Chemical-promoted oxidative polymerization of modified-cobalt salen complexes, efficient catalysts for the dynamic HKR†
Xiang
Hong
a,
Laurianne
Billon
b,
Mohamed
Mellah
*a and
Emmanuelle
Schulz
*ab
aEquipe de Catalyse Moléculaire, ICMMO, UMR CNRS 8182, Université Paris-Sud 11, 91405 Orsay Cedex, France. E-mail: mohamed.mellah@u-psud.fr
bCNRS, Orsay, F-91405, France. E-mail: emmanuelle.schulz@u-psud.fr
First published on 8th November 2012
Abstract
FeCl3 and NOBF4 have been successfully tested as chemical oxidants to promote the polymerization of thiophene- or pyrrole-modified chiral cobalt salen complexes. The iron oxidation procedure particularly delivered an insoluble polymeric species in nearly quantitative yield that was tested for its propensity to promote the dynamic hydrolytic kinetic resolution of epibromohydrin. This catalyst allowed the synthesis of the expected diol in high yield and up to 85% ee. As an insoluble powder, it was either efficiently recycled in a batch reactor via simple filtration or dispersed in a silica support and reused in a flow procedure for up to five cycles. A copolymerization strategy delivered an even more reliable polymerized complex that was repetitively used without showing any loss in activity or in enantioselectivity for five reuses in this transformation.
Introduction
Chiral salen complexes belong unambiguously to one of the main successful catalyst families used for the production of valuable enantioenriched synthons, with both high efficiency and selectivity.1 Even if in some specific cases high TON and TOF were already achieved, large efforts are nonetheless until now always devoted to the search of new methodologies towards the easy recovery and reuse of these catalysts.2In this context, we have described an electrochemical oxidative procedure leading to the synthesis of linear polymers that possessed the chiral metallic salen moiety in the main chain.3 Properly modified salen complexes with thiophene groups at the 5,5′-position were thus synthesized and corresponding chromium- or cobalt complexes were successfully polymerized. Tested as insoluble heterogeneous catalysts, they could promote diverse carbon–carbon or carbon–heteroatom bond formation in hetero Diels–Alder reactions,4 in nitroaldol transformations,5 in dialkylzinc addition to aldehydes, or in the ring-opening of epoxides with various nucleophiles.6
Due to their important chemical and thermal stability associated with a high conductivity, poly(thiophenes) have been developed as organic conducting polymers for numerous applications, such as light-emitting devices, sensor applications, energy storage or thin film transistors.7 These properties are directly linked to the polymeric chain organization and thus to the polymerization conditions. Organometallic couplings leading successfully to structurally well-defined polymers have been described involving Kumada–Corriu,8 Negishi,9 Stille,10 Suzuki11 or Heck-type couplings.12 Oxidative electrochemical polymerization of thiophene derivatives is also well documented,13 and oxidative chemical polymerization reactions have been likewise well reported, which imply monomer oxidation by FeCl3.14 This methodology generally offers a high yield of high molecular weight polymers which have a low amount of irregular couplings.15
Chiral salen cobalt complexes have already demonstrated their undeniable value for promoting the hydrolytic kinetic resolution of epoxides.16 Many efforts were already consequently devoted to their immobilization on various supports for their easy recovery and subsequent reuse.17 Moreover, as detailed mechanistic studies indicated that this transformation was catalysed through a cobalt bimetallic activation,18 targeted oligomeric salen complexes have been prepared aiming at the optimization of the catalyst activity.19 We recently reported that the efficiency of the dynamic hydrolytic kinetic resolution (HKR) of terminal epoxides promoted by electrochemically generated cobalt salen polymers was highly dependent on the polymerization conditions, influencing indeed significantly the polymer structure.20 In relation to these results, we present our efforts towards the synthesis of analogous chiral thiophene–salen cobalt polymers via chemical oxidation and their characterization. These polymers are used here as insoluble, recyclable catalysts to promote the HKR of epibromohydrin not only in a batch reactor but also in a fixed-bed reactor, and preliminary results concerning their use under flow conditions are discussed.
Results and discussion
Monomers 1 and 2 (Fig. 1) were chosen as building blocks to test the chemical polymerization. These chiral ligands were prepared through a simple procedure that involves a Suzuki coupling between accordingly functionalized aromatic rings and a subsequent condensation step with (S,S)-cyclohexane-1,2-diamine as we previously described.3,4,20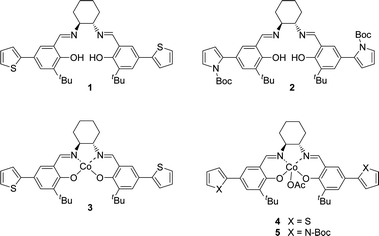
Corresponding cobalt complexes were synthesized according to the reported procedures.21 A Co(II) complex 3 was prepared from ligand 1, which was oxidized in the presence of acetic acid in air to yield Co(III) complex 4, an active catalyst for the dynamic HKR of epibromohydrin.22 Analogously, this procedure, starting from the pyrrole-based ligand 2, afforded Co(III) complex 5 (Fig. 1).
Thiophene–salen cobalt complexes were previously successfully polymerized through electrochemical oxidation. We describe here the ability of complexes 3–5 to be chemically oxidized for the formation of the corresponding polymeric metallic chiral species and their study as catalysts for the dynamic HKR of terminal epoxides.
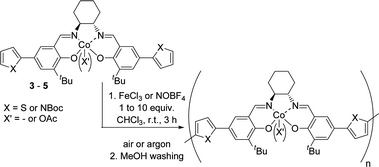
The general procedures for chemical oxidations are depicted in Scheme 1. Two different oxidizing agents have been tested, FeCl3 and NOBF4, both of which already proved their efficiency in promoting the polymerization of thiophene-based monomers.23,24 The reactions have been performed in chloroform at room temperature under an air or argon atmosphere. The results are gathered in Table 1.
 | ||
| Scheme 1 A general chemical procedure for chiral cobalt salen polymerization. | ||
| Entry | Monomer | Polymer | Oxidant | Atmosphere | Yield (%) |
|---|---|---|---|---|---|
| a Monomer 5.0 × 10−2 mol L−1 in CHCl3, 3 h, 10 equiv. oxidants. | |||||
| 1 | 3 | Poly-3a | FeCl3 | Argon | 80 |
| 2 | 3 | Poly-3b | FeCl3 | Air | >99 |
| 3 | 4 | Poly-4a | FeCl3 | Argon | 98 |
| 4 | 5 | Poly-5a | FeCl3 | Argon | 98 |
| 5 | 4 | Poly-4b | NOBF4 | Argon | >99 |
| 6 | 5 | Poly-5b | NOBF4 | Argon | >99 |
Our previous studies have showed that corresponding polymerized Co(III) complexes could be directly prepared from Co(II) precursors under the oxidative electrochemical polymerization conditions.20 Co(II) complex 3 has thus been firstly engaged in the chemical polymerization with an excess of FeCl3 (10 equiv.)25 as an oxidant under an argon atmosphere. After precipitation in MeOH and repeated rinsing with MeOH and THF, the corresponding polymer Poly-3a could be isolated as an insoluble black powder in 80% yield (Table 1, entry 1).
Polymerized 3-alkyl thiophenes with a very high molecular weight have been prepared by Haider et al. by bubbling air through the reaction mixture during the polymerization.26 The polymerization of complex 3 in air has thus been tested, with Poly-3b being recovered with quantitative yield (Table 1, entry 2).
EPR (Electron Paramagnetic Resonance) studies have been carried out to identify the oxidation state of the metal centers in these polymers, with a reported Poly-Co(III) prepared by anodic oxidation as a reference.27 The EPR spectrum of electrogenerated Poly-Co(III) showed a strong signal for the radical (g = 2.002) at 3500 G and also a strong signal for paramagnetic Co(II) at 1500 G, which are in accordance with the reported EPR signals of Jacobsen's Co(III)–OAc complex, as the salen Co(III) complex is in equilibrium with a Co(II) phenoxyl radical species.28 The EPR spectrum of Poly-3b showed no noticeable difference in comparison to that of Poly-Co(III), while a strong signal of Co(II) and a totally negligible signal of radical were observed for Poly-3a. This suggests that metal centers in this polymer are still at the (+II) oxidation state.29
The studies of Poly-3a and Poly-3b by IR spectroscopy have also demonstrated obvious differences in the stretching vibration v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). Compared to complex 3 (v(CN) 1598 cm−1), the v(CN) band in the IR spectrum of Poly-3b splits into two peaks at 1634 and 1592 cm−1, a phenomenon that was also observed after the oxidation of the Co(II) complex 3 to the Co(III) complex 4 (v(CN) 1632 and 1607 cm−1).30 However, only one peak was observed for the stretching vibration of Poly-3a (v(CN) 1609 cm−1), which also tends to prove that metal centers in this polymer stay as Co(II) under the oxidative polymerization conditions.29
N). Compared to complex 3 (v(CN) 1598 cm−1), the v(CN) band in the IR spectrum of Poly-3b splits into two peaks at 1634 and 1592 cm−1, a phenomenon that was also observed after the oxidation of the Co(II) complex 3 to the Co(III) complex 4 (v(CN) 1632 and 1607 cm−1).30 However, only one peak was observed for the stretching vibration of Poly-3a (v(CN) 1609 cm−1), which also tends to prove that metal centers in this polymer stay as Co(II) under the oxidative polymerization conditions.29
The chemical polymerization procedure modifies the oxidation state of the metal centers when a Co(II) complex was used as a monomer, we thus preferred to use preformed Co(III) complexes for the chemical polymerization to ensure the oxidation state of cobalt centers in the polymers.
Complexes 4 and 5 have thus been engaged in chemical polymerization under an argon atmosphere with FeCl3 as an oxidant under the same conditions and Poly-4a and Poly-5a were both obtained with a high yield of 98% (Table 1, entries 3 and 4). NOBF4 has also been proved useful for the polymerization of these two complexes, affording Poly-4b and Poly-5b with nearly quantitative yield (Table 1, entries 5 and 6).
IR spectra of Poly-4a and Poly-4b showed signals similar to those observed for complex 4, two peaks at the stretching vibration region of CN characteristic for Co(III) metal centers in these polymers. However, for Poly-5a and Poly-5b, a complete disappearance of the stretching vibration band at 1737 cm−1 indicates the total decomposition of Boc under the polymerization conditions.29
EDS studies have been performed for Poly-4a and Poly-5a, and signals corresponding to C, N, O, S and Co were also observed. As for Poly-4b and Poly-5b, a relatively high peak of fluorine was also found in the EDS spectrum, which indicated that a part of the Co(III)–OAc complexes might have been transformed to Co(III)–BF4 by anion exchange.31 This has also been confirmed by elemental analysis of Poly-4b, showing that 81% of Co(III)–OAc have been transformed to Co(III)–BF4 during the polymerization.29
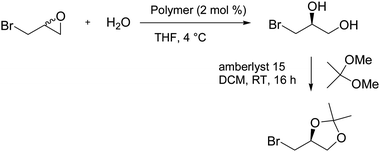
All recovered cobalt polymers were engaged to test their ability as promoters for the dynamic HKR of epibromohydrin (Scheme 2). The reaction was performed in THF at room temperature in the presence of 2 mol% of the polymers for 48 hours. The resulting diol was then protected by 2,2-dimethoxypropane to allow the determination of the yield and the enantiomeric excess.
 | ||
| Scheme 2 Dynamic hydrolytic kinetic resolution of epibromohydrin catalyzed by polymeric cobalt salen complexes. | ||
The efficiency of complexes 3, 4 and 5 has been previously reported,20 and both Co(III) complexes can catalyse the dynamic HKR of epibromohydrin with good activity and high enantioselectivity under homogeneous conditions (Table 2, entries 2 and 3).
| Entry | Catalyst | Yieldb (%) | eec (%) |
|---|---|---|---|
| a 2 mol% cat., 1.5 equiv. H2O, THF, rt, 48 h. b Isolated yield, determined by the yield of corresponding acetals prepared from 2,2-dimethoxypropane. c Determined by chiral GC analysis of corresponding acetals. d No reaction. | |||
| 1 | 3 | NRd | — |
| 2 | 4 | 83 | 92 |
| 3 | 5 | 75 | 97 |
| 4 | Poly-3a | NR | — |
| 5 | Poly-3b | 88 | 83 |
| 6 | Poly-4a | 76 | 85 |
| 7 | Poly-5a | 51 | 67 |
| 8 | Poly-4b | 40 | 25 |
| 9 | Poly-5b | 57 | 17 |
Poly-3b prepared from 3 under an air atmosphere proved to be an efficient heterogeneous catalyst for the HKR of epibromohydrin, showing similar activity and slightly decreased enantioselectivity in comparison to the monomer complexes (Table 2, entry 5). However, no reaction occurred when Poly-3a obtained from an anaerobic polymerization was tested as a catalyst (Table 2, entry 4). This result exactly matched the observation in EPR and IR analyses of this polymer that Co(II) monomer 3 could not be oxidized to active Co(III) species if the polymerization procedure took place under an argon atmosphere, even in the presence of an excess of FeCl3.32
Poly-4a arising from a pre-formed Co(III)–OAc species led to comparable results to those obtained with Poly-3b, indicating that similar polymeric species might have been prepared through both oxidative procedures (Table 2, entries 5 and 6).33 However, Poly-4b prepared from the same precursor with NOBF4 as an oxidant led to less appealing results in catalysis, which might originate from the unexpected anion exchange during the polymerization (Table 2, entry 8).
Complex 5 has showed good activity and great enantioselectivity in the dynamic HKR of epibromohydrin under homogeneous conditions (Table 2, entry 3). However, poor efficiencies were observed when the corresponding polymerized complexes Poly-5a and Poly-5b were tested under heterogeneous conditions (Table 2, entries 7 and 9). The decreased activity and enantioselectivity probably arose from the loss of the labile protecting Boc group of complex 5 during the polymerization, as was confirmed by IR studies.
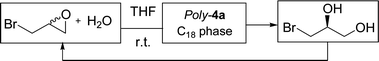
Immobilization of catalysts is a great achievement for sustainable development, considering the use of continuous-flow processes. By this strategy, new synthetic methods are economically viable at an industrial scale.34 Indeed, the confinement of the catalyst facilitates the recycling technique by more efficient purification procedures and catalyst regeneration. Moreover in some cases this catalyst immobilization was proved to increase its stability and enantioselectivity.35 Complex 4 could be polymerized chemically with nearly quantitative yield and the resulting polymer Poly-4a showed good activity and enantioselectivity in heterogeneous catalysis. In this context, the application of a fixed-bed reactor was investigated using Poly-4a. The polymer was immobilized through mixing with an octadecyl carbon chain (C18) bonded silica in a reversed phase mode (1/1 w/w). The reversed phase support was preferred to facilitate the release of the diol resulting from the hydrolytic kinetic resolution of epibromohydrin thus preventing fast reactor saturation. The solid support–catalyst mixture was confined in a short HPLC-type pre-column that was thus used as a flow reactor for a new recycling procedure for dynamic HKR of epibromohydrin (Scheme 3). In this home-made reactor the heterogeneous catalytic reaction and the catalyst are perfectly confined and only the desired product and solvent are recovered at the reactor outlet.
 | ||
| Scheme 3 Dynamic hydrolytic kinetic resolution of epibromohydrin catalyzed by immobilized Poly-4a. | ||
To adopt the fixed-bed reactor, the HKR of epibromohydrin was realized under diluted conditions: a solution of epibromohydrin in THF (1.1 mol L−1, to insure 2 mol% of catalyst) was introduced and maintained as a constant flow at 1 mL min−1. After 24 hours, 89% of the substrate was hydrolysed to the corresponding diol and the enantiomeric excess of the product was determined to be 81%, values which are totally comparable with those obtained under the classic conditions in a batch reactor.36
As Poly-4a is totally insoluble in the reaction mixture, we thus wondered if this polymeric complex could be reused in successive catalytic runs. Recycling studies of Poly-4a were thus performed in both batch and fixed-bed reactors to evaluate its stability. For the recycling studies in the batch reactor, the polymeric species was simply separated from the product by filtration after the first catalytic run, rinsed with THF, dried under vacuum and reused in a renewed catalytic run by adding new substrates. For the recycling studies in the fixed-bed reactor, the immobilized Poly-4a was rinsed with a flow of dry THF for 30 minutes, and then directly engaged in another run. Five successive catalytic cycles were performed in both cases, and the results are gathered in Tables 3 and 4.
| Run | Conv.b (%) | eec (%) |
|---|---|---|
| a 9.08 mmol of epibromohydrin in 8 mL of THF in order to insure 2 mol% of catalyst. b Conversion determined by GC analysis after 24 h, with chlorobenzene as an internal standard. c Determined by chiral GC analysis of corresponding acetals. | ||
| 1 | 89 | 81 |
| 2 | 74 | 78 |
| 3 | 71 | 74 |
| 4 | 54 | 69 |
| 5 | 48 | 71 |
Poly-4a proved delightfully to be an efficient promoter for the repetitive transformation of epibromohydrin in the batch procedure. The desired product could be obtained at each cycle, with high and stable values of enantiomeric excess. However, continuing decreased yields showed that this catalyst deactivated along the recycling studies, although it is still not clear whether the mechanical or the chemical instability of Poly-4a is responsible for the decrease of its activity in this recycling procedure.
As for the recycling studies in the fixed-bed reactor, stable enantiomeric excesses of the expected product were again observed (71% ee after the 5th run). The average enantioselectivity value is however slightly diminished in comparison to the one observed in the batch procedure, and an influence of the catalyst support on the course of the enantioselective transformation cannot be excluded. In this case again, large variations in the conversion (from 89% to 48% at the last run) went along with the recycling. The similarly decreased efficiency of Poly-4a in the fixed-bed reactor indicated that the deactivation of this catalyst should arise from its chemical structural alteration during the HKR.
Mechanistic studies of the HKR by Jacobsen et al. showed that a mixture of salen Co(III) complexes containing both nucleophilic and non-nucleophilic counter anions is more efficient than the classic Co(III)–OAc catalyst,18 and we previously prepared a stable heterogeneous catalyst for the HKR by the electrochemical copolymerization of a salen Co(III)–OAc complex and a salen Co(III)–BF4 complex.20 In order to enhance the stability of the chemically generated polymers, we wondered if the copolymerization strategy could also be applied to the chemical polymerization. Towards this aim, complex 6 was synthesized in high yield from Co(II) complex 3 through oxidation with ferrocenium tetrafluoroborate,37 and a copolymerization test was performed by mixing an equimolar amount of monomers 4 (Co(III)–OAc sites) and 6 (Co(III)–BF4 sites) in the presence of FeCl3 as an oxidant (Scheme 4).
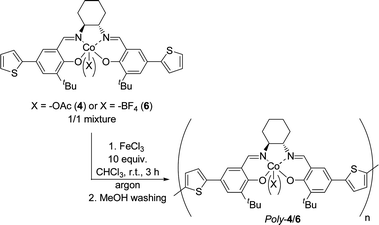 | ||
| Scheme 4 A chemical oxidative procedure for the synthesis of chiral Poly-4/6. | ||
Poly-4/6 was also recovered as an insoluble powder after methanol washings in 90% yield. Elemental analyses were performed for the determination of the Co, B and F contents, and the percentage of Co(III)–BF4 in the resulting copolymer was determined to be 49%. This copolymer was tested for the dynamic HKR of epibromohydrin under the same conditions, allowing the formation of the corresponding diol in good yield (75%) and high enantiomeric excess (80%) after 48 hours. To test the stability of the polymeric structure during the HKR transformation, repetitive experiments following the same procedures as those performed to study the stability of Poly-4a were achieved, and the results obtained are gathered in Tables 5 and 6.
| Run | Conv.b (%) | eec (%) |
|---|---|---|
| a 9.08 mmol of epibromohydrin in 8 mL of THF in order to insure 2 mol% of catalyst. 2 mol% cat., 2 equiv. H2O, rt, 48 h. b Conversion determined by GC analysis after 24 h, with chlorobenzene as an internal standard. c Determined by chiral GC analysis of corresponding acetals. | ||
| 1 | 72 | 74 |
| 2 | 76 | 76 |
| 3 | 81 | 75 |
| 4 | 80 | 78 |
| 5 | 83 | 76 |
In contrast to the catalytic results reported for Poly-4a in Tables 4 and 5, Poly-4/6 allowed the repetitive preparation of the scalemic diol with stable high yields and high enantiomeric excesses for both batch and fixed-bed procedures. These values remained furthermore stable, as a clear differentiation in the catalytic behaviour of both polymers. The yield fluctuation was indeed no more noticeable for the catalysis promoted by Poly-4/6. Even after the last cycle, the product was still isolated in a high yield of more than 80%, whatever the recycling methodology. The structures of Poly-4a and Poly-4/6 only differ in the axial ligands of the cobalt atoms. As already demonstrated by the studies of Jacobsen et al.,18 the cobalt complex bearing acetate as an axial ligand undergoes irreversible hydrolysis to CoIII–OH species in the HKR, which leads to the deactivation of the catalyst during the reaction. This formation of CoIII–OH sites during the catalysis performed by Poly-4a may be an explanation for its deactivation along with its recycling. In the case of Poly-4/6 possessing both nucleophilic and non-nucleophilic active sites, such a deactivation is presumably prevented to allow its repeated efficient use.
To determine the extent of applications of Poly-4/6, this insoluble catalyst was used in a batch procedure for promoting successively the hydrolytic kinetic resolution of different terminal epoxides, by changing the structure of the substrate at each reuse of the polymer. A first run was then devoted to the already studied HKR of epibromohydrin yielding the expected results (see Table 7, run 1). The recovered catalyst was then engaged in the transformation of 2-phenoxymethyloxirane (Table 7, run 2).38
| Run | Epoxide | Time (h) | Yielda (%) | ee (%) |
|---|---|---|---|---|
| a GC yield, determined by achiral GC analysis with chlorobenzene as an internal standard. b 2 mol% cat., 1.5 equiv. H2O, THF, RT. c 0.5 mol% cat., 0.65 equiv. H2O, RT. d 0.8 mol% cat., 0.65 equiv. H2O, THF, RT. e Determined by chiral GC analysis (Chiraldex β-PM, 110 °C isothermal). f Determined by chiral HPLC analysis (Chiralcel OD-H, Hex/iPrOH 95/5, 1.0 mL min−1, 214 nm, 25 °C). g Determined by chiral GC analysis (Chiraldex β-PM, 35 °C isothermal). h Determined by chiral GC analysis (Chiraldex β-PM, 110 °C, 10 min, 10 °C min−1 up to 170 °C). i Theoretical yield = 50%. | ||||
| 1b | Epibromohydrin | 48 | 75 | 80e |
| 2c | 2-Phenoxymethyl-oxirane | 48 | 38i | 99f |
| 3c | 2-Allyloxymethyl-oxirane | 48 | 41i | 99g |
| 4d | 2-Phenyloxirane | 64 | 45i | 85h |
| 5 | Epibromohydrin | 48 | 89 | 85 |
The subsequent recycling procedure engaged consecutively 2-allyloxymethyloxirane and 2-phenyloxirane and the last run was assigned again to the hydrolysis of epibromohydrin. Albeit the heterogeneous catalysis proved to be less efficient in terms of activity for the transformations of the new substrates in comparison to the results obtained under homogeneous conditions (Table 7, runs 2–4), nearly enantiopure products could nevertheless be obtained in two cases with higher substrate conversion after prolonged reaction time. Finally, Poly-4/6 was reused a fifth time for the dynamic HKR of epibromohydrin (Table 7, run 5), delivering interestingly an improved activity and enantioselectivity in comparison to the first run. Detailed kinetic studies were realized for the first run and the fifth reuse of this polymer and no obvious difference was observed.29 Together with a supplementary experiment proving no metal leaching,39 this undoubtedly demonstrated the stability of this easily prepared polymer in the HKR.
Conclusion
Chemical oxidative polymerization has been thus demonstrated as a powerful and practical procedure for the preparation of chiral insoluble cobalt salen complexes in high yield. According to the polymerization conditions and the structure of the engaged monomer, Co(II)- or Co(III)-polymeric derivatives were obtained and characterized. The latter species was tested for its propensity to promote the dynamic hydrolytic kinetic resolution of epibromohydrin, leading to the expected scalemic diol with high yield and enantioselectivity. The polymerized catalyst could be recovered and efficiently reused in both a batch reactor and also a flow procedure. As a convenient and reliable methodology, the chemical oxidation furthermore allowed a copolymerization strategy to be performed, leading to the easy preparation of an even more reliable catalyst. Starting indeed from a salen Co(III)–OAc complex and a salen Co(III)–BF4 complex, a copolymer could be synthesized, showing a dramatically enhanced stability upon the recycling studies, for the HKR of structurally varied terminal epoxides.As far as we know, this report is the first demonstration of the efficient use of a heterogeneous catalyst generated as a polymeric structure through chemical oxidation, involved successfully in repetitive asymmetric transformations. This simple procedure requiring only cheap and easily accessible oxidants could be applied to the heterogenization of other chiral metallic complexes, or even their corresponding ligands, opening the way of an economic and environmentally benign use (and reuse) of valuable asymmetric catalysts. Work towards this end is in progress in our laboratory.
Experimental section
A representative procedure for the chemical oxidative polymerizations
For polymerizations under an argon atmosphere, a solution of a monomer complex (0.25 mmol) in chloroform (2.5 mL) was added to a Schlenk tube charged with a suspension of FeCl3 (407 mg, 2.5 mmol) in chloroform (2.5 mL) or a solution of NOBF4 (293 mg, 2.5 mmol) in chloroform under argon. The resulting suspension was stirred at room temperature under argon for 3 hours, and then added to MeOH (200 mL) dropwise under vigorous stirring. The precipitate was filtrated under reduced pressure, rinsed successively with MeOH, water, MeOH and THF to afford the targeted polymerized species as black powders. The same procedure was performed without argon protection for the polymerization under an air atmosphere.Representative dynamic HKR of epibromohydrin under heterogeneous conditions in a batch reactor
THF (145 μL) and water (33 μL, 1.81 mmol) were added to a suspension of polymerized complexes (2 mol% for Co) in epibromohydrin (104 μL, 1.21 mmol) dropwise at 0 °C with continuous stirring. The resulting suspension was stirred at RT for 48 h, diluted with THF (5 mL) and the products solution removed by filtration. The catalyst residue was rinsed five times with THF (5 mL), and the combined solutions were concentrated at reduced pressure before the addition of DCM (5.4 mL), Amberlyst 15 (16 mg) and 2,2-dimethoxypropane (317 μL, 2.42 mmol). The resulting mixture was stirred at RT for another 18 h, then filtrated on celite and the solvents removed under reduced pressure. The residue was purified by chromatography on silica gel (pentane/diethylether = 95/5) for the determination of the yield of the reaction and the enantiomeric excess of the product. In the Schlenk tube, the remaining catalyst was dried under vacuum and reused directly for the next run.Representative dynamic HKR of epibromohydrin under heterogeneous conditions in a fixed-bed reactor
In a HPLC system, Poly-4a (150 mg, 0.18 mmol for Co) and C18 modified silica (150 mg) were mixed and confined in a short HPLC pre-column. THF (8 mL) containing epibromohydrin (780 μL, 9.08 mmol) and chlorobenzene (375 μL) was introduced into the system and maintained as a constant flow of 1.0 mL min−1. Water (250 μL, 13.6 mmol) was then injected into the system to start the reaction and the conversion of the substrate was monitored by GC analysis of an aliquot of 2 μL at specific times. At the end of the reaction, the catalyst residue was rinsed with a flow of dry THF for 30 minutes and the combined solutions were concentrated at reduced pressure before the addition of DCM (40 mL), Amberlyst 15 (120 mg) and 2,2-dimethoxypropane (2.4 μL, 18.2 mmol). The resulting mixture was stirred at RT for another 18 h, and then filtrated on celite. The solvents were removed under reduced pressure and the residue was purified by chromatography on silica gel (pentane/diethylether = 95/5) for the determination of the yield of the reaction and the enantiomeric excess of the product. The catalyst anchored in the fixed-bed reactor could be reused in another run without further treatment.Acknowledgements
The CNRS, the Ministère de l'Enseignement Supérieur et de la Recherche and the program “Chimie et Développement Durables” du CNRS are acknowledged for financial support.Notes and references
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410 RSC.
- (a) C. Baleizão and H. Garcia, Chem. Rev., 2006, 106, 3987 CrossRef; (b) A. Zulauf, M. Mellah, X. Hong and E. Schulz, Dalton Trans., 2010, 39, 6911 RSC.
- (a) A. Voituriez, M. Mellah and E. Schulz, Synth. Met., 2006, 156, 166 CrossRef CAS; (b) A. Zulauf, X. Hong, F. Brisset, E. Schulz and M. Mellah, New J. Chem., 2012, 36, 1399 RSC.
- A. Zulauf, M. Mellah, R. Guillot and E. Schulz, Eur. J. Org. Chem., 2008, 2118 CrossRef CAS.
- A. Zulauf, M. Mellah and E. Schulz, J. Org. Chem., 2009, 74, 2242 CrossRef CAS.
- A. Zulauf, M. Mellah and E. Schulz, Chem. Commun., 2009, 6574 RSC.
- (a) J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498 CrossRef CAS; (b) A. Kraft, A. C. Grimsdale and A. B. Holmes, Angew. Chem., Int. Ed., 1998, 37, 402 CrossRef; (c) J. Pei, W.-L. Yu, W. Huang and A. J. Heeger, Chem. Commun., 2000, 1631 RSC; (d) H. E. Katz, Z. Bao and S. L. Gilat, Acc. Chem. Res., 2001, 34, 359 CrossRef CAS.
- (a) R. D. McCullough, R. D. Lowe, M. A. Jayaraman and D. L. Anderson, J. Org. Chem., 1993, 58, 904 CrossRef CAS; (b) R. D. McCullough and S. P. Williams, J. Am. Chem. Soc., 1993, 115, 11608 CrossRef CAS; (c) R. S. Loewe and R. D. McCullough, Chem. Mater., 2000, 12, 3214 CrossRef CAS.
- (a) T.-A. Chen, X. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233 CrossRef CAS; (b) J.-G. Kim, S.-H. Kim and R. D. Rieke, Macromol. Res., 2011, 19, 749 CrossRef CAS.
- (a) A. Iraqi and G. W. J. Barker, J. Mater. Chem., 1998, 8, 25 RSC; (b) M. Bouachrine, J.-P. Lere-Porte, J. J. E. Moreau, F. Serein-Spirau and C. Torreilles, J. Mater. Chem., 2000, 10, 263 RSC; (c) J. P. Lere-porte, J. J. E. Moreau and C. Toreilles, Eur. J. Org. Chem., 2001, 1249 CrossRef CAS.
- (a) S. Guillerez and G. Bidan, Synth. Met., 1998, 93, 123 CrossRef CAS; (b) G. Bidan, A. De Nicola and V. Enee, Chem. Mater., 1998, 10, 1052 CrossRef CAS.
- (a) M. Sévignon, J. Papillon, E. Schulz and M. Lemaire, Tetrahedron Lett., 1999, 40, 5873 CrossRef; (b) J. Hassan, E. Schulz, C. Gozzi and M. Lemaire, J. Mol. Catal. A: Chem., 2003, 195, 125 CrossRef CAS.
- (a) J. Roncali, Chem. Rev., 1992, 92, 711 CrossRef CAS; (b) P. Blanchard, P. Leriche, P. Frere and J. Roncali, in Handbook of Conducting Polymers, ed. T. A. Skotheim and J. R. Reynolds, 2007, vol. 1, pp. 13/1–13/77 Search PubMed.
- R. Sugimoto, S. Takeda, H. B. Gu and K. Yoshino, Chem. Express, 1986, 1, 635 CAS.
- M. Leclerc, F. M. Diaz and G. Wegner, Makromol. Chem., 1989, 190, 3105 CrossRef CAS.
- (a) M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 277, 936 CrossRef CAS; (b) E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421 CrossRef CAS.
- For reviews see: (a) C. Baleizao and H. Garcia, Chem. Rev., 2006, 106, 3987 CrossRef CAS; (b) A. Zulauf, M. Mellah, X. Hong and E. Schulz, Dalton Trans., 2010, 39, 6911 RSC ; for some representative recent examples see: ; (c) F.-J. Song, C. Wang, J. M. Falkowski, L.-Q. Ma and W.-B. Lin, J. Am. Chem. Soc., 2010, 132, 15390 CrossRef CAS; (d) Y.-S. Kim, X.-F. Guo and G.-J. Kim, Chem. Commun., 2009, 4296 RSC; (e) T. Belser and E. N. Jacobsen, Adv. Synth. Catal., 2008, 350, 967 CrossRef CAS; (f) H. Yang, L. Zhang, L. Zhong, Q. Yang and C. Li, Angew. Chem., Int. Ed., 2007, 46, 6861 CrossRef CAS.
- L. P. C. Nielsen, C. P. Stevenson, D. G. Blackmond and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 1360 CrossRef CAS.
- (a) J. M. Ready and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 2687 CrossRef CAS; (b) J. M. Ready and E. N. Jacobsen, Angew. Chem., Int. Ed., 2002, 41, 1374 CrossRef CAS; (c) M. A. Kwon and G. J. Kim, Catal. Today, 2003, 87, 145 CrossRef CAS; (d) M. Holbach and M. Weck, J. Org. Chem., 2006, 71, 1825 CrossRef CAS; (e) B. M. Rossbach, K. Leopold and R. Weberskirch, Angew. Chem., Int. Ed., 2006, 45, 1309 CrossRef CAS; (f) X. Zheng, C. W. Jones and M. Weck, J. Am. Chem. Soc., 2007, 129, 1105 CrossRef CAS; (g) X. Zheng, C. W. Jones and M. Weck, Adv. Synth. Catal., 2008, 350, 255 CrossRef CAS; (h) X. Zhu, K. Venkatasubbaiah, M. Weck and C. W. Jones, J. Mol. Catal. A: Chem., 2010, 329, 1 CrossRef CAS; (i) Y. Liu, J. Rawlston, A. T. Swann, T. Takatani, C. D. Sherrill, P. J. Ludovice and M. Weck, Chem. Sci., 2011, 2, 429 RSC.
- X. Hong, M. Mellah, F. Bordier, R. Guillot and E. Schulz, ChemCatChem, 2012, 4, 115 CrossRef.
- S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307 CrossRef CAS.
- M. E. Furrow, S. E. Schaus and E. N. Jacobsen, J. Org. Chem., 1998, 63, 6776 CrossRef CAS.
- M. Chahma, Synth. Met., 2005, 155, 474 CrossRef CAS.
- M. Okutan, Y. Yerli, S. E. San, F. Yilmaz, O. Günaydin and M. Durak, Synth. Met., 2007, 157, 368 CrossRef CAS.
- The quantity of oxidant has been optimized by preliminary studies. Less oxidant can also promote the polymerization, but with a poorer efficiency and a lower isolated yield.
- M. Pomerantz, J. J. Tseng, H. Zhu, S. J. Sproull, J. R. Reynolds, R. Uitz, H. J. Arnott and H. I. Haider, Synth. Met., 1991, 41–43, 825 CrossRef.
- The preparation of Poly-Co(III) has been described in ref. 20, and the oxidation state of the metal centers in this polymer has been verified by cyclic voltammetry studies.
- E. Vinck, D. N. Murphy, I. A. Fallis, R. R. Strevens and S. S. Doorslaer, Inorg. Chem., 2010, 49, 2083–2092 CrossRef CAS.
- See ESI† for details.
- The observation of two stretching vibration bands v(CN) for Co(III) complex 4 might originate from the dissymmetric effect of a strongly binding axial ligand. Similar phenomena have also been observed in the 1H NMR studies of this complex (see ESI† for analysis details). Also see ref: B. Liu, Y. Gao, X. Zhao, W. Yan and X. Wang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 359 CrossRef CAS.
- The polymers have been recovered by precipitation in MeOH in their dedoped forms, and the only possible location of the BF4 anions is on the metal centers.
- A control experiment showed that FeCl3 cannot oxidize Jacobsen's Co(II) complex to an active Co(III) complex under an argon atmosphere, which also confirmed our observation that metal centers in Poly-3a are still Co(II) species. The exact reason for the different oxidative behaviors of FeCl3 according to the atmosphere is still not clear. But as salen Co(II) complexes were known to be able to bind O2 in air (see E. C. Niederhoffer, J. H. Timmons, and A. E. Martell, Chem. Rev., 1984, 84, 137), one can imagine that O2 plays an important role in the oxidation of salen Co(II) complexes to Co(III) ones.
- It must be emphasized here that Poly-4a, which is produced easily with high yield through FeCl3 oxidation, is as efficient, in terms of both activity and enantioselectivity, as its electrogenerated analogue in the HKR of epibromohydrin. We indeed previously reported the formation of 3-bromo-propane-1,2-diol in 81% ee and 76% isolated yield by the catalysis with the electrochemically prepared insoluble catalytic species (see ref. 20).
- (a) G. Jas and A. Kirschning, Chem.–Eur. J., 2003, 9, 5708 CrossRef CAS; (b) W. Solodenko, H. Wen, S. Leue, F. Stuhlmann, G. Sourkouni-Argirusi, G. Jas, H. Schönfeld, U. Kunz and A. Kirschning, Eur. J. Org. Chem., 2004, 3601 CrossRef CAS; (c) A. Kirschning, W. Solodenko and K. Mennecke, Chem.–Eur. J., 2006, 12, 5972 CrossRef CAS.
- A. Kirschning, H. Monenschein and R. Wittenberg, Angew. Chem., Int. Ed., 2001, 40, 650 CrossRef CAS.
- It is also important to note that under these same diluted conditions, when the dynamic hydrolytic kinetic resolution of epibromohydrin was realized in a batch reactor, the conversion was less than 5% after 24 hours of reaction. The catalysis was thus significantly more efficient in the continuous flow procedure.
- G. J. Kim, H. Lee and S. J. Kim, Tetrahedron Lett., 2003, 44, 5005 CrossRef CAS.
- The efficiency of catalyst 4 for the HKR of these three other substrates, namely 2-phenoxymethyl-oxirane, 2-allyloxymethyl-oxirane and 2-phenyl-oxirane, was firstly evaluated under homogeneous conditions, see ref. 20: 2-phenoxymethyl-oxirane (16 h, 46% yield, 99% ee for the epoxide), 2-allyloxymethyl-oxirane (16 h, 47% yield, 98% ee for the epoxide), 2-phenyl-oxirane (72 h, 45% yield, 93% ee for the epoxide).
- The reaction mixture of an incomplete reaction catalyzed by Poly-4/6 was filtrated and the filtrate was further stirred for two days at room temperature, but no further evolution of the conversion was observed, proving that the extent of catalyst leaching between each reuse is negligible.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20644g |
| This journal is © The Royal Society of Chemistry 2013 |