NiO/Ce1−xNixO2−δ as an alternative to noble metal catalysts for CO oxidation†
Sk.
Mahammadunnisa
a,
P.
Manoj Kumar Reddy
a,
N.
Lingaiah
b and
Ch.
Subrahmanyam
*a
aEnergy and Environmental Research Laboratory, Department of Chemistry, Indian Institute of Technology Hyderabad, Andhra Pradesh, 502205, India. E-mail: csubbu@iith.ac.in; Tel: +91-40-2301-6050
bCatalysis Laboratory, I&PC Division, Indian Institute of Chemical Technology, Hyderabad, 500 007, India
First published on 30th October 2012
Abstract
A series of NiO/CeO2 catalysts were prepared by using the combustion method and characterized by using XRD, N2-physisorption, TEM, UV-visible, Raman, XPS, ICP-OES and H2-TPR. Typical results indicated the NiO/CeO2 had a better performance than CeO2 for CO oxidation and 15 wt% NiO/CeO2 is active and stable, whereas catalysts with a higher NiO content of 20 and 30 wt% deactivated at 130 °C onwards. The high activity and excellent stability of 15% NiO/CeO2 catalysts can be ascribed to their high oxygen vacancy, high dispersion of NiO and strong metal–support interaction that leads to synergy between NiO and CeO2.
Introduction
Catalytic oxidation of carbon monoxide (CO) has been receiving attention due to serious health effects associated with the exposure to CO.1 Traditionally, the CO oxidation reaction has been conventionally operated with noble metals (Au, Pd, Pt) supported on either reducible (CeO2, Fe2O3, MnO) or irreducible supports (Al2O3, SiO2) and several reports have highlighted the role of the oxide supports in providing adequate metal–support interaction. Among the reducible oxides, CeO2 due to high thermal stability and redox behavior has been widely used as an important support/catalyst in CO oxidation reactions. The other important parameters of CeO2 which directs the catalytic activity are surface area and oxygen vacancy.2–4 As the noble metals are expensive there is a need to develop low cost alternatives.5,6 Some of the recent studies revealed that the activity of ceria and ceria-containing catalysts in oxidation reactions is greatly enhanced by base metals.5–8Among them NiOx/CeO2 based catalysts found to be less expensive and show excellent catalytic activity for the CO oxidation reaction, CH4 decomposition etc.,9–11 Recently, Kang et al.9 and Marino et al.10 examined the CO oxidation over various CeO2 supported metal oxides such as cobalt, copper, manganese, chromium, iron, nickel and vanadium. Li et al.11 proposed that homogeneous doping of lower valent cations introduces elastic strain that results in extrinsic surface defects and higher performance in CO oxidation. The remarkable performances of Ni-Ceria based systems have been related to the ability of these oxides to show Ce3+/Ce4+ oxidation states, which facilitate the oxygen release and storage in the matrix. NiO/Ce1−xNixO2−δ can also provide strong metal–support interactions (SMSI) between NiO particle and CeO2, whereas CeO2 can modify the shape and electronic state of the metal.3–14 Combustion synthesis is an effective way to make CexM1–xO2−δ with a large variety of tunable parameters such as fuel, oxidant to fuel ratio etc., and has been used for the preparation of different ceramics and perovskites.15–18 When doping with Ni cations an increase in the concentration of oxygen vacancies in the ceria structure may be expected.4 This feature promotes the mobility of oxygen ions through the lattice oxide and provides the material with high oxygen ion conductivity.3
During the present study combustion synthesis was used to prepare a series of NiO/CeO2 catalysts aiming at low temperature CO oxidation. A systematic characterization was done and the resulting systems were tested for CO oxidation.
Experimental
Synthesis of the catalyst
CeO2 and a series of NiO/CeO2 samples were prepared by solution combustion synthesis using citric acid as the fuel. The NiO/CeO2 catalysts with different NiO weight ratios (0%, 5%, 10%, 15%, 20%, and 30%) were synthesized and denoted 0NiCe, 5NiCe, 10NiCe, 15NiCe, 20NiCe and 30NiCe, respectively. In a typical synthesis, appropriate amounts of (NH4)2Ce(NO3)6·6H2O, Ni(NO3)2·H2O and citric acid (oxidant/fuel = 1) were dissolved in a minimum amount of distilled water at room temperature and sonicated for 30 min and transferred into an alumina crucible. The alumina crucible was then placed in a preheated furnace maintained at ∼450 °C for 5 min. The solution undergoes dehydration and a spark appears at one corner, which spreads throughout the mass yielding a voluminous solid product with grey color. Prior to the activity test, the samples were calcined at 600 °C over 4 hours to remove the carbon content.The size and morphology of the nanoparticles were examined by using an FEI model TECNAI G 220 S-Twin TEM instrument. To investigate the surface enrichment, the as-prepared samples were immersed in 50% dilute nitric acid solution for 24 h, filtered, and washed with deionized water thoroughly to remove any dissolved nickel oxide in the sample. Inductively coupled plasma (ICP-OES on a Teledyne prodigy high dispersion) analysis was performed to identify the amount of Ni present on NiCe (5–30 wt%) catalysts before and after enrichment. Ultraviolet–visible (UV-vis) diffuse reflectance spectra in the wavelength range of 200–800 nm were recorded under ambient conditions using a Shimadzu (UV-3600) spectrophotometer equipped with an integrating sphere by using BaSO4 reference. Raman spectra of CeO2 and NiO/CexNi1–xO2−δ catalysts were recorded on a Bruker senterra dispersive Raman microscope with a laser excitation of wavelength of 532 nm.
X-ray Photoelectron Spectroscopy (XPS) data of the combustion synthesized catalysts were recorded by an Axis Ultra instrument under ultra-high vacuum conditions (<10–8 Torr) and using a monochromatic Al Kα X-ray source (1486.6 eV). Temperature Programmed Reduction (TPR), temperature programmed decomposition (TPD) and catalytic activity measurements were carried out in a flow system (Quantachrome autosorb-IQ automated gas sorption Analyzer) equipped with a thermal conductivity detector (TPR-TCD). The data were reconfirmed with an in-built mass spectrometer (RGA Prisma plus-200) controlled by the software Quadera and the second confirmation was done with a pre-calibrated ULTRAMAT 23 multi-gas Analyzer (SIEMENS) that measures CO, CO2 and O2. For TPR measurements, 50 mg of the sample was sandwiched between quartz wool plugs in a U-shaped quartz reactor and flushed with He for 30 min. The TPR profiles were obtained by heating the sample from room temperature to the desired temperature (600 °C) in 10% H2 in Ar (gas flow rates 40 ml min−1 and heating rate of 10 °C min−1) and the gaseous products were sampled through a fine control leak valve to TCD after passing through a cold tarp to remove H2O. Quantitative analysis was done by integrating the reduction signal and comparison was made by pre-calibrated signals.
| % of COconv = 100 × [COin − COout]/[COin] |
Where COin is the amount of the CO in the inlet and COout is that of the outlet.
Results and discussion
XRD
Fig. 1 presents the XRD patterns of the NiO/CexNi1–xO2−δ catalysts prepared by the solution combustion method. It can be seen in Fig. 1 that the diffraction peaks confirm the fluorite structure of CeO2.16–18,24 The absence of NiO diffraction lines for 5 to 15% CeNi catalysts indicates the isomorphic substitution and/or dispersion of NiO on the surface of ceria, whereas with increasing NiO content to 20, 30 wt% the presence of bulk NiO is seen from the characteristic peaks of NiO at 35.6 and 38.8° ascribed to bulk NiO.18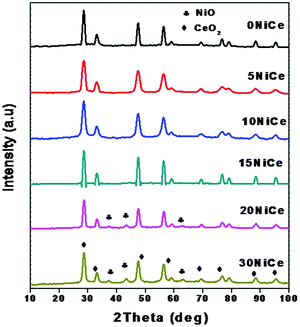 | ||
| Fig. 1 Powder XRD patterns showing the various wt% of Ni loaded ceria support and compared with bare CeO2. | ||
The Ni addition to the CeO2 structure decreased the lattice parameter, because Ni2+ (72 Å) has a smaller ion radius than Ce4+ (92 Å) and introduces additional lattice perturbation and structural stress and causes extrinsic defects both near grain boundaries and on the plane surface.24 From the lattice parameter of the mixed oxides one can determine the magnitude of the modification of the CeO2 structure by Ni2+ and the results are summarized in Table 1. As seen in Table 1 the cell parameter decreases with increasing NiO content up to 15 wt%, and at higher loadings, formation of bulk oxide may take place as indicated by the appearance of characteristic peaks of NiO in XRD (Fig. 1).
| Catalyst | Crystallite size (nm) | Lattice parameter (Å) | Surface area (m2 g−1) | |
|---|---|---|---|---|
| CeO2 | NiO | |||
| 0NiCe | 20 | — | 5.412 | 60 |
| 5NiCe | 15 | — | 5.405 | 50 |
| 10NiCe | 14 | — | 5.395 | 48 |
| 15NiCe | 10 | — | 5.390 | 47 |
| 20NiCe | 12 | 13 | 5.390 | 40 |
| 30NiCe | 12 | 13 | 5.390 | 30 |
The Ni addition also decreased the crystallite size of CeO2 suggesting dispersion of NiO.24 A detailed analysis of corresponding sizes for NiO and CeO2 calculated by using the Scherrer's formula is presented in Table 1. As seen in Table 1, the particle size of Ni is found to be 13 nm in 20NiCe and 30NiCe, whereas for CeO2 it decreases up to 15NiCe and increased further NiO loading.
Nitrogen adsorption
The surface area for the NiO/CexNi1–xO2−δ catalysts with various Ni loading is listed in Table 1. As seen in Table 1, NiCe samples exhibited specific surface area in the range of 30 to 50 m2 g−1 and the CeO2 support showed a slightly higher surface area than the doped samples. The decrease in the surface area for NiCe with Ni doping may be due to penetration of the dispersed metal oxides into the pores of the support and also owing to the formation of specific solid solutions as a result of interaction with the ceria.18 The extent of NiO crystallite growth is greater in the case of 20 and 30NiCe. This indicated that increasing NiO introduction into the CeO2 structure may accelerate the grain growth of ceria.18TEM
The morphology and particle size of the 15NiCe catalyst was measured by a high resolution TEM technique and typical pictures are shown in Fig. 2. Particles of approximately 10 nm of ceria were observed for the 15NiCe sample. The crystallite size observed by the TEM image is consistent with the results from XRD analysis (Table 1). In addition, nanoclusters with a size between 3 and 6 nm corresponding to NiO distributed on the surface of the ceria particles were also observed. The lattice spacing calculated from HRTEM was 0.310 nm and 0.241 nm coinciding with XRD observations of (111) facets of CeO2 fluorite and rock salt NiO crystals (shown in Fig. 2b).24 In Fig. 2b, many lattice line defects located in the crystalline structure of ceria (represented in squares) were observed.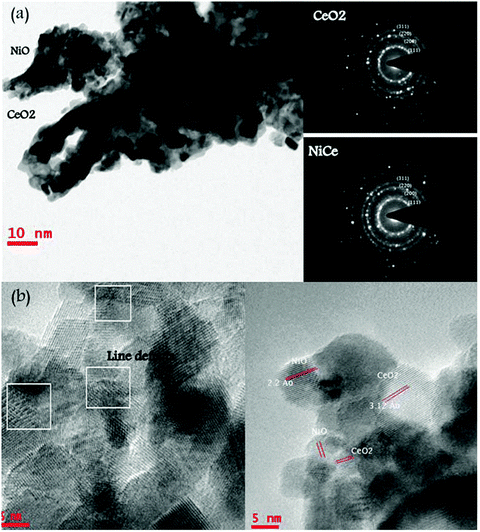 | ||
| Fig. 2 (a) TEM and selected area electron diffraction image (SAED) pattern of 15NiCe; (b) HR-TEM of 15NiCe along with many line defects observed. | ||
UV-visible spectra
The optical absorption spectrum of NiO/CexNi1–xO2−δ is shown in Fig. 3, which indicates the presence of O2− → Ce3+ charge transfer and inter band transition in the wavelength range of 260–350 nm.25–27 The peaks corresponding to 4f → 5d transitions of Ce3+ are observed for all NiO/CexNi1–xO2−δ catalysts between 220 and 320 nm. Also, a broad absorption edge in the 480–550 nm region due to O 2p → Ce 4f charge transfer transitions was also observed.28–30 Broadening of this band for 15NiCe may be assigned due to the increasing number of surface defects.25 The red shift for Ni doped ceria is due to surface oxygen vacancies. Hence, UV-Vis spectra of the synthesized catalysts confirmed the doping of Ni in the CeO2 matrix. Additionally, the weak d–d bands of octahedrally coordinated Ni2+ were also observed at 720 nm for 20, 30NiCe samples confirming the Ni2O3 bulk oxide species.17,18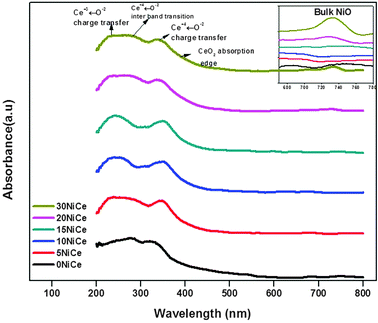
Raman spectra
Fig. 4 presents the Raman spectra of various NiO/CexNi1–xO2−δ catalysts along with the CeO2 support for comparison. Two distinct peaks at 460 and 588 cm−1 are observed for NiO/CexNi1–xO2−δ catalysts. The intense peak at ∼460 cm–1 was assigned to the F2g Raman active interior phonon mode of fluorite structure,27–29 whereas, the second band at ca. 588 cm−1 is widely reported to be due to the defect-induced D band that confirms oxygen vacancies.30 The formation of CexNi1−xO2−δ solid solution was confirmed by the shift of Raman peaks to lower wave numbers, as presented by the solid line. The peak area of the bands at 460 and 588 cm−1 was calculated as shown in Fig. 4 (denoted A1 and A2, respectively). The ratio of A2/A1(values included in Table 2) may be taken as the concentration of oxygen vacancies in the catalyst.23 It is seen from Table 2 that the A2/A1 value for the 15NiCe catalyst (1.5) is highest among the catalysts prepared. It is well-known that the introduction of metal cations to the CeO2 lattice may improve the oxygen vacancy.30 Thus, a higher A2/A1 value for the 15NiCe catalyst confirms the formation of the Ce1−x NixO2−δ solid solution.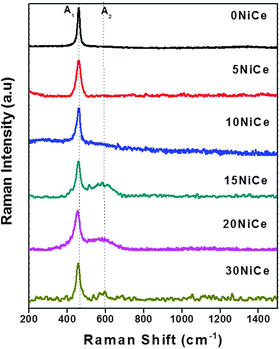 | ||
| Fig. 4 Raman spectra of CeO2 support and NiCe catalysts. | ||
| Catalyst | H2 consumption valuesa (μmole g−1) | NiO contentb (wt%) | (A2/A1)c | |||
|---|---|---|---|---|---|---|
| T 1 | T 2 | T 3 | Total | Free | ||
| a H2 consumption values calculated by integrating the reduction signal and comparison was made by pre-calibrated signals. b Quantified the amount of Ni present on NiCe (5–30 wt%) catalysts before and after enrichment. c The peak area of the bands at 460 (A1) and 588 (A2) cm−1 was calculated from Raman spectra. | ||||||
| 0NiCe | — | — | — | — | — | 0.1 |
| 5NiCe | 200 (218 °C) | 938 (379 °C) | — | 4.5 | 1 | 0.3 |
| 10NiCe | 250 (217 °C) | 1548 (381 °C) | — | 9.6 | 3 | 0.8 |
| 15NiCe | 586 (190 °C) | 1823 (402 °C) | — | 14.6 | 5 | 1.4 |
| 20NiCe | 340 (237 °C) | 3260 (380 °C) | 543 (500 °C) | 19 | 12 | 0.7 |
| 30NiCe | 225 (239 °C) | 3900 (376 °C) | 771 (501 °C) | 28 | 22 | 0.2 |
Temperature programmed reduction
The reducibility of the NiO/CexNi1–xO2−δ catalyst is studied by H2-TPR and the results are presented in Fig. 5. Typical results in the temperature range of 150 to 550 °C indicated three main reduction peaks (denoted T1, T2, and T3).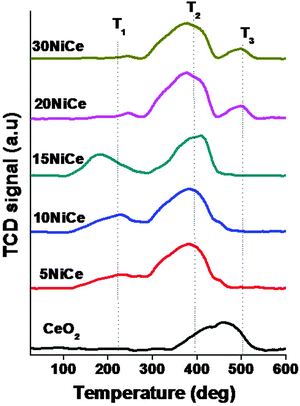
The peak at around 200 °C can be attributed to the replacement of Ce4+ by Ni2+ into the lattice of CeO2. Due to this charge imbalance lattice distortion would occur that leads to the formation of Ni–O–Ce solid solution.22 As a result, reactive oxygen species are generated that may be reduced easily at low temperatures.18–24 The second reduction peak (T2) between 350 and 410 °C can be attributed to the reduction of strongly interactive NiO species with CeO2 supports, whereas, the third reduction peak between 480 and 530 °C may be assigned to the high temperature reduction peak due to bulk NiO (observed only for 20NiCe, 30NiCe).18 The hydrogen consumption and Tmax during the TPR are given in Table 2. As seen in Table 2, for all the cases, the consumption of H2 was higher than the calculated value for the reduction of NiO to metallic Ni0 (based on the Ni amount calculated from ICP-OES). A similar behavior was observed in the literature, which was assigned due to surface reduction of the CeO2 particles along with NiO reduction.23,31–40 This fact is easily understood in terms of a “spillover effect” of hydrogen adsorbed on Ni particles to the CeO2 surface. The Tmax position of the T1 peak decreased up to 15 wt% and increases with increasing Ni loading. The T2 peak temperature and intensity of this peak increased with increasing nickel loading. As mentioned, the T3 peak was observed only for nickel loading higher than 15 wt%. As seen in Table 2, the highest H2 consumption is observed for 586 μmole g−1 for 15NiCe.
X-ray photo electron spectroscopy
The deconvoluted XPS spectra of Ce3d, Ni2p and O1s for 15%, 20% Ni catalysts are shown in Fig. 6a, b and c respectively. The Ce3d core level spectra for all the samples were deconvoluted into eight contributions, as shown in Fig. 5. The four main 3d5/2 features at 882.7, 884.1, 888.5, and 898.3 eV correspond to V, V′,V′′ and V′′′ components, while the 3d3/2 features at 901.3, 903.4, 907.3, and 916.9 eV correspond to U, U′,U′′ and U′′′ components, respectively.41 The presence of the fingerprint U′′′ satellite peak at about 919.9 eV confirms the Ce4+ oxidation state.42 The relative intensity of well separated U′′′ from the remaining signals is often used to assess the degree of reduction of Ce ions in the surface region.42 The appearance of characteristic bands of Ce3+ labeled V′ at 884.1 eV and U′ at 903.4 eV suggests that both oxidation states Ce4+ and Ce3+ co-exist on the surfaces of 15NiCe and 20NiCe samples.24 The presence of Ce3+ contributes to the interaction with surrounding NiO species. The relative amount of Ce3+ is high in the case of 15NiCe (30%) than 20NiCe(15%). The high Ce3+ content in 15NiCe may be due to the reduction of ceria enhanced by the synergic effect of nickel.18Fig. 6b shows Ni 2p3/2 for the 15, 20NiCe samples, having a main peak centered at 849.9 eV assigned to Ni2+ species. In addition, a broad peak at ∼861.5 eV which is assigned as the shake-up satellite peak of Ni2+ (NiO) and Ni3+ (Ni2O3), respectively, is observed for 20NiCe.43,44 The main XPS peak of Ni2+ centered at approximately 849.9 eV relatively higher BE corresponds to NiO (847.9 eV) representing the NiO intimately interacting with the CeO2 support consistent with TPR analysis.45 The XPS spectra of O1s for both catalysts are given in Fig. 6c. The main peak at 529.8 eV has been assigned to the lattice oxygen in ceria and nickel oxides (O2− species). The other peak at 531.5 eV has been assigned for “O− ion” deficiencies in the subsurface layer of metal oxides due to the formation of Ce1−xNixO2−δ solid solution.18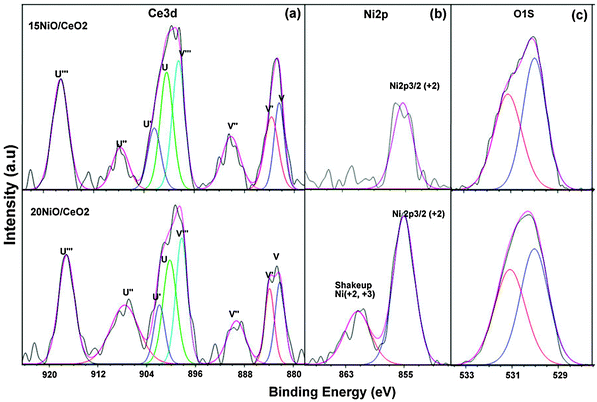 | ||
| Fig. 6 Ce 3d (a), Ni 2p (b), O 1s (c) XPS spectra of 20, 15NiCe catalysts. | ||
CO oxidation
The catalytic activity of various NiCe systems during CO oxidation is shown in Fig. 7a. Even though all the catalysts showed increasing activity with temperature, the promoting effect of NiO on CO oxidation is evident from the better activity of NiO/CeO2 than CeO2 alone. At 100 °C NiO/CeO2 showed close to 80% conversion against CeO2 (15% CO conversion). The enhanced CO activity of NiO/Ce1−xNixO2−δ may be attributed to the synergetic effect of NiO and CeO2. Ni doping may increase the concentration of oxygen vacancies in the ceria structure and promotes the migration of oxygen ions and provides the high oxygen ion conductivity.14 The catalytic performance of NiO/Ce1−xNixO2−δ is not only dependent on the reduction behavior of oxygen-ions of the support, but also depends on the number of surface active NiO centers.18 The superior catalytic activity was shown by 15 wt% NiO/Ce1−xNixO2−δ (50% conversion at 70 °C) due to the formation of NiO/Ce1−xNixO2−δ solid solution containing both free NiO species on the surface and in the form of Ce1−xNixO2−δ solid solution. The presence of mixed Ni–O–Ce–O bonds in such species would modify the properties of Ni–O–Ni groups present in NiO. The content of unexposed NiO in the NiO/Ce1−xNixO2−δ solid solution is much higher compared to that exposed on the surface in 15NiCe (ICP-OES data from Table 2). As can be seen from Fig. 7a, the activity of the catalysts increases with increasing Ni loading up to 15 wt% and with further increase in nickel (20, 30 wt%) loading, the activity decreases. It is reasonable to assume that the catalytic activity decreases with increasing bulk nickel oxide content for 20 and 30 wt% catalysts (as confirmed by XRD, XPS, and TPR). The T50% CO conversion for 15 wt% Ni loading catalyst is at 70 °C, whereas the same activity was achieved only at 160 °C, 110 °C, 120 °C and 130 °C for 5, 10, 20 and 30 wt% NiCe catalysts respectively. Also, as seen in Fig. 7a, the deactivation for 20 and 30% NiCe was faster when compared to other systems.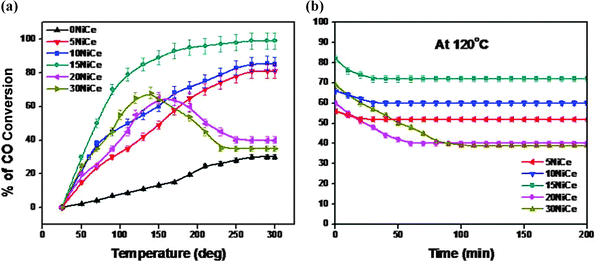 | ||
| Fig. 7 The catalytic activity (a) and stability (b) testing of the NiCe catalysts with different Ni loadings of 0, 5, 10, 15, 20 and 30 wt%. | ||
It is worth mentioning that XRD and TEM findings (Fig. 1 and 2) have also pointed out that the crystallite size of NiO was smaller on the 15NiCe catalyst. The dispersed NiO species on the CeO2 support may be due to strong metal–support interactions. The structural defects enhanced the catalytic properties for 15 wt% NiO/CeO2. Furthermore, as seen from Raman spectra of samples (Fig. 5), a maximum A2/A1 (1.5) seems for 15NiCe, suggesting that the introduction of 15 wt% Ni into the CeO2 lattice creates the highest oxygen vacancy when compared to both lower and higher Ni loading on CeO2. It may be concluded that the enrichment of oxygen vacancies may promote the activation of oxygen species from Ce1−xNixO2−δ to NiO surface and accelerate the reaction. The mixed oxide catalysts may have higher defect density than pure ceria. From TPR data, the highest H2 consumption of 539 μmole g−1 was observed for 15NiCe. On the other hand, the XPS data indicated the increase of the surface coverage with the increasing Ni loading. The relative intensity of the Ni 2p3/2 signal in XPS spectra was too low for 15NiCe than 20NiCe. The excess of the Ni loading above 15 wt% may not contribute to the activity due to the presence of octahedral coordination of Ni (Fig. 6b). In all the cases the long term performance of the catalysts was studied at 120 °C and the results are shown in Fig. 7b. As seen in Fig. 7(b), 5NiCe, 10NiCe and 15NiCe stabilizes within 50 min and shows a stable activity thereafter. Among the catalysts investigated, the catalyst 15NiCe exhibits the highest CO activity than other catalysts even after 200 min with a stable steady state conversion of about 70%. At the same time 20 and 30 NiO/CeO2 showed a relatively lower CO activity (stable steady state conversion of 51, 47% attained after 90 min). In order to understand the species responsible for the decrease in the activity, the spent catalysts were further investigated by XRD and temperature programmed decomposition up to 600 °C and the products were analyzed with an on-line mass spectrometer. No structural collapse or phase segregation has been observed in XRD (ESI-S1†). During the TPD process, CO2 is detected in the effluent indicating that the accumulation of carbonates on the catalyst surface may be one of the causes for the catalyst deactivation (ESI-S2†).
These results indicate that only a fraction of the doped Ni species in CeO2 takes part in the catalytic reaction. The synergy effect may be ascribed to the formation of NiOx species interacting with CeO2. Previous studies showed that at the high O2 (21 vol%) exposure, NiO (111) surfaces provide apex sites (the vertex) for the formation of Ni–O bonds prior to CO chemisorption. The incoming CO spontaneously reacts with the lattice apex ‘O’ to give CO246,47 and the desorption of CO2 on the defective NiO(111) is favorable. The apex oxygen vacancy created by the release of CO2 will be filled by the ‘O’ migration from the Ce1−xNixO2−δ solid solution.
Conclusion
A series of NiO/CeO2 with varying Ni loadings was prepared and characterized. XRD confirmed the dispersion of NiO up to 15 wt%, whereas higher NiO loadings result in bulk oxide formation. Experimental results during CO oxidation indicated that the Ni doping into CeO2 has a significant effect on the catalytic properties of NiO/Ce1−xNixO2−δ catalysts. Increasing Ni loading can affect both catalyst particle sizes and metal–support interaction, which are among the most important factors for CO oxidation. At 15 wt% Ni relatively small particle sizes with high oxygen vacancies may be created and increasing the NiO loading beyond 15% led to bulk NiO formation. From the catalytic activity data, 15 wt% NiO/CeO2 shows the best activity due to highly dispersed Ni2+or NiOx species.Acknowledgements
MU would like to thank the Council of Scientific & Industrial Research (CSIR), New Delhi, for the award of Junior Research Fellowship.References
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935 CrossRef CAS.
- K. Takehira, T. Shishido, P. Wang, T. Kosaka and K. Takaki, J. Catal., 2004, 221, 43–54 CrossRef CAS.
- V. M. Gonzalez-DelaCruz, J. P. Holgado, R. Pereñíguez and A. Caballero, J. Catal., 2008, 257, 307–314 CrossRef CAS.
- C. Courson, L. Udron, D. Świerczyński, C. Petit and A. Kiennemann, Catal. Today, 2002, 76, 75–86 CrossRef CAS.
- G.-C. Wang, J. Jiao and X.-H. Bu, J. Phys. Chem., 2007, 111, 12335–12339 CAS.
- B. M. Reddy, P. Bharali, P. Saikia, G. Thrimurthulu, Y. Yamada and T. Kobayashi, Ind. Eng. Chem. Res., 2008, 48, 453–462 CrossRef.
- C. K. Costello, J. H. Yang, H. Y. Law, Y. Wang, J. N. Lin, L. D. Marks, M. C. Kung and H. H. Kung, Appl. Catal., A, 2003, 243, 15 CrossRef CAS.
- C. E. Taylor, B. H. Howard and C. R. Myers, Ind. Eng. Chem. Res., 2007, 46, 8906–8909 CrossRef CAS.
- M. Kang, M. W. Song and C. H. Lee, Appl. Catal., A, 2003, 251, 143–156 CrossRef CAS.
- F. Marino, C. Descorme and D. Duprez, Appl. Catal., B, 2005, 58, 175–183 CrossRef CAS.
- T. Li, G. Xiang, J. Zhuang and X. Wang, Chem. Commun., 2011, 47, 6060–6062 RSC.
- F. Ocampo, B. Louis and A. Roger, Appl. Catal., A, 2009, 369, 90–96 CrossRef CAS.
- F. Ocampo, J. A. Cunha, M. R. de Lima Santos, J. P. Tessonnier, M. M. Pereira and B. Louis, Appl. Catal., A, 2010, 390, 102–109 CrossRef CAS.
- S. T. Aruna and K. C. Patil, Nanostruct. Mater., 1998, 10, 955–964 CrossRef CAS.
- X. Guo, P. S. Devi, B. G. Ravi, J. B. Parise, S. Sampath and J. C. Hanson, J. Mater. Chem., 2004, 14, 1288–1292 RSC.
- V. M. Gonzalez-Delacruz, F. Ternero, R. Pereñíguez, A. Caballero and J. P. Holgado, Appl. Catal., A, 2010, 384, 1–9 CrossRef CAS.
- J. Xu, B. Xue, Y.-M. Liu, Y.-X. Li, Y. Cao and K.-N. Fan, Appl. Catal., A, 2011, 405, 142–148 CrossRef CAS.
- P. V. R. Rao, V. P. Kumar, G. S. Rao and K. V. R. Chary, Catal. Sci. Technol., 2012, 2, 1665–1673 CAS.
- M. Araquea, L. M. Martínez, T. J. C. Vargas, M. A. Centeno and A. C. Roger, Appl. Catal., B, 2012, 125, 556–566 CrossRef.
- M. Araquea, L. M. Martínez, T. J. C. Vargas and A. C. Roger, Catal. Today, 2011, 176, 352–356 CrossRef.
- B. Koubaissy, A. Pietraszek, A. C. Roger and A. Kiennemann, Catal. Today, 2010, 157, 436–439 CrossRef CAS.
- B. Choudhury and A. Choudhury, Mater. Chem. Phys., 2012, 131, 666–671 CrossRef CAS.
- L. Meng, A.-P. Jia, J.-Q. Lu, L.-F. Luo, W.-X. Huang and M.-F. Luo, J. Phys. Chem. C, 2011, 115, 19789–19796 CAS.
- R. K. Pati, I. C. Lee, S. Hou, O. Akhuemonkhan, K. J. Gaskell, Q. Wang, A. I. Frenkel, D. Chu, L. G. Salamanca-Riba and S. H. Ehrman, ACS Appl. Mater. Interfaces, 2009, 1, 2624–2635 CAS.
- A. Bensalem, J. C. Muller and F. Bozon-Verduraz, J. Chem. Soc., Faraday Trans., 1992, 88, 153–154 RSC.
- R. Li, S. Yabe, M. Yamashita, S. Momose, S. Yoshida, S. Yin and T. Sato, Mater. Chem. Phys., 2002, 75, 39–44 CrossRef CAS.
- V. Sánchez Escribano, E. Fernández López, M. Panizza, C. Resini, J. M. Gallardo Amores and G. Busca, Solid State Sci., 2003, 5, 1369–1376 CrossRef.
- E. Kis, R. M. Neducin, G. Lomic, G. Boskovic, D. Z. Obadovic, J. Kiurski and P. Putanov, Polyhedron, 1998, 17, 27 CrossRef CAS.
- V. Rives and S. Kannan, J. Mater. Chem., 2000, 10, 489 RSC.
- Z.-Y. Pu, J.-Q. Lu, M.-F. Luo and Y.-L. Xie, J. Phys. Chem. C, 2007, 111, 18695–18702 CAS.
- B. M. Reddy, L. Katta and G. Thrimurthulu, Chem. Mater., 2009, 22, 467–475 CrossRef.
- S. Sato, R. Takahashi, T. Sodesawa and N. Yamamoto, Catal. Commun., 2004, 5, 397–400 CrossRef CAS.
- L. A. Bruce, M. Hoang, A. E. Hughes and T. W. Turney, Appl. Catal., A, 1996, 134, 351–362 CrossRef CAS.
- P. Zimmer, A. Tschöpe and R. Birringer, J. Catal., 2002, 205, 339–345 CrossRef CAS.
- H. C. Yao and Y. F. Y. Yao, J. Catal., 1984, 86, 254–265 CrossRef CAS.
- H. Zhu, Z. Qin, W. Shan, W. Shen and J. Wang, J. Catal., 2004, 225, 267 CrossRef CAS.
- R. Pérez-Hernández, A. Gutiérrez-Martínez, J. Palacios, M. Vega-Hernández and V. Rodríguez-Lugo, Int. J. Hydrogen Energy, 2011, 36, 6601–6608 CrossRef.
- I. Kosacki, T. Suzuki, H. U. Anderson and P. Colomban, Solid State Ionics, 2002, 149, 99–105 CAS.
- S. Tsunekawa, T. Fukuda and A. Kasuya, Surf. Sci., 2000, 457, L437–L440 CrossRef CAS.
- J. P. Holgado and G. Munuera, Stud. Surf. Sci. Catal., 1995, 96, 109 CrossRef CAS.
- J. Jia, J. N. Kondo, K. Domen and K. Tamaru, J. Phys. Chem. B, 2001, 105, 3017–3022 CrossRef CAS.
- G. Samjeské, K.-i. Komatsu and M. Osawa, J. Phys. Chem. C, 2009, 113, 10222–10228 Search PubMed.
- S. A. Krasnikov, A. B. Preobrajenski, T. Chasse and R. Szargan, Thin Solid Films, 2003, 428, 201–205 CrossRef CAS.
- D. Srinivas, C. V. V. Satyanarayana, H. S. Potdar and P. Ratnasamy, Appl. Catal., A, 2003, 246, 323–334 CrossRef CAS.
- M. M. Barroso-Quiroga and A. E. Casto-Luna, Int. J. Hydrogen Energy, 2010, 35, 6052–6056 CrossRef CAS.
- X. Wang, M. Shen, J. Wang and S. Fabris, J. Phys. Chem. C, 2010, 114, 10221–10228 CAS.
- G. Peng, L. R. Merte, J. Knudsen, R. T. Vang, E. Lægsgaard, F. Besenbacher and M. Mavrikakis, J. Phys. Chem. C, 2010, 114, 21579–21584 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20641b |
| This journal is © The Royal Society of Chemistry 2013 |