Investigation of the physicochemical properties and catalytic activities of Ce0.67M0.33O2 (M = Zr4+, Ti4+, Sn4+) solid solutions for NO removal by CO
Xiaojiang
Yao
a,
Changjin
Tang
a,
Zeyang
Ji
a,
Yue
Dai
a,
Yuan
Cao
a,
Fei
Gao
*b,
Lin
Dong
*ab and
Yi
Chen
a
aKey Laboratory of Mesoscopic Chemistry of MOE, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, PR China. E-mail: donglin@nju.edu.cn; Fax: +86 25 83317761; Tel: +86 25 83592290
bJiangsu Key Laboratory of Vehicle Emissions Control, Center of Modern Analysis, Nanjing University, Nanjing 210093, PR China. E-mail: gaofei@nju.edu.cn; Fax: +86 25 83317761; Tel: +86 25 83596545
First published on 20th November 2012
Abstract
NO removal by CO model reaction was investigated over a series of ceria-containing solid solutions, prepared by an inverse co-precipitation method, to explore the relationship between the physicochemical properties and catalytic performances of these catalysts. The synthesized samples were studied in detail by means of XRD, Raman, TEM, UV-Vis spectroscopy, N2-physisorption, H2-TPR, OSC, XPS and in situ FT-IR technologies. These results indicate that the incorporation of Zr4+, Ti4+ and Sn4+ into the lattice of CeO2 leads to a smaller grain size and enhanced reduction behavior. Furthermore, the catalytic performance test shows that the activities and selectivities of these solid solutions are higher than pure CeO2 and that the Sn4+-doped sample shows the best results. The reason may be that: (1) the decrease in grain size results in an enlargement of the BET specific surface area and an increase of surface Ce3+. The former is conducive for sufficient contact between the catalyst and reactant molecules and the latter contributes to the adsorption of COx species; (2) the enhanced reduction behavior is beneficial in generating more surface oxygen vacancies during the reaction process, which can weaken the N–O bond to promote the dissociation of NOx effectively. Finally, in order to further understand the nature of the catalytic performances for these samples, a possible reaction mechanism is tentatively proposed.
1. Introduction
Nitrogen oxides, such as NO, NO2, and N2O5 (hereafter denoted as NOx), which originate from various combustion processes, are a major kind of air pollutants and are very harmful to human health due to the formation of photochemical smog, acid rain, ozone depletion and greenhouse effects. Fortunately, catalytic reduction is a powerful technology for handling many environmental problems and the most important one among them is the removal of NOx by using different reducing agents, such as CO, H2, NH3 and CxHy.1Cerium oxide has been the subject of numerous investigations in recent years due to its wide application in environmental catalytic areas, including deNOx catalysis, three-way catalysis (TWC), catalytic wet oxidation, oxygen permeation membrane system, fuel cell process and exhaust combustion catalysis.2–7 The success of ceria in pollution abatement and other technologies is mainly due to its high oxygen storage/release efficiency associated with the formation of oxygen vacancies and the low redox potential between Ce3+ and Ce4+.8–10 Despite widespread applications, the use of pure ceria is highly discouraged because of its poor thermal stability and low specific surface area.11 As a solution, some transition and non-transition metal ions (Zr4+, Si4+, Ti4+, Hf4+, Al3+, Mg2+, Cox+, Mnx+, etc.) are normally introduced into the ceria cubic structure and have been investigated systematically with the aim of overcoming these disadvantages.8,11–16 The formation of ceria-based mixed oxides could modify the structural, textural, redox, oxygen migration and catalytic properties via the incorporation of dopant ions into the lattice of ceria to generate defects. Furthermore, ceria can also easily form solid solutions with transition and non-transition metal ions, such as Zr4+, Ti4+, Al3+, Cu2+, and Mnx+, and each has its own uniqueness.1,13,16–18 When designing a ceria-containing solid solution, several factors should be considered, such as the influence of the dopant concentration (a high concentration of ions with redox character is generally preferred) and the presence of a single phase (a fluorite-structured material is favored).19
In recent years, the preparation and characterization of ceria-containing solid solutions have attracted much attention due to their outstanding textural properties, improved oxygen storage capacity and excellent redox properties in comparison with pure ceria. Among the ceria-containing solid solutions, special attention has been focused on the CeO2–ZrO2 solid solution, mainly because of its industrial utilization in a three-way catalyst to eliminate exhaust gases. From the reported results, the CeO2–ZrO2 solid solution shows enhanced redox and catalytic properties compared with pure ceria.17,20–23 However, the research process of other ceria-containing solid solutions is still very slow and a direct comparison between them is highly lacking.
Since the electron can exchange easily between Ce4+/Ce3+ and Ti4+/Ti3+ (or Sn4+/Sn2+) redox couples, the redox properties of CeO2–TiO2 and CeO2–SnO2 solid solutions may be better than the CeO2–ZrO2 solid solution, which is beneficial to their catalytic activities.24,25 Luo et al.11 synthesized a series of CexTi1−xO2 (x = 0.1–0.9) mixed oxides and found that CexTi1−xO2 solid solutions with a cubic phase could be formed when x ≥ 0.6 and that these solid solutions are a promising new material for three-way catalysis or other forms of catalysis for oxidation reactions due to their excellent redox properties. In addition, Perrichon et al.26 reported that the redox property of a CeO2–SnO2 solid solution is improved in comparison to the single oxides CeO2 and SnO2 and it could be used in some catalytic reactions. However, as far as we know, a comparative study of the physicochemical properties of ceria-containing solid solutions and their application in a specified reaction has not been reported.
In the present work, a series of Ce0.67M0.33O2 (M = Zr4+, Ti4+, Sn4+) solid solutions (Ce![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M = 2:1 mol ratio) were prepared by an inverse co-precipitation method and characterized by means of XRD, Raman, TEM, UV-Vis spectroscopy, N2-physisorption, H2-TPR, OSC, XPS and in situ FT-IR technologies. Moreover, NO removal by CO as a model reaction was carried out to evaluate the catalytic performances of these samples. This study is mainly focused on: (i) exploring the relationship between the catalytic performances and physicochemical properties of these ceria-containing solid solutions; (ii) approaching the nature of NO removal by CO reaction through investigating the interaction of CO and/or NO with these catalysts by an in situ FT-IR technique in the temperature range of 25–300 °C.
:M = 2:1 mol ratio) were prepared by an inverse co-precipitation method and characterized by means of XRD, Raman, TEM, UV-Vis spectroscopy, N2-physisorption, H2-TPR, OSC, XPS and in situ FT-IR technologies. Moreover, NO removal by CO as a model reaction was carried out to evaluate the catalytic performances of these samples. This study is mainly focused on: (i) exploring the relationship between the catalytic performances and physicochemical properties of these ceria-containing solid solutions; (ii) approaching the nature of NO removal by CO reaction through investigating the interaction of CO and/or NO with these catalysts by an in situ FT-IR technique in the temperature range of 25–300 °C.
2. Materials and methods
2.1. Catalysts preparation
The Ce0.67M0.33O2 (M = Zr4+, Ti4+, Sn4+) solid solutions (Ce:M = 2:1 mol ratio) were prepared from their salt solutions as precursors by an inverse co-precipitation method.27 The precursors of Ce4+, Zr4+, Ti4+ and Sn4+ were ammonium cerium(IV) nitrate, zirconium(IV) nitrate, titanium(IV) tetrachloride and tin(IV) tetrachloride, respectively. To prepare the Ce0.67Sn0.33O2 solid solution, briefly, the desired quantities of ammonium cerium(IV) nitrate and tin(IV) tetrachloride were separately dissolved in distilled water, mixed together with magnetic stirring for 2 h and then slowly added into excess ammonia (25%) with vigorous stirring until pH = 10. The resulting suspension was kept stirring for another 3 h, aged for 24 h and then filtered, washed several times with distilled water until no pH change was seen and no Cl− was detected by a solution of AgNO3. The obtained cake was oven dried at 110 °C for 12 h and finally calcined at 550 °C in flowing air for 5 h. The Ce0.67Zr0.33O2 and Ce0.67Ti0.33O2 solid solutions were obtained via the same procedure. In addition, pure CeO2 was prepared by a similar method for comparison. These synthesized samples were the CeO2, Ce0.67Zr0.33O2 (hereafter denoted as CZ), Ce0.67Ti0.33O2 (hereafter denoted as CT), and Ce0.67Sn0.33O2 (hereafter denoted as CS) solid solutions.
2.2. Catalysts characterization
X-ray diffraction (XRD) patterns were recorded on a Philips X'pert Pro diffractometer using Ni-filtered Cu–Kα radiation (λ = 0.15418 nm). The X-ray tube was operated at 40 kV and 40 mA. The intensity data were collected over a 2θ range of 10–80°. The scan speed was set at 10° min−1 with a step size of 0.02°. The mean grain sizes (Dβ) from the (111) plane of these samples were determined from XRD line broadening measurements using the Debye–Scherrer equation, Dβ = Kλ/βcos θ, where K is the particle shape factor, usually taken as 0.89, λ is the X-ray wavelength, β is the full-width at half maximum height (FWHM) in radians and θ is the diffraction angle.Raman spectra were collected on a Renishaw inVia Laser Raman spectrometer using an Ar+ laser beam. The Raman spectra were recorded with an excitation wavelength of 514 nm and a laser power of 20 mW.
Transmission electron microscopy (TEM) images of these samples were obtained by a JEM-2100 instrument at an acceleration voltage of 200 kV. The samples were dispersed in ethanol and kept in an ultrasonic bath for 15 min and then deposited on a carbon-covered copper grid for each measurement.
UV-Visible diffuse reflection spectra (UV-Vis DRS) were performed on a Shimadzu UV-2401 spectrophotometer. The spectra were recorded in the range of 200 to 700 nm using BaSO4 as the reference for the baseline emendation.
BET specific surface areas of these samples were obtained by N2-physisorption at 77 K on a Micromeritics ASAP-2020 analyzer. Prior to each analysis, the sample was degassed under vacuum at 300 °C for 4 h.
H2-temperature programmed reduction (H2-TPR) experiments were performed in a quartz U-type reactor connected to a thermal conductivity detector (TCD) with an Ar–H2 mixture (7.0% of H2 by volume, 70 ml min−1) as a reductant. Prior to the reduction, the sample (50 mg) was pretreated in a high purity N2 stream at 300 °C for 1 h and then cooled to room temperature. After that, the TPR started from 50 °C to the target temperature at a rate of 10 °C min−1.
Oxygen storage capacity (OSC) experiments were measured over a sample (50 mg) previously reduced in an Ar–H2 mixture (7.0% of H2 by volume, 70 ml min−1) from room temperature to 900 °C at a rate of 10 °C min−1. When the sample cooled to ambient temperature in high purity N2, a pure oxygen stream at 70 ml min−1 was admitted into the reactor at 300 °C for 1 h. After purging the oxidizing gas with high purity N2 at room temperature, the Ar–H2 mixture was switched on and the hydrogen consumption was monitored by a thermal conductivity detector (TCD). OSC was evaluated by hydrogen consumption.
X-ray photoelectron spectroscopy (XPS) analysis was performed on a PHI 5000 VersaProbe system, using monochromatic Al–Kα radiation (1486.6 eV) operating at an accelerating power of 15 kW. Before the measurement, the sample was outgassed at room temperature in a UHV chamber (<5 × 10−7 Pa). The sample charging effects were compensated for by calibrating all the binding energies (BE) with the adventitious C 1s peak at 284.6 eV. This gives BE values with an accuracy at ±0.1 eV.
In situ Fourier transform infrared (in situ FT-IR) spectra were collected from 400 to 4000 cm−1 at a spectral resolution of 4 cm−1 (number of scans, 32) on a Nicolet 5700 FT-IR spectrometer equipped with a DTGS as the detector. The sample was pressed into a self-supporting wafer (about 15 mg) and mounted in a commercial controlled environment chamber (HTC-3). The wafer was pretreated with high purity N2 at 300 °C for 1 h. After cooling to ambient temperature, the sample was exposed to a controlled stream of CO–Ar (10% of CO by volume) and/or NO–Ar (5% of NO by volume) at a rate of 5.0 ml min−1 for 40 min in order to be saturated. Desorption/reaction studies were performed by heating the adsorbed species and the spectra were recorded at various target temperatures at a rate of 10 °C min−1 from room temperature to 300 °C by subtraction of the corresponding background reference (collected from the gas data at each target temperature without the sample).
2.3. Catalytic activity test
The catalytic activities of these catalysts for NO removal by CO were determined under steady state, involving a feed steam with a fixed composition, 5% NO, 10% CO and 85% He by volume as diluents. The sample (50 mg) was fitted in a quartz tube, pretreated in a high purity N2 stream at 300 °C for 1 h, cooled to room temperature and afterwards the mixed gases were switched on. The reactions were carried out at different temperatures with a space velocity of 12000 ml g−1 h−1. Two columns (length, 1.75 m; diameter, 3 mm) and a thermal conductivity detector (T = 100 °C) were used to analyze the products. Column A used Paropak Q to separate CO2 and N2O, column B was packed with 5A and 13X molecule sieves (40–60 M) to separate N2, NO and CO.
3. Results and discussion
3.1. Catalytic activity and selectivity of NO removal by CO
NO removal by CO is taken as a model reaction to evaluate the catalytic performances of these samples. For this model reaction, the reaction products are N2O (by-product), N2 and CO2. The NO conversion and N2 selectivity of these samples as a function of reaction temperature are presented in Fig. 1. At low temperatures (≤250 °C), both the NO conversion and N2 selectivity are identical (close to 0%) for all of the samples except CS. While at high temperatures (>250 °C), the values are dependent on the compositions. From Fig. 1a and b, we can find that the NO conversion and N2 selectivity are enhanced with the introduction of Zr4+ and Ti4+. Strikingly, when Sn4+ is doped into CeO2 (i.e., the CS sample), it shows an obviously improved catalytic performance, with full N2 selectivity achieved at temperature as low as 325 °C and accompanied by NO conversion of ca. 80%. In order to explore whether the extraordinary catalytic performance of the CS sample is from the contribution of SnO2, a pure SnO2 sample has been prepared by a precipitation method for comparison. The NO conversion and N2 selectivity of SnO2 are also shown in Fig. 1a and b. We find that the activity of SnO2 is negligible during the reaction temperature range, which proves SnO2 is not an active component for the NO + CO reaction and prompts us to suppose that the distinct activity enhancement of CS is actually related to certain changes in the structure of ceria or the strong interaction between Ce4+/Ce3+ and Sn4+/Sn2+. In order to gain a clear idea of the reason for the different catalytic performances, comprehensive characterizations of these samples were carried out and the results are listed and discussed in the following sections.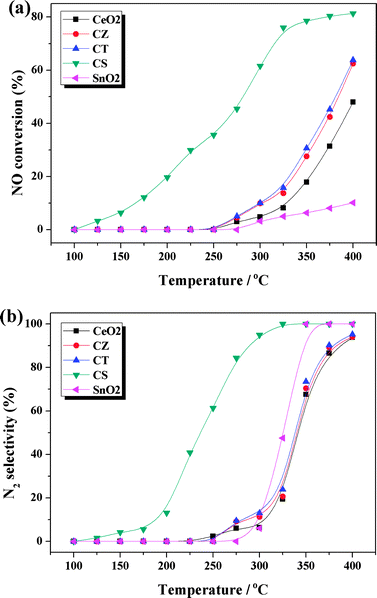 | ||
| Fig. 1 (a) NO conversion and (b) N2 selectivity of these samples. | ||
3.2. Structural characteristics (XRD, Raman, TEM and UV-Vis DRS)
The XRD patterns of these samples are shown in Fig. 2a. For all samples, only the broad diffraction lines, attributed to a cubic fluorite-type phase CeO2 (PDF-ICDD 34-0394), are detected without extra characteristic lines assigned to ZrO2, TiO2 and SnO2. Furthermore, the diffraction peaks of CZ, CT and CS are slightly shifted in the high angle direction compared with CeO2, indicating that Zr4+, Ti4+ and Sn4+ have been successfully incorporated into the lattice of CeO2 to form uniform solid solutions whilst maintaining the fluorite-type structure. Compared with CeO2, the diffraction peaks of CZ, CT and CS are broadened due to the crystallite nanodimensions effect,28 as evidenced by the grain size of these samples determined from the diffraction peak of the (111) plane by Debye–Scherrer equation (listed in Table 1). In general, the grain size of these ceria-containing solid solutions is smaller than that of CeO2. In particular, the CS sample has the smallest grain size among these samples (i.e., CeO2 > CZ > CT > CS). This phenomenon may be caused by the incorporation of Zr4+, Ti4+ and Sn4+ into the lattice of CeO2 which inhibits the crystal growth of the cubic phase.11 According to the literatures,29–34 the decrease in grain size results in an enlargement of the BET specific surface area and an increase of surface Ce3+. The former is conducive to sufficient contact between the catalyst and reactant molecules and the latter contributes to the adsorption of COx species, which further promotes the catalytic performance. In addition, it can be seen from Table 1 that the lattice parameters of CZ, CT and CS are smaller than that of CeO2 (5.4217 Å), which is primarily due to the fact that the ionic radius of Zr4+ (0.80 Å), Ti4+ (0.68 Å) and Sn4+ (0.71 Å) is smaller than that of Ce4+ (0.92 Å) and therefore, the incorporation of Zr4+, Ti4+ and Sn4+ into the lattice of CeO2 leads to contraction and distortion of the lattice.8,14| Samples | BET surface area (m2 g−1) | Grain size by XRDa (nm) | Grain size by TEMb (nm) | Lattice parameterc (Å) | Position of raman line (cm−1) | FWHM of raman line (cm−1) | (AI + AIII)/AII |
|---|---|---|---|---|---|---|---|
| a Determined from the XRD peak of the (111) plane by the Debye–Scherrer equation. b Estimated from the TEM results based on statistical analysis. c Calculated from the characteristic XRD peak of the (111) plane by Bragg's law. | |||||||
| CeO2 | 36.5 | 20.2 | 21.9 | 5.4217 | 464 | 42.3 | 0.0376 |
| CZ | 70.1 | 7.7 | 8.5 | 5.3211 | 473 | 107.5 | 0.5235 |
| CT | 72.3 | 5.3 | 6.1 | 5.3838 | 460 | 44.8 | 0.2705 |
| CS | 82.9 | 3.0 | 3.7 | 5.3849 | 456 | 49.4 | 0.3087 |
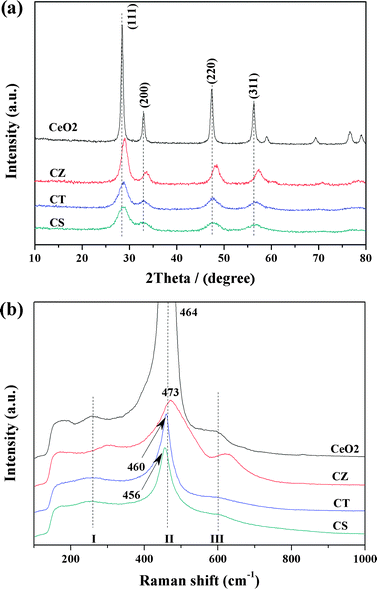 | ||
| Fig. 2 The results of the (a) XRD and (b) Raman spectra for these samples. | ||
Raman spectra can complement the XRD results very well and detect the changes in the vibrational structure of CeO2 caused by the incorporation of Zr4+, Ti4+ and Sn4+. Fig. 2b displays the Raman spectra of these samples. All of the Raman spectra include the main bands at 456–473 cm−1 (labeled II), corresponding to the F2g vibration model of the cubic fluorite type CeO2 lattice.12 Two weak bands at 260–300 cm−1 (labeled I) and 600–628 cm−1 (labeled III) are linked to the presence of defects such as oxygen vacancies in the CeO2 lattice35 and the sum of their peak area divided by the peak area of band II (i.e., (AI + AIII)/AII) can reflect the concentration of defects such as oxygen vacancies, which increase with the introduction of Zr4+, Ti4+ and Sn4+ (Table 1). It can be observed from Fig. 2b that the characteristic bands for MO2 (M = Zr4+, Ti4+, Sn4+) are absent and in addition, it is important to notice that the main band has a slight shift with the addition of Zr4+, Ti4+ and Sn4+ (Table 1), indicating that these foreign metal cations have been incorporated into the lattice of CeO2, which is in agreement with XRD results. Moreover, the full-width at half maximum height (FWHM) values of the ceria F2g bands are ranked in the order: CeO2 < CT < CS < CZ, which may be related to the concentration of defects such as oxygen vacancies and/or the grain size of these samples.1 Summarily, the incorporation of Zr4+, Ti4+ and Sn4+ into the CeO2 lattice can induce a smaller grain size and more defects such as oxygen vacancies.
The TEM technique is performed to ascertain the morphology and crystallite growth of these samples. Fig. 3 exhibits the low and high resolution TEM (HRTEM) and selected area electron diffraction (SAED) images of each sample. The average grain size of these samples, obtained by statistical analysis of the TEM images (Fig. 3a, d, g and j), is summarized in Table 1, which is in good agreement with the results of the XRD (i.e., CeO2 > CZ > CT > CS). Based on the observation of the TEM global view of these samples, we can find that all the samples are composed of smoothly interfaced particles with irregular shapes and have a disordered wormhole-like mesoporous structure, formed by the agglomeration of the nanoparticles. Moreover, it can be seen that the CS sample has the highest degree of agglomeration due to the smallest grain size among these samples. As can be seen from the HRTEM images (Fig. 3b, e, h and k), only one kind of periodicity of the lattice fringes, with a d spacing of ca. 0.31 nm, can be observed for the CeO2, CZ, CT and CS samples, which is compatible with the distance expected between the (111) reticular plane of these samples. These results indicate that the most frequently exposed crystal plane of these samples should be the (111) plane. In addition, we notice that the lattice fringes of MO2 (M = Zr4+, Ti4+, Sn4+) are not visible. Furthermore, according to the continuous diffraction rings in the SAED pattern (Fig. 3c, f, i and l), there is no evidence for the presence of MO2 (M = Zr4+, Ti4+, Sn4+), probably due to the incorporation of these foreign metal cations into the lattice of CeO2, which is consistent with XRD and Raman results.
 | ||
| Fig. 3 TEM, HRTEM and SAED images of (a, b and c) CeO2, (d, e and f) CZ, (g, h and i) CT and (j, k and l) CS. | ||
Information on the surface coordination and electronic states of the metal ions by measuring the d–d and f–d electron transitions and oxygen–metal ion charge transfer bands can be acquired from the UV-Vis DRS measurement. As shown in Fig. 4, all these samples exhibit three adsorption bands at 255, 295 and 340 nm, which could be attributed to O2− → Ce3+ charge transfer transitions, O2− → Ce4+ charge transfer and interband transitions, respectively.8,36,37 However, no adsorption band is observed above 500 nm in wavelength. Furthermore, it can be seen that the adsorption bands of ZrO2, TiO2 and SnO2 are not detected during the UV-Vis DRS experiment and that the adsorption edges of the CZ, CT and CS samples are shifted in the higher wavelength direction from 445 nm to 489, 502 and 509 nm compared with CeO2. The reason may be that the incorporation of Zr4+, Ti4+ and Sn4+ into the lattice of CeO2 to form solid solutions leads to a decrease of the symmetry and consequently the strain development at the cerium sites.36 Interestingly, the adsorption bands of the CZ, CT and CS samples are broadened and weakened compared with those of CeO2, which could be due to the decrease in grain size. In summary, the observations from the UV-Vis DRS are very consistent with the XRD, Raman and TEM results.
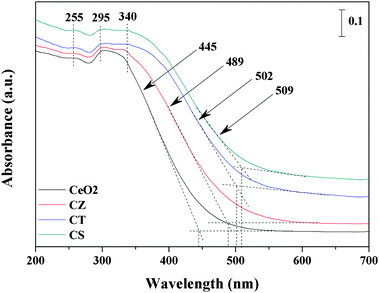
The BET specific surface area of these samples, obtained from N2-physisorption, is also summarized in Table 1. Compared with pure CeO2, when Zr4+, Ti4+ and Sn4+ are doped into the lattice of CeO2, the BET specific surface area increases from 36.5 to 70.1, 72.3, and 82.9 m2 g−1, respectively, which is related to the grain size of these samples to some extent. In other words, the grain size of CeO2 could be greatly decreased by the incorporation of Zr4+, Ti4+ and Sn4+ into the lattice of CeO2, which leads to an increase in the BET specific surface area and consequently the improvement of the catalytic performance due to sufficient contact with the reactant molecules.
3.3. Reduction properties and oxygen storage capacity (H2-TPR and OSC)
Fig. 5 shows the H2-TPR profiles of the synthesized samples. The reduction of CeO2 exhibits two broad peaks around 500 and 825 °C, which may be assigned to surface species and bulk particle reduction, respectively, according to the literature.12 It can be observed that CZ and CT present a similar reduction behavior and are indeed very similar to the behaviour observed over CeO2, i.e., all of them display surface species reduction in zone(I) and bulk particle reduction in zone(II). It is noteworthy that the H2 consumption of CZ and CT is larger than that of CeO2 while the fraction of surface species reduction increases with the incorporation of Zr4+ and Ti4+ into the lattice of CeO2 (seen from Table 2). Therefore, this result corroborates the widely reported findings concerning the zirconium or titanium promotional role on the bulk reduction of solid solutions.11,38 By reducing CeO2, oxygen atoms are removed from the surface, creating vacancies. Then, oxygen atoms migrate from the bulk to the surface while the oxygen vacancies move in the opposite direction. By inserting zirconium or titanium, the amount of defects is increased (according to the results of the Raman spectra) and the oxygen mobility in the lattice is enhanced, which facilitates the CeO2 reduction process. Furthermore, we can see that the H2 consumption of CT is larger than CZ, which may be due to the establishment of Ce4+/Ce3+ and Ti4+/Ti3+ redox couples, according to the results of Madras et al.25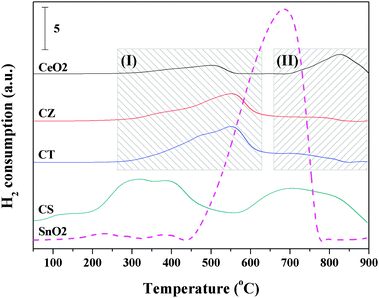 | ||
| Fig. 5 The H2-TPR results of the synthesized samples. | ||
| Samples | H2 consumption (μmol g−1) | A I/(AI + AII) | OSC from H2-TPR (μmol g−1 of H2) | |
|---|---|---|---|---|
| Experimental amount | Estimated amount | |||
| a Estimated amount of CS was determined from the H2 consumption of CeO2 and SnO2: 0.67 × 773 + 0.33 × 9746. | ||||
| CeO2 | 773 | — | 0.3839 | 876 |
| CZ | 1329 | — | 0.8435 | 1593 |
| CT | 1870 | — | 0.8848 | 1622 |
| CS | 4248 | 3764a | — | 2443 |
| SnO2 | 9746 | — | — | — |
Interestingly, it can be seen from Fig. 5 that CS presents four reduction peaks and its reduction peak temperatures are obviously lower than the above-mentioned three samples, indicating that the oxygen atoms of CS are easier to migrate in order to generate oxygen vacancies during the reduction process. In addition, Hegde et al.24 has previously investigated Ce0.6Sn0.4O2 before and after H2 reduction up to 550 °C by XPS, the results showed that Sn4+ was reduced, probably completely to the Sn2+ state, and only a part of the Ce4+ ion was reduced to the Ce3+ state. They concluded that the two reduction peaks before 550 °C were mainly attributed to the reduction of Sn4+ to the Sn2+ state and the partial reduction of Ce4+ to the Ce3+ state. Therefore, in this work, we have the same attribution for the reduction peaks of CS before 550 °C. Chen et al.39 reported that SnO2 exhibited a major reduction peak at about 670 °C and that the final product was Sn0 metal. Combined with the reduction peak temperatures for CeO2 and SnO2, we speculate that the reduction peaks after 550 °C may be related to the reduction of Sn2+ to the Sn0 state and the reduction of bulk CeO2. As can be seen from Table 2, the H2 consumption of CS is larger than the other three samples. Furthermore, the experimental H2 consumption is greater than the estimated theoretical H2 consumption for CS, indicating that the introduction of Sn4+ can effectively promote the reduction of CeO2 due to the strong interaction between Ce4+/Ce3+ and Sn4+/Sn2+ through the redox equilibrium of 2Ce4+ + Sn2+↔2Ce3+ + Sn4+.
Recently, many researchers have systematically investigated the oxygen storage capacity (OSC) of ceria-containing solid solutions and concluded that the improved OSC can enhance the catalytic activity for many reactions.24,26,34,38 Therefore, in the present work, the total OSC values, as measured from the H2 uptake up to 900 °C in the TPR experiment, are also summarized in Table 2. In addition, the OSC values are given in micromoles of H2 per gram of the catalyst for ready comparison. As can be seen from Table 2, the OSC values of these ceria-containing solid solutions are uniformly higher than that of CeO2. Furthermore, it is found that the OSC value of CS is higher than those for the corresponding CZ and CT because the Sn4+/Sn2+ redox couple can provide two electrons exchange compared to only one electron in the Ti4+/Ti3+ couple and that no Zrm+/Zrn+ redox couple can be established, which is associated with the ones reported by Hegde et al.24
In summary, the incorporation of Zr4+, Ti4+ and Sn4+ into the lattice of CeO2 can markedly enhance the reduction behavior and the total OSC of CeO2. In particular, the CS sample has the lowest reduction peak temperature, the largest H2 consumption and total OSC, which is beneficial to the enhancement of the catalytic activity for some redox reactions, such as deNOx catalysis and three-way catalysis.
3.4. Chemical states analysis (XPS)
The synthesized samples are further analyzed by XPS to verify the elementary oxidation states and surface compositions. Due to the charging effects during XPS analysis, the binding energy scale is calibrated using adventitious carbon (284.6 eV).16 The complex spectrum of Ce 3d is decomposed into eight components, with the assignment defined in Fig. 6a. The chemical valence of cerium on the surface of these samples is mainly in a +4 oxidation state (u′′′, v′′′) and a small amount of Ce3+ (u′, v′) co-exists,37,40,41 which is consistent with the results of the UV-Vis DRS. For all the samples, the relative integrated intensities of u′ and v′ to the total eight bands are listed in Table 3. It is worth noting that the value of CZ, CT and CS is larger than that of CeO2, indicating that the ratio of Ce3+/(Ce3+ + Ce4+) on the surface of these samples is enhanced by the incorporation of Zr4+, Ti4+ and Sn4+ into the CeO2 lattice and is ranked as: CeO2 < CZ< CT < CS. There are two possible reasons, the first is that the substitution of Ce4+ (r = 0.92 Å) by Zr4+ (r = 0.80 Å), Ti4+ (r = 0.68 Å) and Sn4+ (r = 0.71 Å) in the lattice of CeO2 will lead to the contraction of the lattice while the spontaneous transformation of Ce4+ (r = 0.92 Å) into the larger Ce3+ (r = 1.03 Å) ion can compensate for this lattice contraction.37 The second is that the fraction of Ce3+ ions in the particles increases when the grain size decrease,31,32 which is supported by the XRD and TEM results.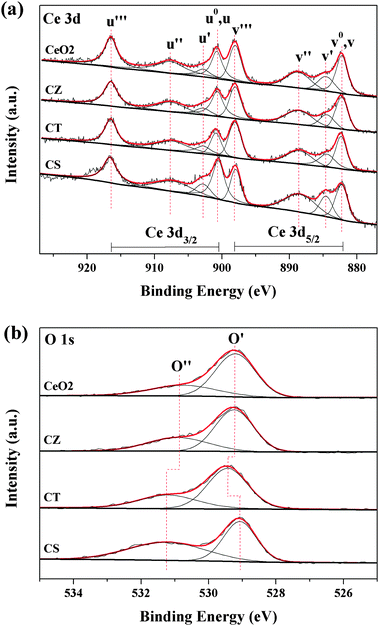 | ||
| Fig. 6 XPS spectra of (a) Ce 3d and (b) O 1s for these samples. | ||
| Sample | (Au′ + Av′)/Atotal | Atomic concentration | O/(Ce + M) | A O′′/(AO′ + AO′′) | |||
|---|---|---|---|---|---|---|---|
| C (at%) | Ce (at%) | M (at%) | O (at%) | ||||
| CeO2 | 0.1144 | 35.46 | 17.42 | — | 47.12 | 2.70 (2.00) | 0.31 |
| CZ | 0.1225 | 32.54 | 11.49 | 6.98 | 48.99 | 2.65 (2.00) | 0.34 |
| CT | 0.1307 | 29.39 | 14.21 | 5.10 | 51.30 | 2.66 (2.00) | 0.35 |
| CS | 0.1632 | 30.18 | 12.22 | 6.94 | 50.66 | 2.64 (2.00) | 0.51 |
The high-resolution spectrum for the O 1s ionization features of the synthesized samples is numerically fitted, with two components representing the primary O 1s ionization feature and chemically shifted O 1s feature from the chemisorbed surface species, which is exhibited in Fig. 6b. The main peak at 529.1–529.4 eV (O′) is attributed to the characteristic lattice oxygen bonding to the metal cations while the shoulder with the higher binding energy at 530.9–531.2 eV (O′′) is considered as the adsorbed oxygen and the oxygen in the carbonate and hydroxyl groups.16,37,40,42,43 Furthermore, the surface compositions are calculated from the XPS results and are summarized in Table 3. We can find that the O/(Ce + M) ratios for all of the samples are higher than the nominal ratio (2.00) of the full oxidation state and that the excess surface oxygen may be assigned to a high concentration of surface oxygen as an adsorbed layer of CO2, CO or water.40,42 Recently, some researchers reported that CO2 and CO could be adsorbed onto the reduced Ce3+ sites to form carbonate-like species which were more effective than those on the oxidized Ce4+ sites.33,34 In addition, as can be noted from Table 3, the relative percentage of these two oxygen species is quantified based on the area ratio of O′ and O′′ and the proportion of AO′′/(AO′ + AO′′) for these samples follows the order: CeO2 < CZ < CT < CS, which is in accordance with the order of the relative content of surface Ce3+, indicating that CO2 and CO can interact with Ce3+ sites easier than with Ce4+ sites.
3.5. CO and/or NO interaction with these samples (in situ FT-IR)
The in situ FT-IR spectra of the CO adsorption are performed at different temperatures to understand the adsorption and reduction behaviors of these samples (Fig. 7). Exposure of CeO2 to CO at room temperature gives IR bands at 1064, 1305 and 1534 cm−1 for the formation of bidentate carbonate associated with CeO2.15,43,44 Increasing the temperature to 250 °C results in the decrease of these bands due to their weak adsorption at elevated temperature. Moreover, a band at 2360 cm−1 for gaseous CO2 can be detected and its intensity gradually increases when the temperature increases from 25 to 250 °C, which may be resulted from the reduction of CeO2 by CO and/or the desorption of bidentate carbonate during the heating process.17,28,45 Interestingly, further increasing the temperature up to 300 °C leads to an increase of the bands for bidentate carbonate and the appearance of new bands at 1380 and 1474 cm−1 for polydentate carbonate coordinated with the reduced ceria.15,43,46 Simultaneously, the band from gaseous CO2 decreases with the temperature from 250 to 300 °C and the reason may be that the formation of Ce3+ is beneficial for adsorbing CO2 to form bidentate carbonate and polydentate carbonate during the heating treatment.33,34 For the CZ, CT and CS samples, the bands assigned to the adsorption of bidentate carbonate at 1060–1064, 1305, and 1530–1534 cm−1 decrease and the band for gaseous CO2 at 2360 cm−1 increases from 25 to 200 °C. In addition, the bands for bidentate carbonate increase, the bands attributed to polydentate carbonate appear at 1380–1384 and 1470–1474 cm−1 and the band assigned to gaseous CO2 decreases when the temperature further increases up to 300 °C, indicating that these ceria-containing solid solutions are more easily reduced than CeO2 by CO.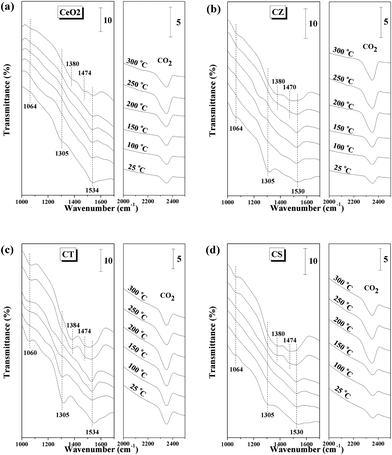 | ||
| Fig. 7 In situ FT-IR spectra of the 10% CO/Ar interaction with (a) CeO2, (b) CZ, (c) CT and (d) CS at different temperatures. | ||
In order to obtain information on the adsorbed NO species on CeO2, CZ, CT, and CS, the NO adsorption FT-IR spectra of these samples have been recorded, as shown in Fig. 8. For the CeO2, CZ and CT samples, the bands at 1263–1270 and 1473–1505 cm−1 could be attributed to the NO2 asymmetric vibration band of linear nitrite and monodentate nitrate, respectively.28,47–49 When the temperature increases to 200 °C, these bands disappear due to their poor stability. The band for physically adsorbed NO species at 1749 cm−1 is lost at 100 °C because of the weak adsorption behavior.46 The bridging bidentate nitrate exhibits a remarkable NO2 symmetric vibration band at 1002–1005 cm−1 and a weak N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching model at 1604–1616 cm−1 during the whole temperature range and the intensity of these bands weakens when the temperature is raised to 300 °C but do not disappear.22,28 The increase in the temperature up to 200 °C leads to the enhancement of the band at 1220–1227 cm−1 and the appearance of a new band at 1560–1564 cm−1 for the NO2 symmetric and asymmetric vibration of chelating bidentate nitrate and raising the temperature further up to 300 °C leads to the decrease of these bands (but it is difficult to be completely desorbed/transformed/decomposed).28,46 For the CS sample, besides the adsorption behaviors of the bands at 1005, 1604, 1227, and 1749 cm−1 for bridging bidentate nitrate, chelating bidentate nitrate and physically adsorbed NO species are very similar to the above-mentioned three samples. The bands attributed to the adsorption of linear nitrite and monodentate nitrate at 1263 and 1473 cm−1 are eliminated at a lower temperature of 150 °C and then a new band appears at 1564 cm−1 for chelating bidentate nitrate, indicating that the NO adsorbed species on the surface of CS could be desorbed/transformed/decomposed more easily than those on the CeO2, CZ and CT samples.
O stretching model at 1604–1616 cm−1 during the whole temperature range and the intensity of these bands weakens when the temperature is raised to 300 °C but do not disappear.22,28 The increase in the temperature up to 200 °C leads to the enhancement of the band at 1220–1227 cm−1 and the appearance of a new band at 1560–1564 cm−1 for the NO2 symmetric and asymmetric vibration of chelating bidentate nitrate and raising the temperature further up to 300 °C leads to the decrease of these bands (but it is difficult to be completely desorbed/transformed/decomposed).28,46 For the CS sample, besides the adsorption behaviors of the bands at 1005, 1604, 1227, and 1749 cm−1 for bridging bidentate nitrate, chelating bidentate nitrate and physically adsorbed NO species are very similar to the above-mentioned three samples. The bands attributed to the adsorption of linear nitrite and monodentate nitrate at 1263 and 1473 cm−1 are eliminated at a lower temperature of 150 °C and then a new band appears at 1564 cm−1 for chelating bidentate nitrate, indicating that the NO adsorbed species on the surface of CS could be desorbed/transformed/decomposed more easily than those on the CeO2, CZ and CT samples.
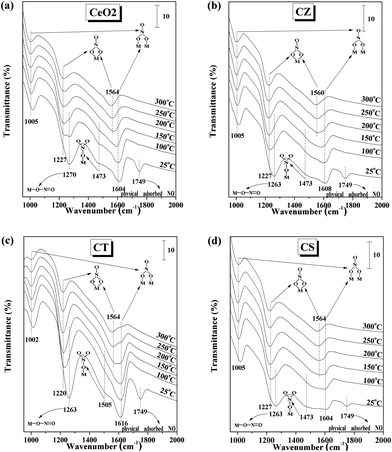 | ||
| Fig. 8 In situ FT-IR spectra of the 5% NO/Ar interaction with (a) CeO2, (b) CZ, (c) CT and (d) CS at different temperatures (the models of these adsorbed NO species were displayed in the figure). | ||
The nature and population of the adsorbed NOx/COx species are identified by in situ FT-IR spectra under simulated reaction conditions, with the purpose of further understanding the difference in the activities and selectivities for the synthesized samples. Fig. 9 displays a representative spectra of these samples at various temperatures. It can be seen from this figure that NO preferentially interacts with these samples due to its unpaired electron as a result of producing complex types of nitrite-/nitrate-like species which are chemisorbed on the surface of these samples.50,51 For the CeO2, CZ and CT samples, the spectra are dominated by the bands for bridging bidentate nitrate located at 1004–1005 cm−1 and 1604–1616 cm−1. The intensity of these bands weakens during the heating process but they do not disappear even at 300 °C. The increase of the band at 1227 cm−1 and the appearance of a new band at 1564 cm−1 for chelating bidentate nitrate can be observed when the temperature is increased up to 150 °C but further increasing the temperature up to 300 °C leads to a decrease of these bands. Furthermore, the bands at 1266–1270 and 1473–1505 cm−1 for linear nitrite and monodentate nitrate, respectively, disappear at 250 °C due to their weak adsorption. At the same time, a new band appears at 1531 cm−1 for bidentate carbonate, which is a result from the desorbed/transformed/decomposed of NOx species leading to the exposure of some adsorption sites to adsorb COx species. Interestingly, when the temperature is increased up to 300 °C, the appearance of a new band for gaseous CO2 at 2360 cm−1 can be observed, indicating that a reaction between the NOx and COx species has happened,37 which is consistent with the results of the activity and selectivity. For the CS sample, all the different NOx adsorption species completely disappear at 250 °C and then several new bands appear at 1008, 1531, 1384, 1464 and 2360 cm−1 for bidentate carbonate, polydentate carbonate and gaseous CO2, respectively. In particular, the band at 1531 cm−1 can be observed even at 200 °C. These results indicate that the initial temperature of the reaction between the NOx and COx species for CS is lower than the CeO2, CZ and CT samples, suggesting that the catalytic performance of CS is better than the other three samples, which agrees with the activity and selectivity results.
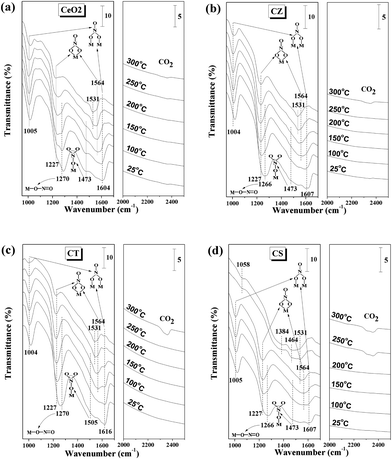 | ||
| Fig. 9 In situ FT-IR spectra of the 10% CO/Ar and 5% NO/Ar co-interaction with (a) CeO2, (b) CZ, (c) CT and (d) CS at different temperatures. | ||
3.6. Possible reaction mechanism for NO removal by CO
According to the obtained results, a possible reaction mechanism for NO removal by CO is tentatively proposed to further understand the nature of this reaction, as shown in Fig. 10. Taking the CS solid solution as an example, when exposing it to a simulated reaction condition of NO and CO at room temperature (25 °C), NO is preferentially adsorbed onto the surface of CS to generate several types of nitrite and nitrate species. Raising the temperature up to 200 °C leads to a small quantity of adsorbed NOx species being desorbed/transformed/decomposed and therefore releasing a few sites to adsorb CO. Furthermore, trace amounts of N2O, N2 and CO2 are produced during this process. When the temperature is further increased up to 250 °C, the CS solid solution can be reduced by CO to form a large quantity of surface Ce3+ and oxygen vacancies. Since the surface Ce3+ can provide more adsorbed-sites to adsorb COx species,33,34 the oxygen vacancy can weaken the N–O bond to promote the dissociation of NOx.52 As a result, a considerable amount of N2 and CO2 can be observed. Overall, on the one hand, the smallest grain size of the CS sample results in the highest amount of surface Ce3+. On the other hand, the best reduction behavior of the CS sample is beneficial to generate more surface Ce3+ and oxygen vacancies during the reaction process. The synergistic effect of these aspects promotes the catalytic performance of NO elimination efficiently.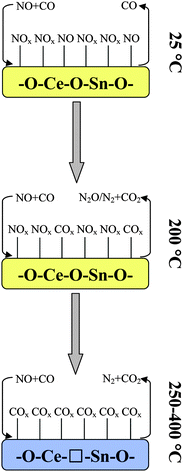 | ||
| Fig. 10 Possible reaction mechanism for NO removal by CO over the CS solid solution, □: oxygen vacancy. | ||
4. Conclusions
In this paper, we prepared a series of ceria-containing solid solutions (CZ, CT and CS) and CeO2 as a reference by an inverse co-precipitation method with the purpose of investigating the structural, reduction, adsorption properties and catalytic activities of the synthesized samples for NO removal by CO. Combined with the above-mentioned characterizations, several conclusions can be obtained as follows:(1) The crystal growth of the cubic phase can be inhibited by the introduction of Zr4+, Ti4+ and Sn4+, i.e., the grain size will be decreased, which leads to the enlargement of surface Ce3+ content and an increase of the BET specific surface area and consequently improves the catalytic performance.
(2) The reduction properties and total OSC of these ceria-containing solid solutions are enhanced compared with pure CeO2, which is conducive to the formation of more surface Ce3+ and oxygen vacancies during the reaction process and further upgrades the catalytic performance.
(3) The catalytic activities and selectivities of these solid solutions are higher than those of CeO2, particularly for CS which has the optimal catalytic performance due to its smallest grain size, best reduction behavior and suitable adsorption intensity for NOx and COx species. The most essential reason is that the surface Ce3+ and oxygen vacancies play a significant role in NO removal by CO model reaction.
Acknowledgements
The financial supports of the National Natural Science Foundation of China (Nos. 20973091, 21273110), the National Basic Research Program of China (973 program, Nos. 2009CB623500, 2010CB732300) and Jiangsu Province Science and Technology Support Program (Industrial, BE2011167) are gratefully acknowledged.References
- L. Ilieva, G. Pantaleo, I. Ivanov, A. M. Venezia and D. Andreeva, Appl. Catal., B, 2006, 65, 101–109 CrossRef CAS.
- G. S. Qi and R. T. Yang, Chem. Commun., 2003, 848–849 RSC.
- J. Kašpar, P. Fornasiero and M. Graziani, Catal. Today, 1999, 50, 285–298 CrossRef.
- M. A. Centeno, M. Paulis, M. Montes and J. A. Odriozola, Appl. Catal., A, 2002, 234, 65–78 CrossRef CAS.
- F. Larachi, J. Pierre, A. Adnot and A. Bernis, Appl. Surf. Sci., 2002, 195, 236–250 CrossRef CAS.
- M. Sahibzada, B. C. H. Steele, K. Zheng, R. A. Rudkin and I. S. Metcalfe, Catal. Today, 1997, 38, 459–466 CrossRef CAS.
- A. Trovarelli, C. de Leitenburg, M. Boaro and G. Dolcetti, Catal. Today, 1999, 50, 353–367 CrossRef CAS.
- Q. Yu, X. X. Wu, C. J. Tang, L. Qi, B. Liu, F. Gao, K. Q. Sun, L. Dong and Y. Chen, J. Colloid Interface Sci., 2011, 354, 341–352 CrossRef CAS.
- B. M. Reddy, P. Lakshmanan and A. Khan, J. Phys. Chem. B, 2004, 108, 16855–16863 CrossRef CAS.
- C. Shi, Y. Y. Ji, U. M. Graham, G. Jacobs, M. Crocker, Z. S. Zhang, Y. Wang and T. J. Toops, Appl. Catal., B, 2012, 119–120, 183–196 CrossRef CAS.
- M. F. Luo, J. Chen, L. S. Chen, J. Q. Lu, Z. C. Feng and C. Li, Chem. Mater., 2001, 13, 197–202 CrossRef CAS.
- S. Letichevsky, C. A. Tellez, R. R. D. Avillez, M. I. P. D. Silva, M. A. Fraga and L. G. Appel, Appl. Catal., B, 2005, 58, 203–210 CrossRef CAS.
- W. T. Chen, K. B. Chen, M. F. Wang, S. F. Weng, C. S. Lee and M. C. Lin, Chem. Commun., 2010, 46, 3286–3288 RSC.
- B. M. Reddy, A. Khan, P. Lakshmanan, M. Aouine, S. Loridant and J. Volta, J. Phys. Chem. B, 2005, 109, 3355–3363 CrossRef CAS.
- J. Y. Luo, M. Meng, Y. Q. Zha and L. H. Guo, J. Phys. Chem. C, 2008, 112, 8694–8701 CAS.
- T. Y. Li, S. J. Chiang, B. J. Liaw and Y. Z. Chen, Appl. Catal., B, 2011, 103, 143–148 CrossRef CAS.
- M. Adamowska, S. Muller, P. Da Costa, A. Krzton and P. Burg, Appl. Catal., B, 2007, 74, 278–289 CrossRef CAS.
- Y. Liu, C. Wen, Y. Guo, G. Z. Lu and Y. Q. Wang, J. Phys. Chem. C, 2010, 114, 9889–9897 CAS.
- B. M. Reddy, A. Khan, Y. Yamada, T. Kobayashi, S. Loridant and J. C. Volta, J. Phys. Chem. B, 2003, 107, 11475–11484 CrossRef CAS.
- P. Fornasiero, G. Balducci, R. Di Monte, J. Kašpar, V. Sergo, G. Gubitosa, A. Ferrero and M. Graziani, J. Catal., 1996, 164, 173–183 CrossRef CAS.
- A. Trovarelli, F. Zamar, J. Llorca, C. D. Leitenburg, G. Dolcetti and J. T. Kiss, J. Catal., 1997, 169, 490–502 CrossRef CAS.
- I. Atribak, B. Azambre, A. Bueno López and A. García-García, Appl. Catal., B, 2009, 92, 126–137 CrossRef CAS.
- Z. K. Zhao, X. L. Lin, R. H. Jin, G. R. Wang and T. Muhammad, Appl. Catal., B, 2012, 115–116, 53–62 CrossRef CAS.
- T. Baidya, A. Gupta, P. A. Deshpandey, G. Madras and M. S. Hegde, J. Phys. Chem. C, 2009, 113, 4059–4068 CAS.
- T. Baidya, A. Marimuthu, M. S. Hegde, N. Ravishankar and G. Madras, J. Phys. Chem. C, 2007, 111, 830–839 CAS.
- T. B. Nguyen, J. P. Deloume and V. Perrichon, Appl. Catal., A, 2003, 249, 273–284 CrossRef CAS.
- Z. G. Liu, R. X. Zhou and X. M. Zheng, J. Nat. Gas Chem., 2008, 17, 125–129 CrossRef CAS.
- L. J. Liu, B. Liu, L. H. Dong, J. Zhu, H. Q. Wan, K. Q. Sun, B. Zhao, H. Y. Zhu, L. Dong and Y. Chen, Appl. Catal., B, 2009, 90, 578–586 CrossRef CAS.
- M. F. Luo, J. M. Ma, J. Q. Lu, Y. P. Song and Y. J. Wang, J. Catal., 2007, 246, 52–59 CrossRef CAS.
- L. X. Yin, Y. Q. Wang, G. S. Pang, Y. Koltypin and A. Gedanken, J. Colloid Interface Sci., 2002, 246, 78–84 CrossRef CAS.
- R. K. Hailstone, A. G. DiFrancesco, J. G. Leong, T. D. Allston and K. J. Reed, J. Phys. Chem. C, 2009, 113, 15155–15159 CAS.
- L. J. Wu, H. J. Wiesmann, A. R. Moodenbaugh, R. F. Klie, Y. M. Zhu, D. O. Welch and M. Suenaga, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 125415 CrossRef.
- Z. X. Song, W. Liu and H. Nishiguchi, Catal. Commun., 2007, 8, 725–730 CrossRef CAS.
- M. Boaro, F. Giordano, S. Recchia, V. Dal Santo, M. Giona and A. Trovarelli, Appl. Catal., B, 2004, 52, 225–237 CrossRef CAS.
- R. Kydd, W. Y. Teoh, K. Wong, Y. Wang, J. Scott, Q. H. Zeng, A. B. Yu, J. Zou and R. Amal, Adv. Funct. Mater., 2009, 19, 369–377 CrossRef CAS.
- B. M. Reddy, P. Bharali, P. Saikia, S. E. Park, M. W. E. van den Berg, M. Muhler and W. Grünert, J. Phys. Chem. C, 2008, 112, 11729–11737 CAS.
- L. J. Liu, Z. J. Yao, B. Liu and L. Dong, J. Catal., 2010, 275, 45–60 CrossRef CAS.
- F. Fally, V. Perrichon, H. Vidal, J. Kaspar, G. Blanco, J. M. Pintado, S. Bernal, G. Colon, M. Daturi and J. C. Lavalley, Catal. Today, 2000, 59, 373–386 CrossRef CAS.
- Y. Z. Chen, B. J. Liaw and C. W. Huang, Appl. Catal., A, 2006, 302, 168–176 CrossRef CAS.
- M. Alifanti, B. Baps, N. Blangenois, J. Naud, P. Grange and B. Delmon, Chem. Mater., 2003, 15, 395–403 CrossRef CAS.
- Q. G. Dai, S. X. Bai, Z. Y. Wang, X. Y. Wang and G. Z. Lu, Appl. Catal., B, 2012, 126, 64–75 CrossRef CAS.
- G. Avgouropoulos and T. Ioannides, Appl. Catal., B, 2006, 67, 1–11 CrossRef CAS.
- L. J. Liu, Z. J. Yao, Y. Deng, F. Gao, B. Liu and L. Dong, ChemCatChem, 2011, 3, 978–989 CrossRef CAS.
- X. Q. Cheng, A. M. Zhu, Y. Z. Zhang, Y. Wang, C. T. Au and C. Shi, Appl. Catal., B, 2009, 90, 395–404 CrossRef CAS.
- A. Kotsifa, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2007, 72, 136–148 CrossRef CAS.
- L. J. Liu, J. G. Cai, L. Qi, Q. Yu, K. Q. Sun, B. Liu, F. Gao, L. Dong and Y. Chen, J. Mol. Catal. A: Chem., 2010, 327, 1–11 CrossRef CAS.
- K. I. Hadjiivanov, Catal. Rev. Sci. Eng., 2000, 42, 71–144 CAS.
- L. J. Liu, Y. Chen, L. H. Dong, J. Zhu, H. Q. Wan, B. Liu, B. Zhao, H. Y. Zhu, K. Q. Sun, L. Dong and Y. Chen, Appl. Catal., B, 2009, 90, 105–114 CrossRef CAS.
- A. Martinez-Arias, J. Soria, J. C. Conesa, X. L. Seoane, A. Arcoya and R. Catalufia, J. Chem. Soc., Faraday Trans., 1995, 91, 1679–1687 RSC.
- M. Haneda, K. Shinoda, A. Nagane, O. Houshito, H. Takagi, Y. Nakahara, K. Hiroe, T. Fujitani and H. Hamada, J. Catal., 2008, 259, 223–231 CrossRef CAS.
- L. J. Liu, Q. Yu, J. Zhu, H. Q. Wan, K. Q. Sun, B. Liu, H. Y. Zhu, F. Gao, L. Dong and Y. Chen, J. Colloid Interface Sci., 2010, 349, 246–255 CrossRef CAS.
- S. H. Oh, G. B. Fisher, J. E. Carpenter and D. W. Goodman, J. Catal., 1986, 100, 360–376 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |