Molecular ruthenium complexes anchored on magnetic nanoparticles that act as powerful and magnetically recyclable stereospecific epoxidation catalysts†
Lydia
Vaquer
a,
Paola
Riente
a,
Xavier
Sala
b,
Susanna
Jansat‡
a,
Jordi
Benet-Buchholz
a,
Antoni
Llobet
*ab and
Miquel A.
Pericàs
*ac
aInstitute of Chemical Research of Catalonia, Av. Països Catalans, 16, 43007 Tarragona, Spain. E-mail: mapericas@iciq.es; allobet@iciq.es; Fax: +34 977 920 222; Tel: +34 977 920 201
bDepartment de Química, Universitat Autònoma de Barcelona, Cerdanyola del Vallès, E-08193, Barcelona, Spain
cDepartment de Química Orgánica, Universitat de Barcelona, c/Martí I Franqués 1-11, 08080, Barcelona, Spain
First published on 6th November 2012
Abstract
Two new Ru-aqua complexes containing a phosphonated trpy ligand with the general formula [Ru(trpy-P)(B)(H2O)]2+ (trpy-P is diethyl [2,2′:6′,2′′-terpyridin]-4′-ylphosphonate (1); B = bpm (5a) is 2,2′-bipyrimidine; (or) B = azpy (cis- and trans-5b) is 2-phenylazopyridine) are reported. These complexes and their synthetic intermediates have been characterized by the usual analytic techniques and by spectroscopic and electrochemical methods. X-ray structures of the cis and trans Ru–Cl complexes that are synthetic intermediates to the corresponding Ru-aqua complexes have also been obtained. The phosphonated complexes 4a and trans-4b have been anchored onto MNPs of Fe3O4 (6.2 ± 2 nm), via covalent bonds to generate 6a and trans-6b, respectively. These new materials have been characterized by UV-vis, IR, TEM and CV. The Ru-aqua complexes were generated in situ by dissolving the Ru–Cl complexes in water. The Ru-aqua complexes 5a and trans-5b are excellent catalysts for the epoxidation of olefins and are stereoselective for cis-alkenes such as cis-β-methylstyrene and cis-stilbene. Remarkably, the analog supported onto MNPs, 7a, displays practically the same behavior as its homogeneous counterpart. Multiple recycling experiments for the epoxidation of cis-β-methylstyrene involving magnetic decantation of the catalyst were carried out with 7a without significant loss of activity.
Introduction
In recent years magnetic nanoparticles (MNPs) have been gaining increasing interest due to their valuable intrinsic properties such as high surface area, low toxicity and superparamagnetism,1 the latter allowing an easy separation using an external magnetic field. In addition, the presence of a large number of hydroxyl groups on their surface allows the immobilization of a variety of catalysts via covalent bonds.2 Because of the combination of these properties MNPs have become a very attractive material to support molecules whose homogeneous counterparts display catalytic activity.3 Following this strategy a significant number of modified MNPs have been prepared that display high catalytic activity and that can be easily recovered and recycled. Examples of catalytic reactions performed using this strategy include acylations,4 Michael additions,5,6 asymmetric hydrogenations,7,8 epoxidations,9 C–C coupling reactions,10 reduction of epoxy ketones11 and hydroformylation12 among others.3Olefin epoxidation is a key chemical transformation for the synthesis of highly functionalized organic species13 and has been extensively developed in the homogeneous phase using transition metal complexes as catalysts.14 However it is surprising that very few examples have been described in the literature using MNPs. For this purpose two radically different strategies have been used: the first one involves the use of oxides as catalysts such as Ag–Fe3O4 nanocomposites15 or Mo oxides,16 whereas the second one involves the use of well-defined molecular Mo catalysts supported onto MNPs.
With the aim to further develop the field of epoxidation catalysis using MNPs as a support, we have prepared a series of MNPs containing well-defined molecular Ru complexes via a phosphonic acid anchoring group. The latter has been chosen because of its high chemical stability and also its high affinity for oxide surfaces forming strong covalent bonds.17 We have also chosen to anchor well-defined metal complexes instead of simply oxides, because the former have a much higher potential to control stereospecific reactions through the ligand design tool kit.
The chemistry of the polypyridyl RuII–OH2/RuIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group has been extensively developed over the past 30 years.18–20 Ru(IV)O higher oxidation state complexes can be easily reached through sequential proton coupled electron transfer type of reactions from the initial RuII–OH2 complex. Once generated, the Ru(IV)O complex is a powerful oxidant that is capable of carrying out a range of oxidative transformations including the olefin epoxidation reaction.21,22 In this scenario, the auxiliary polypyridyl ligand/s that complete the normally octahedral type of coordination around the Ru metal center allow fine tuning of the reactivity of the Ru(IV)O group both from a steric perspective and also from an electronic viewpoint. For the latter how the auxiliary ligands can control the relative stability of the different oxidation states has been shown. In particular, a few examples have been described where the electronic perturbation exerted from the auxiliary ligands to the metal center provokes the instability of Ru oxidation state III toward disproportion to II and IV.23–25 This is particularly relevant in the epoxidation reactions since one electron transfer type of oxidations will lead to radical reactions whereas a direct two electron transfer will lead to concerted epoxidation reactions and thus stereoselective epoxidations.
O group has been extensively developed over the past 30 years.18–20 Ru(IV)O higher oxidation state complexes can be easily reached through sequential proton coupled electron transfer type of reactions from the initial RuII–OH2 complex. Once generated, the Ru(IV)O complex is a powerful oxidant that is capable of carrying out a range of oxidative transformations including the olefin epoxidation reaction.21,22 In this scenario, the auxiliary polypyridyl ligand/s that complete the normally octahedral type of coordination around the Ru metal center allow fine tuning of the reactivity of the Ru(IV)O group both from a steric perspective and also from an electronic viewpoint. For the latter how the auxiliary ligands can control the relative stability of the different oxidation states has been shown. In particular, a few examples have been described where the electronic perturbation exerted from the auxiliary ligands to the metal center provokes the instability of Ru oxidation state III toward disproportion to II and IV.23–25 This is particularly relevant in the epoxidation reactions since one electron transfer type of oxidations will lead to radical reactions whereas a direct two electron transfer will lead to concerted epoxidation reactions and thus stereoselective epoxidations.
In the present paper we present the anchoring of two Ru-aqua complexes, whose Ru(IV)O species behave as direct two electron donors, onto well-defined MNPs in order to benefit from the properties of both types of compounds in a single material. Additionally, we also report their activity as catalysts for the olefin epoxidation reaction.
Synthesis and structure
The synthetic pathway followed for the immobilization of the ruthenium complexes is shown in Scheme 1. The phosphonate-functionalized terpyridine (1 or trpy-P) was coordinated to ruthenium using RuCl3 as a metal precursor in refluxing ethanol for four hours. The octahedral complex [RuCl3(trpy-P)], 2, precipitates as a brown solid in high yield (93%) and was used as a starting material for subsequent ligand exchange. Complex 2 was characterized by the usual analytic and spectroscopic techniques together with cyclic voltammetry (see ESI†). Subsequently 2 was treated with the bidentate ligand, 2,2′-bipyrimidine (bpm) or 2-phenylazopyridine (azpy), in the presence of triethylamine. This produced the substitution of two chloro ligands by the bidentate ligand together with the reduction of Ru(III) to Ru(II), forming complexes [RuCl(bpm)(trpy-P)]+, 3a, and cis- and trans-[RuCl(azpy)(trpy-P)]+, cis- and trans-3b. The latter generates two isomers due to the non-symmetric nature of the azpy ligand. Cis and trans here refer to the position of the coordinating pyridyl N atom from azpy with regard to the central N of the trpy ligand. Fractional recrystallization of the mixture in CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Et2O allows obtaining trans-3b in 97% purity. The X-ray crystal structure of both Ru-chloro isomers cis-3b and trans-3b is depicted in Fig. 1. Both complexes present a distorted octahedral geometry around the Ru metal center. The Ru–Cl distances for cis-3b and trans-3b differ by less than 0.01 Å thus indicating a similar trans influence by either the N-aza group or the N-pyridyl group. The rest of the bond distances and angles are unremarkable and comparable with related Ru complexes previously described in the literature.26–30
:Et2O allows obtaining trans-3b in 97% purity. The X-ray crystal structure of both Ru-chloro isomers cis-3b and trans-3b is depicted in Fig. 1. Both complexes present a distorted octahedral geometry around the Ru metal center. The Ru–Cl distances for cis-3b and trans-3b differ by less than 0.01 Å thus indicating a similar trans influence by either the N-aza group or the N-pyridyl group. The rest of the bond distances and angles are unremarkable and comparable with related Ru complexes previously described in the literature.26–30
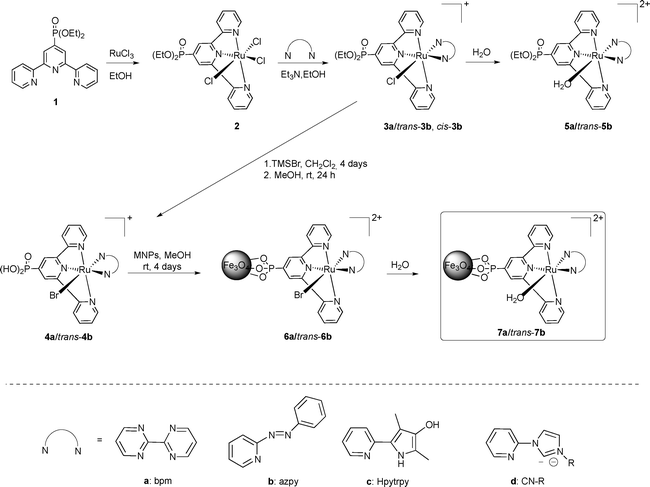 | ||
| Scheme 1 (top) Synthetic strategy for the preparation of Ru complexes and their immobilization on MNPs. (bottom) Bidentate N–N ligands used and discussed in this work. | ||
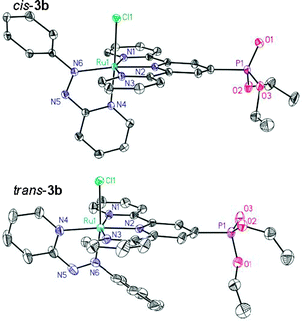 | ||
| Fig. 1 Ortep drawing (30 and 50% of probability, respectively) for the cationic moieties of the X-ray structures of cis-3b and trans-3b. Hydrogen atoms have been omitted for the sake of clarity. | ||
A complete set of 1D and 2D-NMR spectra was recorded for complexes 3a and trans-3b and is reported in Fig. 2 and in the ESI.†Fig. 2 presents the aromatic region of the 1H-NMR of 3a and trans-3b together with their assignment. The most interesting resonance in the spectra is that of H16 because it is substantially deshielded by the chloro ligand and end up appearing above 10 ppm. In the particular case of the 3b isomers, due to the geometry of the azpy ligand, only the trans-3b presents this interaction and thus can be used to identify it.
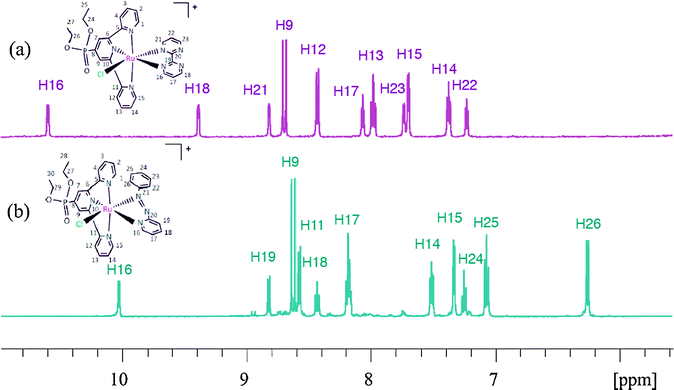 | ||
| Fig. 2 Aromatic region of the 1H-NMR spectra of (a) 3a and (b) trans-3b registered in 400 MHz NMR using CD2Cl2. | ||
The electrochemical properties of complexes 3a and trans-3b were studied by cyclic voltammetry. The stronger π-acceptor character of the azpy ligand with regard to that of bpm is manifested in the 270 mV anodic shift for the Ru(III/II) redox potential of trans-3b with regard to that of 3a (1.37 V for trans-3b and 1.10 V for 3a; redox potentials are reported vs. SSCE). Dissolving 3a or trans-3b in water induces the substitution of the Ru–Cl ligand generating the corresponding Ru-aqua complex, 5a or trans-5b, instantaneously as detected by the MLCT shifts in the UV-vis spectra (see ESI†).
The deprotection of the phosphonate group of 3a and trans-3b was carried out using an excess of trimethylbromosilane (TMSBr) in dry CH2Cl2 and also involved the substitution of the chloro ligand by the bromo. After solvent removal the resulting silane derivative could be easily hydrolyzed in methanol at room temperature (Scheme 1) generating complexes 4a and trans-4b in excellent yields. These complexes were then ready to be anchored onto MNPs. The MNPs of Fe3O4 were prepared by thermal decomposition of iron(III) acetylacetonate in the presence of oleylamine and oleic acid as surfactants.31,32 Complexes 4a and trans-4b were anchored by dissolving them in MeOH and adding the MNPs. The resulting mixture is stirred at room temperature for 4 days. This generates the materials 6a and trans-6b, that, upon addition of water in the reaction media, undergo Br/H2O ligand exchange and generate in situ the active catalysts 7a and trans-7b. Transmission electron microscopy (TEM) micrographs of the bare MNPs showed spherical and monodisperse nanoparticles (6.2 ± 2 nm) (Fig. 3). Very interestingly, the new materials 6a and trans-6b with the anchored catalyst precursors did not lead to a significant increase in the size distribution or to agglomeration of the nanoparticles. For 6a the loading of the catalyst onto the surface of the MNPs was 0.61 mmol g−1 as determined by nitrogen analysis. In the case of trans-6b the catalyst loading was 0.23 mmol g−1. The anchoring of the catalyst onto the MNPs surface was followed by FT-IR (See ESI†) and also confirmed by UV-Vis and cyclic voltammetry. The high solubility of the resulting functionalized MNPs in methanol enabled their characterization by UV-Vis spectroscopy, where the characteristic metal-to-ligand charge transfer (MLCT) bands at around 500 nm were observed. The UV-Vis spectra of the homogeneous and heterogeneous systems were compared, confirming the effective immobilization of 4a on the MNPs surface (Fig. 4). Similar to the homogenous system, the active Ru-aqua catalyst 7a and trans-7b can be obtained from the corresponding Ru–Cl precursors 6a and trans-6b respectively, upon dissolving them in water. This transformation can be observed by UV-vis spectroscopy where a hypsochromic shift of the MLCT bands from 520 to 480 nm is observed when the Ru–Cl bond is replaced by a Ru–aqua bond (Fig. 4). This phenomenon is due to the relative destabilization of dπ(Ru) levels provoked by the chloro ligand with regard to the aqua.33
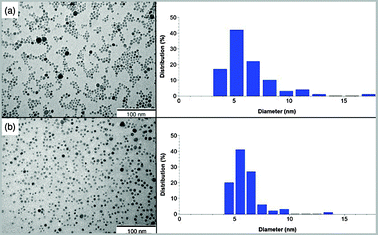 | ||
| Fig. 3 TEM micrographs and size distribution of (a) MNPs and (b) 6a. | ||
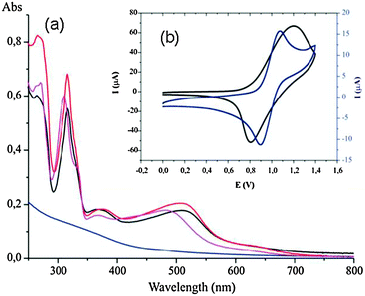 | ||
| Fig. 4 (a) UV-Vis spectra of 3a in MeOH (red), 6a in MeOH (black), 7a in H2O (purple) and MNPs in hexane (blue). (b) Cyclic voltammograms of 3a (blue) and the immobilized Ru complex 6a (black). | ||
The electrochemical properties of the functionalized MNPs were investigated using cyclic voltammetry techniques. For this purpose a methanolic solution of 6a was deposited on the surface of the electrode. After solvent evaporation a film of the MNPs 6a was strongly absorbed at the graphite surface. The cyclic voltammogram obtained for 6a under these conditions is depicted in the inset of Fig. 4. A quasi-reversible wave at E1/2 = 1.0 V vs. SSCE (Ep,a = 1.20 V, Ep,c = 0.80 V, ΔE = 400 mV) is observed which is assigned to the Ru(III)/Ru(II) redox couple. This value is in very good agreement with the corresponding E1/2 = 0.98 V (Ep,a = 1.07 V, Ep,c = 0.90 V, ΔE = 170 mV) obtained for the related complex 3a in the homogeneous phase.
The cyclic voltammetry of the aqua complex 5a was registered in H2O (pH 7) and a single wave at 0.66 V was observed, which corresponds to the 2-electron transition of Ru(IV/II). This is in concordance with the electrochemical properties observed for the analogous non-functionalized complex [Ru(trpy)(bpm)(H2O)]2+.34 Similarly complex trans-5b also shows a single two electron wave at 0.78 V that is also in agreement with its analogous non-functionalized complex [Ru(trpy)(azpy)(H2O)]2+ and also in agreement with the stronger π-acceptor capacity of azpy with regard to bpm.35
Catalytic epoxidation of alkenes
The catalytic activity of the ruthenium aqua complexes described in this work was investigated for the epoxidation of a diversity of alkenes in homogeneous and heterogeneous phases given the interest of the epoxidation reaction in both bulk and fine chemicals that use them as starting materials for a variety of reactions.36–42 The results obtained are reported in Tables 1 and 2 and in Fig. 5 and 6.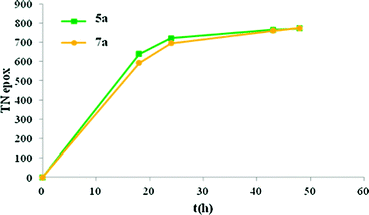 | ||
| Fig. 5 Conversion vs. time plot for the epoxidation of cis-β-methylstyrene using catalysts 5a and 7a. Reaction conditions: catalysts, 2.34 mM/substrate, 1.282 M/PhI(OAc)2 1.923 M/H2O, 1.923 M in CH2Cl2 up to a total volume of 1.95 mL at 25 ± 1 °C. Dodecane (1.923 M) was added as an internal standard. | ||
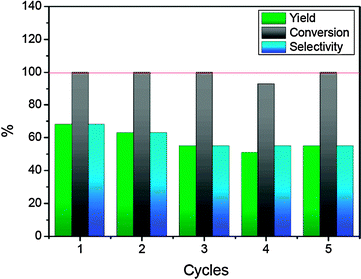 | ||
| Fig. 6 Recycling and reuse of 7a in the epoxidation of cis-β-methylstyrene. Reaction conditions: 7a, 2.34 mM/substrate, 0.234 M/PhI(OAc)2, 0.468 M/H2O, 0.468 M in CH2Cl2 up to a total volume of 1.07 mL at 25.0 ± 1 °C. Dodecane (0.468 M) was added as an internal standard. | ||
| Entry | Catalyst | Epoxide Yieldb (%) | Conv.c (%) | E 1/2 IV/II |
|---|---|---|---|---|
| a Catalyst, 2.34 mM/substrate, 0.234 M/PhI(OAc)2, 0.468 M/H2O, 0.468 M in CH2Cl2 up to a total volume of 1.07 mL at 25 ± 1 °C. Dodecane 0.468 M was added as an internal standard. b Yield of epoxide with regard to total conversion. c Substrate conversion. d trans-5c is trans-[Ru(trpy)(CN-Me)(H2O)]2+.54 e trans-5d is trans-[Ru(trpy)(pyrpy-OH)(H2O)]+.21 f cis- and trans-5e is [Ru(CNC)(CN-nBu)(H2O)]2+; H2CNCBr2 is 1,1′-(pyridine-2,6-diyl)bis(3-butyl-1H-imidazol-3-ium) bromide.25,55 | ||||
| 1 | Trans-5b | 52 | 100 | 0.78 |
| 2 | 5a | 68 | 100 | 0.66 |
| 3 |
Trans-5cd54 |
94 | 81 | 0.58 |
| 4 |
Trans-5de21 |
69 | 55 | 0.55 |
| 5 |
Trans-5ef25 |
100 | 50 | 0.52 |
| 6 |
Cis-5ef25 |
100 | 40 | 0.42 |
| 7 | Trans-7b | 52 | 100 | — |
| 8 | 7a | 71 | 100 | — |
| 9 | MNPs | — | — | — |
| Entry | Alkene | Epoxide Yieldb (%) | Conversionc (%) | Selectivityd (%) |
|---|---|---|---|---|
| a 7a, 2.34 mM/substrate, 0.234 M/PhI(OAc)2, 0.468 M/H2O, 0.468 M in CH2Cl2 up to a total volume of 1.07 mL at 25.0 ± 1 °C. Dodecane 0.468 M was added as an internal standard. b Yield of epoxide with regard to total conversion. c Substrate conversion. d Chemoselectivity towards epoxide. e Only cis-epoxide was detected. | ||||
| 1 |
|
71 | 100 | 71 |
| 2 |

|
75 | 100 | 75 |
| 3 |
|
80 | 100 | 80 |
| 4 |
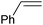
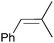
|
50 | 90 | 56 |
| 5 |
|
40 | 90 | 44 |
| 6 |

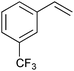
|
42e | 100 | 42 |
| 7 |
|
40 | 100 | 40 |
| 8 |
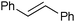
|
30 | 60 | 50 |
| 9 |
|
90 | 100 | 90 |

Within the epoxidation area a particularly interesting reaction is the stereoselective epoxidation of cis-alkenes.22,40,43–51 This is an important issue specially because the well known and efficient Jacobsen's Mn–salen complexes generally give mixtures of cis- and trans-epoxides.52,53
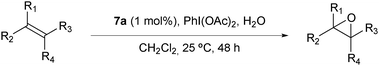
Several pathways have been proposed to operate in the oxo transfer reaction from an MO species to the alkene involving a concerted process, carbon radical intermediates, carbocation intermediates, π-radical intermediates and metalaoxetanes.40,49 Among these pathways radical intermediates are expected to give mixtures of cis- and trans-epoxides whereas the concerted process guarantees a stereoselective epoxidation reaction. The concerted process involves an oxygen atom transfer from the MO complex that is formally related to a two electron process. Therefore Ru-aqua complexes whose oxidation state III is unstable with regard to disproportionation25 are ideal candidates for the stereoselective epoxidation reaction. Indeed we and others have previously shown the success of using such a strategy.21,25,54 In all these cases the ligands used besides generating the instability of the Ru(III) oxidation state were also strong sigma-donor ligands. In the present paper we extend the number of this type of complexes but now using strong electron withdrawing ligands like the bpm and azpy. As a consequence of this electronic effect induced by the ligand, the Ru(VI)/Ru(II) redox potential increases substantially (see Table 1) than in the previous examples. Therefore the thermodynamic driving force increases accordingly.
cis-β-Methylstyrene was the substrate we have chosen to test the performance of the catalysts described in the present work. Table 1 shows the epoxidation results obtained in the homogeneous phase. As an example (entry 1, Table 1), the system trans-5b, 2.34 mM/cis-β-methylstyrene, 0.234 M/H2O, 0.468 M/PhI(OAc)2 0.468 M in CH2Cl2 with a total volume of 1.07 mL at 25 ± 1 °C yields 122 mM cis-epoxide with no detectable amount of trans-epoxide and with 100% conversion of the initial substrate. Under similar conditions (entry 2, Table 1) 5a gives 159 mM cis-epoxide also stereoselectively. Thus complexes 5a and trans-5b add to the list of two-electron Ru-aqua type of complexes capable of carrying out stereoselective epoxidation of cis-alkenes. Entries 3–7 of Table 1 present the performance of the above-mentioned two-electron type of Ru-aqua complexes that contain strong σ-donating type of bidentate ligands. An interesting trend is observed in the Table: as the Eo(IV/II) redox potential increases, the epoxide yield decreases but the substrate conversion increases. This can be interpreted in the sense that as the redox potential increases the thermodynamic driving force for the oxidation also increases and thus it becomes a much powerful catalyst that is capable of reaching 100% conversion in entries 1 and 2. However this higher oxidation power can also enhance oxidations involving C–C scission and secondary oxidation reactions. The nature of these compounds has not been pursued in the present work. This hypothesis is further supported by the trans nature of all the complexes compared in Table 1 (except for cis-5f). In all of these cases the Ru–N bond of the pyridyl moiety of the bidentate ligand is situated in the equatorial plane perpendicular to the Ru–OH2 group. On the other hand the other moiety of the bidentate ligand is situated trans with regard to the Ru-aqua group and thus no steric effects are exerted by these ligands to this bond (see Fig. 1). Therefore for complexes 5a and trans-5b–e, the steric interaction between the reactive RuO group and the alkene substrate is basically identical and thus the nature of the different reactivity will be entirely electronic. We then proceeded to evaluate the performance of these complexes in the heterogeneous phase anchored on MNPs. In the case of 7a and trans-7b, it was very rewarding to observe that their reactivity basically paralleled that of their analogues in the homogeneous phase under similar conditions and thus also behaved in a stereoselective manner. No product formation was observed with unfunctionalized MNPs under the same conditions (entry 9 of Table 1). We then increased the catalyst/substrate ratio to 1/1000 in order to explore the limits and scalability of the system for both 5a and 7a and the results are shown in Fig. 5. In both homogeneous and anchored cases the epoxidation was stereoselective with epoxide yield in the 77–78% range, which constitute an impressive TN of 770–780 in 24 hours at which point substrate conversion was completed. Additionally, it is remarkable to observe that the rates of epoxide formation in homogeneous and anchored are very similar. This puts forward the value of the concept of anchoring molecular catalysts in MNPs.
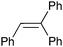
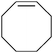
Given the excellent performance of the anchored catalysts with cis-β-methylstyrene, we also studied the performance of 7a with a series of alkenes and the results obtained are presented in Table 2. For trans-β-methylstyrene (entry 2, Table 2) the results are very similar to that of the cis isomer and thus indicate that the sterics of the olefin, in this particular case, do not play a major role. This is in agreement with the trans-configuration of the catalyst that provides a non-encumbered access of the olefin to the RuO group as can be observed in Fig. 1. It is interesting to see that the trisubstituted olefin, (2-methylprop-1-en-1-yl)benzene, gives the best yield among all the studied aromatic alkenes and actually better than cis and trans-β-methylstyrene (entry 3, Table 2). On the other hand, styrene and 3-trifluoromethylstyrene, which are comparatively much more electronically poor, give significantly lower performance (see entries 4 and 5, Table 2). These two phenomena are in agreement with an electrophilic character of the RuO active site. This character arises because of the presence of the highly electron-withdrawing bpm ligand. Finally and in agreement with all the steric and electronic arguments discussed above octene gives the best results of all the olefins tested (entry 9, Table 2). Entries 6 and 7 show the performance of cis- and trans-stilbene, respectively, where now sterics can play an important role in these electron poor olefins. This is even more pronounced in entry 8 with 1,1,2-triphenylethylene that gives the worst performance of all the olefins tested. Also remarkable is the fact that the more sterically demanding cis-stilbene stereospecifically leads to the corresponding cis-epoxide. This is in sharp contrast with what was observed with Mn–Salen catalytic species.
One of the main advantages of supporting a catalyst onto MNPs is the possibility of recovery by simple magnetic decantation applying an external magnetic field. We also studied the possibility of reusing 7a in the epoxidation of cis-β-methylstyrene. For each cycle the MNPs were decanted using a neodymium magnet and the supernatant was separated and analyzed by gas chromatography. Then more solvent and reactants were added to the recovered MNPs in order to perform another catalytic run. The catalytic system could be reused for 5 consecutive runs, with only a slight decrease in selectivity observed after the second run. This could be due to some agglomeration of MNPs after the first cycle.
Experimental
General
All commercial reagents were purchased from Sigma-Aldrich and TCI were directly used without further purification. Diethyl 2,2′:6′,2′′-terpyridine-4′-phosphonate (1) supplied by HetCat was purified by flash chromatography using alumina with ethyl acetate. NMR spectra were registered in a Bruker Advance 400 Ultrashield spectrometer in CDCl3, CD2Cl2 or CD3OD at room temperature, operating at 400.13 MHz (1H) and 100.63 MHz (13C{1H}). The numeration used for 1H and 13C assignment is shown in Fig. S1 of the ESI.† Cyclic Voltammetry (CV) experiments were performed on an IJ-Cambria CHI-660 potentiostat using a three-electrode cell. Typical CV experiments were carried out at a scan rate of 100 mV s−1. A glassy carbon electrode (2 mm diameter) was used as a working electrode, platinum wire as an auxiliary electrode, and a SSCE as a reference electrode. Working electrodes were polished with 0.05 micron alumina paste and washed with distilled water and acetone before each measurement. The complexes were dissolved in CH2Cl2 containing the necessary amount of n-Bu4NPF6 (TBAH) as a supporting electrolyte to yield a 0.1 M ionic strength solution. E1/2 values reported in this work were estimated from CV experiments as the average of the oxidative and reductive peak potentials (Ep,a + Ep,c)/2. The complex concentration was always approx. 1 μM. UV-Vis spectroscopy was performed on a Cary 50 (Varian) UV-Vis spectrophotometer in 1 cm quartz cuvettes. Mass spectrometry analyses were performed in a mass spectrometer with matrix assisted laser desorption ionization (MALDI-TOF, Bruker Autoflex). FT-IR spectra were recorded on a Thermo Nicolet 5700 FTIR spectrometer, using KBr pellets. Potassium bromide used in the preparation of the pellets was kept in an oven at 50 °C. Elemental analyses (C; H; N) were performed in LECO CHNS-model 932 by C.A.I. microanálisis elemental, Universidad Complutense de Madrid, Madrid, Spain. Catalytic experiments were analyzed in an Agilent 6890N gas chromatograph coupled to a mass selective detector with ionization by electronic impact and in an Agilent 6890N with a FID detector. TEM images were recorded using a JEOL JEM 1011 microscope equipped with a lanthanum hexaboride filament, operated at an acceleration voltage of 100 kV, at Microscopy Units, Universitat Rovira i Virgili, Tarragona, Spain. A drop of the magnetic nanoparticles (MNPs) suspension was added to a holey-carbon coated 200 mesh copper grid allowing the solvent to evaporate before being introduced into the microscope. X-Ray diffraction patterns were collected in the θ–θ mode using a Bruker D8 Advance X-ray diffractometer: Cu Kα1 irradiation, λ = 1.5406 Å; room temperature (25 °C); 2θ = 4–70°. X-ray single crystal data collection was performed on a Bruker Nonius FR 591 system equipped with a multilayer Montel 200 mirror monochromator Mo Kα (λ = 0.71073 Å) radiation and an Apex II CCD detector. The molecular structure was resolved by direct methods and refined of F2 by full matrix least squares techniques using the SHELX TL package with anisotropic thermal parameters. CCDC numbers for cis-3b and trans-3b are 896921 and 896922 respectively.Synthesis of the complexes
:1), under an argon atmosphere, was heated to reflux. A dispersion of 2 (300 mg, 0.51 mmol) in ethanol (15 mL) was added slowly for a period of 45 minutes. The resulting solution was heated to reflux for 30 minutes more. Diethyl ether was added to the red solution until precipitation of a solid. This solid was filtered and purified by re-precipitation with dichloromethane and diethyl ether. Yield: 45% (180 mg, 0.23 mmol). 1H-NMR (CD2Cl2): δ = 10.6 (dd, J1 = 5.6 Hz, J2 = 2.0 Hz, 1H, H16), 9.39 (dd, J1 = 5.1 Hz, J2 = 2.2 Hz, 1H, H18), 8.82 (dd, J1 = 4.6 Hz, J2 = 2.1 Hz, 1H, H21), 8.70 (d, J = 13 Hz, 2H, H7, H9), 8.44 (d, J = 8.1 Hz, 2H, H4, H12), 8.06 (t, J = 5.3 Hz, 1H, H17), 7.99 (t, J = 7.7 Hz, 2H, H3, H13), 7.74 (dd, J1 = 6.0 Hz, J2 = 2.1 Hz, 1H, H23), 7.69 (d, J = 4.9 Hz, 2H, H1, H15), 7.38 (t, J = 6.7 Hz, 2H, H2, H14), 7.24 (t, J = 5.3 Hz, 1H, H22), 4.41 (m, 4H, H24, H26), 1.52 (t, J = 7.1 Hz, 6H, H25, H27). 13C-NMR (CD2Cl2): δ = 160 (C23), 159 (C16), 158 (C6, C10), 157 (C18), 156 (C21), 152 (C1, C15), 137 (C3, C13), 128 (C2, C14), 124.2 (C4, C12), 124.0 (C7, C9), 127.7 (C17), 123.3 (C22), 63.7 (C24, C26), 16.2 (C25, C27). 31P {1H}-NMR (CD2Cl2): δ = 15.7. CV (CH2Cl2, TBAH): E1/2 = 1.1 V vs. SSCE; Ep,a = 1.25 V; Ep,c = 0.90 V, ΔE = 350 mV. MALDI+ HRMS: [M − Cl]+, calculated for C27H26ClN7O3PRu: 664.0561; found: 664.0677. Elem. anal. (%) calculated for C27H26Cl2N7O3PRu·5H2O: C 41.07, H 4.60, N 12.42; found: C 40.81, H 4.53, N 12.10. UV-Vis (CH2Cl2): λmax, nm (ε, M−1 cm−1) 320 (23809), 370 (7465), 523 (8674); (H2O) 360 (6219), 501 (8205); (CH3OH) 364 (6791), 513 (8314).
:1), under an argon atmosphere, and Et3N (54 μL, 0.38 mmol) were added and the reaction mixture was heated to reflux for 1 hour. Diethyl ether was added until the precipitation of a dark solid. After filtration, 20 mL of dichloromethane were added to dissolve this solid partially. Solution and solid were separated. Diethyl ether was added to the solution and 138 mg of 3b as a mixture of cis/trans isomers (0.24:1) was obtained. Upon further re-precipitation of the remaining dark solid with dichloromethane–diethyl ether 68 mg of the pure trans-3b isomer were obtained (Yield: 26%). Global yield: 82%. 1H-NMR (CD2Cl2): δ = 10.0 (d, J = 6.0 Hz, 1H, H16), 8.82 (d, J = 7.7 Hz, 1H, H19), 8.63 (d, J = 13 Hz, 2H, H7, H9), 8.58 (d, J = 7.9 Hz, 2H, H4, H11), 8.44 (dt, J1 = 7.7 Hz, J2 = 1.4 Hz, 1H, H18), 8.18 (m, 3H, H3, H13, H17), 7.52 (t, J = 6.6 Hz, 2H, H2, H14), 7.33 (d, J = 5.5 Hz, 2H, H1, H15), 7.25 (d, J = 7.2 Hz, 1H, H24), 7.07 (t, J = 8.0 Hz, 2H, H23, H25), 6.26 (d, J = 8.7 Hz, 2H, H22, H26), 4.35 (m, 4H, H27, H29), 1.45 (t, J = 6.6 Hz, 6H, H28, H30). 13C-NMR (CD2Cl2): δ = 165.9 (C20), 157.1 (C5, C11), 155.8 (C6, C8, C10), 155.0 (C21), 152.2 (C1), 150.3 (C16), 139.6 (C3, C13), 139.0 (C18), 129.9 (C24), 128.9 (C23, C25), 128.5 (C2, C14), 126.6 (C17), 125.8 (C19), 124.8 (C4, C12), 124.5 (C7, C9), 120.4 (C22, C26), 63.9 (C27, C29), 16.3 (C28, C30). 31P {1H}31-NMR (CD2Cl2): δ = 14.5. CV (CH2Cl2, TBAH): E1/2 = 1.37 V vs. SSCE; Ep,a = 1.46 V; Ep,c = 1.29 V, ΔE = 170 mV. MALDI+ HRMS: [M − Cl]+, calc. for C30H29ClN6O3PRu: 689.0771; found: 689.0789. Elem. anal. (%) calculated for C30H29Cl2N6O3PRu·H2O: C 48.52, H 4.21, N 11.32; found: C 48.24, H 4.44, N 11.09.
Synthesis of the MNPs of Fe3O431,32
Iron(III) acetylacetonate (1.8 g, 5 mmol), 1,2-dodecanediol (5.6 g, 25 mmol), oleic acid (5.3 mL, 15 mmol), oleylamine (7 mL, 15 mmol) and benzyl ether (20 mL) were mixed at room temperature under an argon atmosphere. The reaction mixture was warmed at 265 °C for 3 hours and was cooled at room temperature. The MNPs were removed using an external magnetic field, washed several times with MeOH and acetone and dried under vacuum.Preparation of 4a and trans-4b anchored onto MNPs
General procedure for the epoxidation of alkenes
To a solution of the corresponding Ru catalyst (2.5 μmol) in CH2Cl2 (1 mL) in a glass tube, cis-β-methylstyrene (33 μL, 250 μmol), dodecane (20 μL) and iodobenzene diacetate (165 mg, 500 μmol) were added. The solution was stirred in a shaker at 25 ± 1 °C and an aliquot for analysis of t0 was taken. H2O (9 μL, 500 μmol) was added to the solution and the mixture was stirred for 24 h at 25 ± 1 °C.Aliquots were taken for the analysis by GC-FID and/or GC-MS and were filtered through a Pasteur pipette filled with celite and diethyl ether was added in order to elute the organic compounds. For the heterogeneous system, the amount of catalyst was calculated taking into account the functionalization of the MNPs (mmol Ru g−1 MNPs). For the recycling experiments, after 48 h of reaction an aliquot was taken to be analysed. Then, diethyl ether was added to the solution mixture to allow a faster separation using a magnet. When all the MNPs were trapped aside using the magnet the solution was removed and the MNPs were washed twice with CH2Cl2 and another portion of reactants was added. All epoxides related to this work are known and were characterized by comparison of their physical and spectroscopic properties with those commercially available samples.
Conclusion
We have reported the synthesis and spectroscopic and electrochemical characterization of two Ru-aqua complexes containing electron withdrawing ligands whose higher oxidation states behave as two electron oxidants. These complexes act as excellent catalysts for the epoxidation of olefins. In particular, it is remarkable that their stereoselectivity with cis-olefins generates only the corresponding cis-epoxides. In addition, we have anchored these catalysts onto MNPs and have demonstrated that they present practically the same behaviour as their homogeneous counterparts, in terms of activity, selectivity and rate of product formation. Thus, the present systems represent a very successful example of using anchored catalysts in MNPs that can behave exactly like the homogeneous phase but that can benefit from the easy separation of MNPs using a simple magnet. The combination of all these well-behaved properties enables the present system to be used in multiple recycling experiments without significant degradation.Acknowledgements
This work was supported by MINECO (CTQ2008-00947, CTQ2010-21497 and CTQ2011-26440), Consolider Ingenio 2010 (grant CSD2006-0003), DEC (grants 2009SGR623, 2009SGR69), and the ICIQ foundation. P.R. thanks MINECO for a Torres Quevedo post-doctoral grant. L.V. thanks AGAUR for a doctoral grant. We also thank ICIQ Support Units and Universitat Rovira i Virgili for TEM images.Notes and references
- S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS
.
- E. McCafferty and J. P. Wightman, Surf. Interface Anal., 1998, 26, 549–564 CrossRef CAS
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS
- O. Gleeson, R. Tekoriute, Y. K. Gun’Ko and S. J. Connon, Chem.–Eur. J., 2009, 15, 5669–5673 CrossRef CAS
- P. Riente, C. Mendoza and M. A. Pericàs, J. Mater. Chem., 2011, 21, 7350–7355 RSC
- B. G. Wang, B. C. Ma, Q. Wang and W. Wang, Adv. Synth. Catal., 2010, 352, 2923–2928 CrossRef CAS
- A. Hu, S. Liu and W. Lin, RSC Adv., 2012, 2, 2576–2580 RSC
- F. Michalek, A. Lagunas, C. Jimeno and M. A Pericàs, J. Mater. Chem., 2008, 18, 4692–4697 RSC
- M. Shokouhimehr, Y. Piao, J. Kim, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 7039–7043 CrossRef CAS
- M.-J. Jin and D.-H. Lee, Angew. Chem., Int. Ed., 2010, 49, 1119–1122 CrossRef CAS
- H.-J. Xu, X. Wan, Y.-Y. Shen, S. Xu and Y.-S. Feng, Org. Lett., 2012, 14, 1210–1213 CrossRef CAS
- T.-J. Yoon, W. Lee, Y.-S. Oh and J.-K. Lee, New J. Chem., 2003, 27, 227–229 RSC
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS
-
H. Adolfsson, in Modern Oxidation Methods, ed. J.-E. Bäckvall, Wiley-VCH, Weinheim, 2010, pp. 37–84 Search PubMed
- D.-H. Zhang, G.-D. Li, J.-X. Li and J.-S. Chen, Chem. Commun., 2008, 3414–3416 RSC
- S. Shylesh, J. Schweizer, S. Demeshko, V. Schünemann, S. Ernst and W. R. Thiel, Adv. Synth. Catal., 2009, 351, 1789–1795 CrossRef CAS
- P. H. Mutin, G. Guerrero and A. Vioux, J. Mater. Chem., 2005, 15, 3761–3768 RSC
- R. A. Binstead, M. E. McGuire, A. Dovletoglou, W. K. Seok, L. E. Roecker and T. J. Meyer, J. Am. Chem. Soc., 1992, 114, 173–186 CrossRef CAS
-
(a) E. L. Lebeau, R. A. Binstead and T. J. Meyer, J. Am. Chem. Soc., 2001, 123, 10535–10544 CrossRef CAS
-
(a) A. Llobet, Inorg. Chim. Acta, 1994, 221, 125–131 CrossRef CAS
- M. Dakkach, M. I. López, I. Romero, M. Rodríguez, A. Atlamsani, T. Parella, X. Fontrodona and A. Llobet, Inorg. Chem., 2010, 49, 7072–7079 CrossRef CAS
- O. Hamelin, S. Ménage, F. Charnay, M. Chavarot, J.-L. Pierre, J. Pécaut and M. Fontecave, Inorg. Chem., 2008, 47, 6413–6420 CrossRef CAS
- K. J. Takeuchi, M. S. Thompson, D. W. Pipes and T. J. Meyer, Inorg. Chem., 1984, 23, 1845–1851 CrossRef CAS
- F. R. Keene, Coord. Chem. Rev., 1999, 187, 121–149 CrossRef CAS
- E. Masllorens, M. Rodríguez, I. Romero, A. Roglans, T. Parella, J. Benet-Buchholz, M. Poyatos and A. Llobet, J. Am. Chem. Soc., 2006, 128, 5306–5307 CrossRef CAS
- F. Laurent, E. Plantalech, B. Donnadieu, A. Jimenez, F. Hernandez, M. Martinez-Ripoll, M. Biner and A. Llobet, Polyhedron, 1999, 18, 3321–3331 CrossRef CAS
- I. Romero, M. Rodríguez, A. Llobet, M.-N. Collomb-Dunand-Sauthier, A. Deronzier, T. Parella and H. Stoeckli-Evans, J. Chem. Soc., Dalton Trans., 2000, 1689–1694 RSC
- X. Sala, E. Plantalech, I. Romero, M. Rodríguez, A. Llobet, A. Poater, M. Duran, M. Solà, S. Jansat, M. Gómez, H. Stoeckli-Evans and J. Benet-Buchholz, Chem.–Eur. J., 2006, 12, 2798–2807 CrossRef CAS
- C. Sens, M. Rodríguez, I. Romero, A. Llobet, T. Parella and J. Benet-Buchholz, Inorg. Chem., 2003, 42, 8385–8394 CrossRef CAS
- N. Planas, G. Christian, S. Roeser, E. Mas-Marzá, M.-R. Kollipara, J. Benet-Buchholz, F. Maseras and A. Llobet, Inorg. Chem., 2012, 51, 1889–1901 CrossRef CAS
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS
- M. Rodríguez, I. Romero, A. Llobet, A. Deronzier, M. Biner, T. Parella and H. Stoeckli-Evans, Inorg. Chem., 2001, 40, 4150–4156 CrossRef
- J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462–16463 CrossRef CAS
- N. C. Pramanik and S. Bhattacharya, Transition Met. Chem., 1997, 22, 524–526 CrossRef CAS
- J. G. Smith, Synthesis, 1984, 629–656 CrossRef CAS
- C. Schneider, Synthesis, 2006, 3919–3944 CrossRef CAS
-
K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, VCH, Weinheim, 1997 Search PubMed
- Q.-H. Xia, H.-Q. Ge, C.-P. Ye, Z.-M. Liu and K.-X. Su, Chem. Rev., 2005, 105, 1603–1662 CrossRef CAS
- C.-M. Che and J.-S. Huang, Chem. Commun., 2009, 3996–4015 RSC
- D. Chatterjee, Coord. Chem. Rev., 2008, 252, 176–198 CrossRef CAS
- K. C. Gupta, A. K. Sutar and C.-C. Lin, Coord. Chem. Rev., 2009, 253, 1926–1946 CrossRef CAS
- W. Mägerlein, C. Dreisbach, H. Hugl, M. K. Tse, M. Klawonn, S. Bhor and M. Beller, Catal. Today, 2007, 121, 140–150 CrossRef
- M. K. Tse, S. Bhor, M. Klawonn, G. Anilkumar, H. Jiao, A. Spannenberg, C. Döbler, W. Mägerlein, H. Hugl and M. Beller, Chem.–Eur. J., 2006, 12, 1875–1888 CrossRef CAS
- J. Benet-Buchholz, P. Comba, A. Llobet, S. Roeser, P. Vadivelu, H. Wadepohl and S. Wiesner, Dalton Trans., 2009, 5910–5923 RSC
- X. Sala, N. Santana, I. Serrano, E. Plantalech, I. Romero, M. Rodríguez, A. Llobet, S. Jansat, M. Gomez and X. Fontrodona, Eur. J. Inorg. Chem., 2007, 5207–5214 CrossRef CAS
- I. Serrano, X. Sala, E. Plantalech, M. Rodríguez, I. Romero, S. Jansat, M. Gomez, T. Parella, H. Stoeckli-Evans, X. Solans, M. Font-Bardia, B. Vidjayacoumar and A. Llobet, Inorg. Chem., 2007, 46, 5381–5389 CrossRef CAS
- G. A. Barf and R. A. Sheldon, J. Mol. Catal. A: Chem., 1995, 102, 23–39 CrossRef CAS
- A. D. Chowdhury, A. Das, K. Irshad, S. M. Mobin and G. K. Lahiri, Inorg. Chem., 2011, 50, 1775–1785 CrossRef
- M. K. Tse, C. Döbler, S. Bhor, M. Klawonn, W. Mägerlein, H. Hugl and M. Beller, Angew. Chem., Int. Ed., 2004, 43, 5255–5260 CrossRef CAS
- D. Chatterjee, J. Mol. Catal. A: Chem., 2009, 310, 174–179 CrossRef CAS
- E. N. Jacobsen, W. Zhang and M. L. Guler, J. Am. Chem. Soc., 1991, 113, 6703–6704 CrossRef CAS
- L. Cavallo and H. Jacobsen, J. Org. Chem., 2003, 68, 6202–6207 CrossRef CAS
- M. Dakkach, X. Fontrodona, T. Parella, A. Atlamsani, I. Romero and M. Rodríguez, Adv. Synth. Catal., 2011, 353, 231–238 CrossRef CAS
-
L. F. Forcada, Ruthenium Complexes with Polynucleating Ligands and their Capacity to Catalytically Oxidize Water to Dioxygen, PhD thesis, Universitat Autònoma de Barcelona, 2011 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 896921 and 896922. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cy20616a |
| ‡ Current address: ACAL Energy Ltd. The Heath Bus. & Tech. Park, Runcorn, Cheshire WA7 4QX, UK. |
| This journal is © The Royal Society of Chemistry 2013 |