system composed of an amino pyridine FeII complex and hydrogen peroxide.
Recyclability study
The catalyst (−)-3 was tested for its reusability in the epoxidation of styrene with H2O2–AcOH as an oxidant under the optimized reaction conditions. After the first catalytic run, the solvent was removed under reduced pressure and the residue was washed with cold hexane to remove the epoxide product, dried in a vacuum, and used as such for the subsequent two catalytic runs without further purification (Fig. 11). The FT-IR spectra of the initial catalyst and the recycled catalyst (after the 3rd cycle) are given in the ESI† and clearly show the integrity of the catalyst after three catalytic cycles (Fig. 12).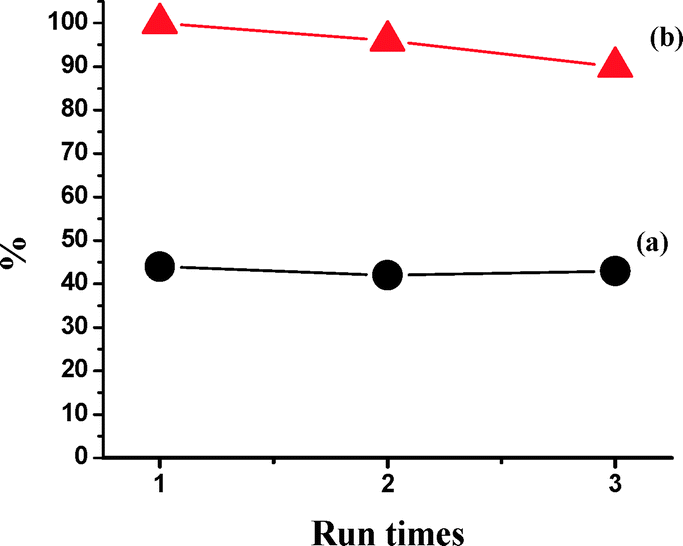 | ||
| Fig. 11 The reuse of catalyst (−)-3 in the asymmetric epoxidation of styrene with H2O2 (1.2 equiv.) and AcOH (3 equiv.) as the oxidant system at 0 °C (a: ee value; b: conversion). | ||
 | ||
| Fig. 12 FT-IR spectrum of fresh (−)-3 catalyst (green in color) and recycled (−)-3 catalyst (black in color). | ||
Conclusions
Out of all the complexes synthesized for the present study, the complex (−)-3 was found to be a robust and recyclable catalyst in the epoxidation of chalcones, styrenes, chromenes and 1-hexene to give the respective epoxides with decent enantioselectivity (up to 88% in selected cases) and conversion (up to 99% with a combined turnover number of >2500) using H2O2 as an oxidant in the presence of 3 equiv. of AcOH at moderate temperature within 1 h. On the basis of kinetic studies, the order of the reaction was found to be second order, dependent on the concentration of catalyst and oxidant. Further structural modifications in the chiral ligand system are underway to enhance the enantioselectivity and also to expand the scope of these catalysts for various other oxidative reactions.Experimental section
General
1H and 13C NMR spectra were recorded on a Bruker Avance III 200 or 400 MHz spectrometer. The chemical shifts (δ) are reported in ppm and coupling constants (J) in Hz. GC were measured on a Bruker 450-GC or a Shimadzu GC 2010. TOF-Mass was obtained on a Micromass Q-TOF-micro instrument. HPLC analysis was performed on a Shimadzu UFLC. Chiralpak OD, IC and IA columns were purchased from Daicel Chemical Industries, LTD. Column chromatography was generally performed on silica gel (100–200 mesh) and TLC inspections were on Merck F254 plates. All chemicals and solvents were purchased from commercial sources and were used as received unless otherwise stated. For the crystal structure determination of the Λ-(−)-2 and Δ-(+)-2, intensity data from a suitable crystal was collected using MoKα (λ = 0.71073 Å) radiation on a Bruker SMART APEX diffractometer equipped with a CCD area detector at 100 K. FT-IR spectra were recorded on a Perkin Elmer Spectrum GX spectrophotometer in the KBr window. Electronic spectra of the complexes were recorded in HPLC grade acetonitrile (CH3CN) on a Shimadzu UV-Vis-NIR spectrophotometer (serial no. A108446). Optical rotations of chiral complexes and their ligand precursors were recorded on an automatic polarimeter (Digipol 781, Rudolph) instrument. Microanalysis of the products was carried out on a vario MICRO cube (Elementar) CHNS analyzer. Circular dichronism (CD) spectra were obtained on a JASCO J-815 CD spectrometer (serial no. A002360843).Synthesis of 1R,2R-C3′′′ and 1S,2S-C3′′′
Improvisation over the method of Yoshida et al.37 was made to synthesize 1R,2R-C3′′′ and 1S,2S-C3′′′. Accordingly, in a flame dried three necked round bottom flask, (R,R)-(−)- or (S,S)-(+)-1,2-diaminocyclohexane (0.454 g, 3.98 mmol), freshly dried 4 Å molecular sieves (~0.02 g), K2CO3 (10 mg) and dry toluene (30 mL) were taken under nitrogen atmosphere, to which 2-acetylpyridine (0.891 mL, 7.96 mmol) was added at RT with stirring. The resulting mixture was stirred for 24 h at 85 °C. Subsequently the reaction mixture was cooled to RT, filtered and the solvent was removed from the filtrate under reduced pressure. The viscous residue was dissolved in hexane and filtered. From the filtrate the desired product of sufficient purity was obtained as white crystals at −20 °C. Noticeably, the product took a longer time to crystallized out from the solvent (typically 4–5 days) whereas white crystals of 1R,2R-C3′′′ were obtained in ~12 h.1R,2R-C3′′′: yield 0.866 g, 68%; lit.37 42%.
1S,2S-C3′′′: yield 0.840 g, 66%; m.p: 126 °C (lit. reported 126.5 °C); [α]30D = +334 (c = 0.08 in CH3CN); 1H NMR (CDCl3, 200 MHz, TMS): δ = 8.51 (d, J = 4.6, 2H), 7.93 (d, J = 8, 2H), 7.62 (ddd, J = 1.6, 1.6, 7.6, 2H), 7.23–7.12 (m, 2H), 3.98–3.85 (m, 2H), 2.38 (s, 6H), 1.90–1.52 (m, 8H). 13C NMR (CDCl3, 50 MHz, TMS): δ = 164.5, 158.2, 147.9, 136.2, 123.7, 120.9, 65.6, 31.5, 24.5, 14.3. FT-IR (KBr): 3435, 3264, 3053, 3003, 2924, 2879, 2857, 1639, 1583, 1564, 1461, 1437, 1358, 1288, 1240, 1102, 1040, 992, 851, 784, 574 cm−1; TOF-Mass (ES+) (m/z): 321.19 (M + H+); anal. calcd. for C20H24N4: C, 74.97; H, 7.55; N, 17.48. Found: C, 74.4; H, 7.3; N, 17.6.
1R,2R-C1′′′: yield 0.951 g, 82%; m.p: 137 °C; [α]30D = −93° (c = 0.35, CH3CN); 1H NMR (CDCl3, 500 MHz, TMS): δ = 8.55 (d, J = 5, 2H); 8.31 (s, 2H), 7.88 (d, J = 8, 2H), 7.64 (ddd, J = 1.5, 2, 7.5, 2H), 7.24–7.21 (m, 2H), 3.56–3.51 (m, 2H), 1.89–1.85 (m, 6H), 1.54–1.50 (m, 2H). 13C NMR (CDCl3, 125 MHz, TMS): δ = 161.4, 154.5, 149.2, 136.5, 124.5, 121.3, 73.5, 32.7, 24.3. FT-IR (KBr): 3428, 2930, 2861, 1644, 1585, 1567, 1467, 1441, 1366, 1339, 1286, 1229, 1079, 991, 937, 776, 617, 434 cm−1; TOF-Mass (ES+) (m/z): 293.28 (M + H+); anal. calcd. for C18H20N4: C, 73.94; H, 6.89; N, 19.16. Found: C, 74.24, H, 7.3; N, 18.8. (Synthesized according to the lit.17 procedure).
Synthesis of 7R,1R,2R,7′S-C3′′, 7S,1S,2S,7′R-C3′′ and 1R,2R-C1′′
1R,2R-C3′′′, 1S,2S-C3′′′ or 1R,2R-C1′′′ (1.25 mmol) was dissolved in dry methanol (5 mL) under inert and dry conditions. To the resulting solution, solid NaBH4 (0.094 g, 2.50 mmol) was added over 2 h and then the reaction was stirred for another 4 h. After that the solvent was removed under pressure and the residue thus obtained was extracted with chloroform (3 × 5 mL). The organic layer was washed with water (3 × 10 mL) and brine (3 × 10 mL) and dried over anhydrous Na2SO4. The solvent was removed under pressure to obtain the ligand 7R,1R,2R,7′S-C3′′ or 7S,1S,2S,7′R-C3′′ as a colorless viscous liquid.7R,1R,2R,7′S-C3′′: 1H NMR data match with the reported lit.38
7S,1S,2S,7′R-C3′′: yield 0.385 g, 95%; [α]30D = +39° (c = 0.333, CH3CN); 1H NMR (CDCl3, 200 MHz, TMS): δ = 8.53 (d, J = 4.6, 2H), 7.63 (ddd, J = 1.4, 1.6, 7.8, 2H), 7.39 (d, J = 7.8, 2H), 7.14 (t, J = 5, 2H), 4.09–3.88 (m, 2H), 2.58 (bs, 2H, D2O exchange), 2.41–2.32 (m, 2H), 1.71–1.50 (m, 2H), 1.38 (d, J = 6.8, 6H), 1.15–0.99 (m, 4H); 13C NMR (CDCl3, 50 MHz): δ = 166.1, 148.8, 136.4, 121.6, 121.0, 60.9, 57.7, 32.4, 24.9, 22.8; FT-IR (KBr): 3432, 2924, 2852, 1631, 1382, 1161, 1102, 1021, 785, 671, 530, 467 cm−1; TOF-Mass (ES+) (m/z): 325.99 (M + H+); anal. calcd. for C20H28N4: C, 74.03; H, 8.70; N, 17.27. Found: C, 74.4; H, 9.0; N, 17.0.
1R,2R-C1′′: yield 0.32 g, 87%; [α]30D = −40° (c = 0.31, CH3CN); 1H NMR (CDCl3, 500 MHz, TMS): δ = 8.52 (d, J = 4.5, 2H), 7.63 (ddd, J = 1.5, 2, 7.5, 2H), 7.39 (d, J = 7.5, 2H), 7.162–7.137 (m, 2H), 4.05 (d, J = 14, 2H), 3.86 (d, J = 14, 2H), 2.44 (bs, 2H, D2O exchange), 2.33–2.29 (m, 2H), 1.73–1.70 (m, 2H), 1.29–1.05 (m, 6H). 13C NMR (CDCl3, 125 MHz): δ = 160.3, 148.9, 136.4, 122.4, 121.8, 61.2, 52.2, 31.4, 24.9; FT-IR (KBr): 3438, 2924, 1633, 1386, 1025, 670, 531, 466 cm−1; TOF-Mass (ES+) (m/z): 297.13 (M + H+); anal. calcd. for C18H24N4: C, 72.94; H, 8.16; N, 18.90. Found: C, 73.2; H, 8.64; N, 18.40. (Synthesized according to the lit.17 procedure).
Synthesis of 7S,1S,2S,7′R-C3′, 7R,1R,2R,7′S-C3′ and 1R,2R-C1′
Ligands 7S,1S,2S,7′R-C3′ and 7R,1R,2R,7′S-C3′ were synthesized by the modified procedure of Ottenbacher et al.39 Accordingly, to the solution of 7S,1S,2S,7′R-C3′′, 7R,1R,2R,7′S-C3′′ or 1R,2R-C1′′ (1.23 mmol) in a mixture of CH3CN (4 mL) and CH3COOH (0.461 mL), 37% aqueous H2CO (0.996 g) was added and the resulting solution was stirred for 20 min at RT. NaBH4 (0.186 mg, 4.92 mmol) was added portion-wise and the mixture was stirred for 12 h at room temperature (caution: the NaBH4 addition reaction is exothermic). CH3CN was removed in a vacuum and 2N KOH was added to the resulting solution until the pH of the solution reached >10. The aqueous phase was extracted with CHCl3 (3 × 10 mL), the extract was washed with H2O and saturated NaCl and finally dried over anhydrous Na2SO4 to get a colorless viscous liquid as the final product.7R,1R,2R,7′S-C3′: yield 0.396 g, 91%; [α]30D = +7.1° (c = 0.280, CH3CN); 1H NMR (CDCl3, 500 MHz, TMS): δ = 8.49 (d, J = 3.5, 2H), 7.60 (t, J = 7.5, 2H), 7.51 (d, J = 7.5, 2H), 7.12 (t, J = 6, 2H), 4.03 (bs, 2H), 2.90 (d, J = 6, 2H), 2.25 (s, 6H), 1.79–1.68 (m, 4H), 1.36 (d, J = 6, 6H), 1.16–1.10 (m, 4H); 13C NMR (CDCl3, 125 MHz, TMS): δ = 167.3, 148.6, 136.7, 121.7, 121.4, 63.7, 59.5, 33.5, 26.0, 25.9, 20.7; FT-IR (KBr): 3432, 2939, 2872, 1647, 1607, 1574, 1480, 1450, 1382, 1287, 1247, 1166, 1031, 956, 896, 870, 782, 761, 639, 576, 516 cm−1; TOF-Mass (ES+) (m/z): 353.3 (M + H+); anal. calcd. for C22H32N4: C, 74.96; H, 9.15; N, 15.89. Found: C, 75.4; H, 9.7; N, 15.3.
7S,1S,2S,7′R-C3′: yield 0.391 g, 90%; [α]30D = −6.3° (c = 0.260, CH3CN); 1H NMR (CDCl3, 200 MHz, TMS): δ = 8.49 (d, J = 3.8, 2H), 7.65–7.50 (m, 4H), 7.13 (t, J = 5.6, 2H), 4.05 (bs, 2H), 2.92 (d, J = 5.6, 2H), 2.25 (s, 6H), 1.82–1.71 (m, 4H), 1.36 (d, J = 6.6, 6H), 1.17–0.85 (m, 4H); 13C NMR (CDCl3, 125 MHz, TMS): δ = 168.8, 150.0, 138.0, 123.0, 122.8, 65.2, 60.9, 34.9, 31.3, 27.4, 22.2; TOF-Mass (ES+) (m/z): 353.6 (M + H+); FT-IR (KBr): 3433, 2925, 2855, 1628, 1591, 1434, 1381, 1112, 1021, 790, 672, 467 cm−1; anal. calcd. for C22H32N4: C, 74.96; H, 9.15; N, 15.89. Found: C, 75.3; H, 9.5; N, 15.2.
1R,2R-C1′: yield 0.358 g, 90%; [α]30D = −13.3° (c = 0.27, CH3CN); 1H NMR (CDCl3, 200 MHz, TMS): δ = 8.49 (d, J = 3.4, 2H), 7.59–7.58 (m, 4H), 7.15–7.09 (m, 2H), 3.97–3.77 (m, 4H), 2.68 (d, J = 7.8, 2H), 2.29 (s, 6H), 2.02–1.78 (m, 4H), 1.28–1.17 (m, 4H); 13C NMR (50 MHz): δ = 161.2, 148.6, 136.3, 122.8, 121.6, 64.5, 60.4, 38.7, 36.6, 25.8; FT-IR (KBr): 3437, 2923, 2854, 1637, 1459, 1023, 670, 588, 460 cm−1; TOF-Mass (ES+) (m/z): 325.21 (M + H+); anal. calcd. for C20H28N4: C, 74.03; H, 8.70; N, 17.27. Found: C, 74.5; H, 9.1; N, 16.9. (Synthesized according to the lit.17 procedure).
General method for the preparation of manganese complexes
Under argon atmosphere, Mn(CF3SO3)2 (0.40 mmol) dissolved in dry CH3CN (1 mL) was added to a stirred solution of chiral ligands 1′, 2′ and 3′ (0.39 mmol) in freshly prepared dry acetonitrile (1 mL) via cannula. The reaction mixture was stirred for 24 h and then the whole reaction mixture was put in a centrifuge to remove any particulate matter. The clear CH3CN reaction mixture was evaporated to dryness in a rotor vapor. The white residue was washed with cold diethyl ether (3 × 5 mL) and finally dried in a vacuum desiccator to yield a foamy white solid.Δ-(+)-2: yield 0.239 g, 94%; [α]30D = +47° (c = 0.180 g, CH3CN); FT-IR (KBr): 3430, 2943, 2870, 1611, 1448, 1254, 1170, 1032, 640, 518 cm−1; TOF-Mass (ES+) (m/z): 528.77 (M-OTf); anal. calcd. for C22H28F6MnN4O6S2·H2O: C, 37.99; H, 4.35; N, 8.05. Found: C, 38.2; H, 4.46; N, 8.10.
Λ-(−)-2: yield 0.231 g, 92%; [α]30D = −46.3° (c = 0.173 g, CH3CN); FT-IR (KBr): 3413, 3266, 3221, 2947, 2876, 1610, 1487, 1445, 1382, 1307, 1222, 1163, 1028, 893, 786, 636, 574, 517 cm−1. TOF-Mass (ES+) (m/z): 528.07 (M-OTf); anal. calcd. for C22H28F6MnN4O6S2·H2O: C, 37.99; H, 4.35; N, 8.05. Found: C, 38.3; H, 4.55; N, 7.90.
(−)-3: yield 0.272 g, 92%; [α]30D = −45° (c = 0.23 g, CH3CN); FT-IR (KBr): 3429, 2926, 2857, 1638, 1543, 1458, 1262, 1163, 1023, 669, 462 cm−1; TOF-Mass (ES+) (m/z): 556.69 (M-OTf); anal. calcd. for C24H32F6MnN4O6S2·H2O: C, 39.84; H, 4.74; N, 7.74. Found C, 40.1, H, 4.92, N, 7.52.
(+)-3: yield 0.275 g, 93%; [α]30D = +44.6° (c = 0.227 g, CH3CN); FT-IR (KBr): 3431, 2925, 2862, 1656, 1629, 1584, 1381, 1265, 1164, 1022, 671, 466 cm−1; TOF-Mass: 556.36 [M-OTf]; anal. calcd. for C24H32F6MnN4O6S2·H2O: C, 39.84; H, 4.74; N, 7.74. Found C, 40.2; H, 4.93; N, 7.81.
General method for epoxidation
In a double jacketed glass reactor connected to a circulating chiller at 0 °C, catalyst (1.0 mg, 0.0014 mmol, 0.1 mol%) taken from stock solution of the catalyst in CH3CN and made up to 1 mL with CH3CN, substrate (1.4 mmol) and 10 mg of n-dodecane as an internal standard were charged. Once the reaction mixture temperature reached 0 °C, AcOH (0.240 mL, 4.2 mmol) was added and the mixture was stirred for 10 minutes. To the above stirring mixture, 50% aqueous H2O2 (1.68 mmol, 1.2 equiv.) mixed with an equal volume of CH3CN was added over 30–45 min by a syringe pump. The reaction mixture was stirred for another 15 minutes and then the solvent was removed under reduced pressure. The residue was purified by a short silica gel column using EtOAc:hexane (4:1) as eluent. The conversions were evaluated by 1H NMR analysis (after removing the solvent under vacuum an internal standard (mesitylene) was added, finally the residue was dissolved in CDCl3) and on a GC after passing the aliquot through a pad of basic alumina. The ee of the products was determined on a chiral GTA GC column or on OD, IC, OB and OD-H Daicel HPLC columns.
Procedure for kinetic experiment
Kinetic studies were carried out using 0.1 mol% catalyst loading and at −10 °C for convenience in reaction monitoring (the reaction is too fast to monitor manually at 0 °C). Styrene was taken as a model substrate with dodecane as an internal standard during the kinetics studies. The reaction was quenched at the desired time by the addition of triethyl amine to an aliquot of the reaction mixture, which was then diluted with diethyl ether or CH3CN, filtered through a basic alumina plug, and analyzed by GC.Acknowledgements
Nabin Ch. Maity and S. H. R. Abdi are thankful to DST and CSIR Indus Magic Project (CSC-0123) on Catalysis for financial assistance. Authors are also thankful to “Analytical Discipline and Centralized Instrument Facility” for providing instrumental facilities.Notes and references
- G. D. Faveri, G. Ilyashenko and M. Watkinson, Chem. Soc. Rev., 2011, 40, 1722–1760 RSC.
- A. Correa, O. G. Mancheno and C. Blom, Chem. Soc. Rev., 2008, 37, 1108–1117 RSC.
- M. Wu, B. Wang, S. Wang, C. Xia and W. Sun, Org. Lett., 2009, 11, 3622–3625 CrossRef CAS PubMed.
- X. Wang, C. Miao, S. Wang, C. Xia and W. Sun, ChemCatChem, 2013, 5, 2489–2494 CrossRef CAS.
- M. Wu, C. X. Miao, S. Wang, X. Hu, C. Xia, F. E. Kuhn and W. Sun, Adv. Synth. Catal., 2011, 353, 3014–3022 CrossRef CAS.
- B. Wang, S. Wang, C. Xia and W. Sun, Chem.–Eur. J., 2012, 18, 7332–7335 CrossRef CAS PubMed.
- B. Wang, C. X. Miao, S. F. Wang, F. E. Kühn, C. G. Xia and W. Sun, J. Organomet. Chem., 2012, 715, 9–12 CrossRef CAS PubMed.
- S. Yu, C. X. Miao, D. Wang, S. Wang, C. Xia and W. Sun, J. Mol. Catal. A: Chem., 2012, 353–354, 185–191 CrossRef CAS PubMed.
- W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, J. Am. Chem. Soc., 1990, 112, 2801–2803 CrossRef CAS.
- R. Irie, K. Noda, Y. Ito, N. Matsumoto and T. Katsuki, Tetrahedron Lett., 1990, 31, 7345–7348 CrossRef CAS.
- K. Matsumoto and T. Katsuki, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 3rd edn, 2010, pp. 839–890 Search PubMed.
- M. Palucki, G. J. McCormick and E. N. Jacobsen, Tetrahedron Lett., 1995, 36, 5457–5460 CAS.
- Z. X. Wang, W. Tu, M. Frohn, J.-R. Zhang and Y. Shi, J. Am. Chem. Soc., 1997, 119, 11224–11235 CrossRef CAS.
- H. Tian, X. She, L. Shu, H. Yu and Y. Shi, J. Am. Chem. Soc., 2000, 122, 11551–11552 CrossRef CAS.
- T. Niwa and M. Nakada, J. Am. Chem. Soc., 2012, 134, 13538–13541 CrossRef CAS PubMed.
- W. Dai, J. Li, G. Li, H. Yang, L. Wang and S. Gao, Org. Lett., 2013, 15, 4138 CrossRef CAS PubMed.
- A. Murphy, G. Dubois and T. Stack, J. Am. Chem. Soc., 2003, 125, 5250–5251 CrossRef CAS PubMed.
- A. Murphy and D. P. T. Stack, J. Mol. Catal. A: Chem., 2006, 251, 78–88 CrossRef CAS PubMed.
- M. C. White, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 7194 CrossRef CAS.
- L. Gόmez, I. G. Bosch, A. Company, X. Sala, X. Fontrodona, X. Ribas and M. Costas, Dalton Trans., 2007, 5539–5545 RSC.
- I. Garcia-Bosch, L. Gόmez, A. Polo, X. Ribas and M. Costas, Adv. Synth. Catal., 2012, 354, 65–70 CrossRef CAS.
- G. Guillemot, M. Neuburger and A. Pfaltz, Chem.–Eur. J., 2007, 13, 8960–8970 CrossRef CAS PubMed.
- R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, Adv. Synth. Catal., 2011, 353, 885–889 CrossRef CAS.
- E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418–1434 CrossRef CAS PubMed.
- O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2012, 2, 1196–1202 CrossRef CAS.
- E. J. Corey and F. Y. Zhang, Org. Lett., 1999, 1, 1287–1290 CrossRef CAS.
- C. Ng, M. Sabat and C. L. Fraser, Inorg. Chem., 1999, 38, 5545–5556 CrossRef CAS PubMed.
- The CIF data for complex Δ-(+)-2 (CCDC 933759) and Λ-(−)-2 (CCDC 945142) are deposited. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- A. Murphy, P. Allyson and T. D. P. Stack, Org. Lett., 2004, 6, 3119–3122 CrossRef CAS PubMed.
- C. Linde, B. Åkermark, P. Norrby and M. Svensson, J. Am. Chem. Soc., 1999, 121, 5083 CrossRef CAS.
- K. Srinivasan, P. Michaud and J. K. Kochi, J. Am. Chem. Soc., 1986, 108, 2309–2320 CrossRef CAS PubMed.
- T. Linker, Angew. Chem., Int. Ed. Engl., 1997, 36, 2060–2062 CrossRef CAS.
- S. Groni, P. Dorlet, G. Blain, S. Bourcier, R. Guillot and E. Anxolabéhère-Mallart, Inorg. Chem., 2008, 47, 3166–3172 CrossRef CAS PubMed.
- L. Prat, J. S. Mathieson, M. Güell, X. Ribas, J. M. Luis, L. Cronin and M. Costas, Nat. Chem., 2011, 3, 788–793 CrossRef PubMed.
- O. Y. Lyakin, K. P. Bryliakov and E. P. Talsi, Inorg. Chem., 2011, 50, 5526–5538 CrossRef CAS PubMed.
- Y. Wang, D. Janardanan, D. Usharani, K. Han, L. Que and S. Shaik, ACS Catal., 2013, 3, 1334–1341 CrossRef CAS.
- S. Yoshida, K. Fukui, S. Kikuchi and T. Yamada, J. Am. Chem. Soc., 2010, 132, 4072–4073 CrossRef CAS PubMed.
- M. Dalai and M. Periasamy, Tetrahedron: Asymmetry, 2009, 20, 1247–1253 CrossRef CAS PubMed.
- R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, Inorg. Chem., 2010, 49, 8620–8628 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 933759 and 945142. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cy00528c |
| This journal is © The Royal Society of Chemistry 2014 |