Catalytically active structures of SiO2-supported Au nanoparticles in low-temperature CO oxidation
Kun Qianabc, Liangfeng Luoabc, Huizhi Baoabc, Qing Huaabc, Zhiquan Jianga and Weixin Huang*abc
aHefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei 230026, China
bCAS Key Laboratory of Materials for Energy Conversion, University of Science and Technology of China, Hefei 230026, China
cDepartment of Chemical Physics, University of Science and Technology of China, Hefei 230026, China. E-mail: huangwx@ustc.edu.cn
First published on 26th October 2012
Abstract
Various Au/SiO2 catalysts have been prepared by the deposition–precipitation method followed by calcination in air or reduction in H2. The structures of supported Au nanoparticles were characterized in detail by XRD, TEM, XPS, in situ XANES and operando DRIFTS of CO chemisorption, and their catalytic activity in CO oxidation was evaluated. Calcined in air, the gold precursor decomposes into Au(I) species at low temperatures and further to Au(0) at elevated temperatures, forming supported Au nanoparticles mostly larger than 4.5 nm. Reduced in H2, the gold precursor can be facilely reduced to Au(0) at low temperatures, forming supported Au nanoparticles with different size distributions depending on the reduction temperature. Supported Au nanoparticles around 3–4.5 nm with both abundant low-coordinated Au atoms and bulk Au-like electronic structure effectively chemisorb CO and catalyze CO oxidation at room temperature (RT). Larger supported Au nanoparticles with bulk Au-like electronic structure but few low-coordinated Au atoms do not chemisorb CO and catalyze CO oxidation at RT, and finer supported Au nanoparticles with abundant low-coordinated Au atoms but bulk Au-unlike electronic structure also do not chemisorb CO and catalyze CO oxidation at RT. These results provide solid and comprehensive experimental evidence that supported Au nanoparticles with both abundant low-coordinated Au atoms and bulk Au-like electronic structure are the catalytic active structures for catalyzing CO oxidation at RT without the involvement of oxide supports. The density of low-coordinated Au atoms increases with the decrease of their size, but their electronic structure eventually deviates from bulk Au-like electronic structure; therefore, the catalytic activity of SiO2-supported Au nanoparticles in low-temperature CO oxidation inevitably exhibits a volcano-shaped dependence on their size with the optimum size between 3 and 4.5 nm.
Introduction
Heterogeneous catalysis by gold has advanced greatly since Haruta et al. found that gold was catalytically very active for low-temperature CO oxidation when dispersed on base metal oxides as nanoparticles below 5 nm.1 In addition to low-temperature CO oxidation, supported Au catalysts also exhibit unusual activity and selectivity in a wide array of reactions, particularly in catalytic reactions involving molecular oxygen.2 However, the fundamental understandings of gold catalysis, particularly concerning the active structure of supported Au nanoparticles and the catalytic reaction mechanism, still remain ambiguous even for low-temperature CO oxidation reaction.3 The periphery hypothesis is popular for low-temperature CO oxidation catalyzed by Au nanoparticles supported on semi-conductive or reducible metal oxides, in which CO is considered to adsorb on the gold surface (most likely at edges and corners) and the oxygen molecule is activated at the Au–metal oxide perimeter interfaces.4,5 Another popular reaction mechanism only involves gold, in which CO chemisorbed on the gold surface is proposed to insert into the Au–OH bond to form a hydroxycarbonyl that is oxidized to bicarbonate, which then decomposes into Au–OH and CO2.6,7The size-dependent catalytic activity of supported Au nanoparticles in low-temperature CO oxidation has been the core issue under investigation. Haruta et al. firstly reported that the catalytic activity of Au nanoparticles supported on TiO2, Fe2O3 and Co3O4 sharply increased when their diameter decreases below 5 nm.8,9 Later Goodman et al. observed that Au nanoparticles supported on the TiO2(110) surface with a size of 3.5 nm exhibited the maximum catalytic activity.10 Thereafter, studies exploring Au nanoparticles supported on TiO2 and TiO2-coated silica aerogel smaller than 2 nm also demonstrated the existence of an optimal size (2–2.5 nm) for their catalytic activity.11,12 Recently Laoufi et al. confirmed the existence of an optimal size (2.1 ± 0.3 nm) for the catalytic activity in the Au/TiO2(110) model catalyst system by an operando study.13 The structure of supported Au nanoparticles with the optimal size and the interpretation of size-dependent catalytic activity of supported Au nanocatalysts remain controversial. The decrease in the size of supported Au nanoparticles could increase the Au–metal oxide perimeter interface length and thus enhance the catalytic activity of supported Au catalysts following the periphery reaction mechanism.3 The size of supported Au nanoparticles also significantly affects their structure and morphology. Tai et al. revealed that lattice contraction and structural changes of Au nanoparticles supported on TiO2-coated silica aerogel became prominent at Au diameters less than 4–5 nm, consistent with the enhanced catalytic activity.12 Fine Au nanoparticles normally expose a high density of low-coordinated Au atoms at their corners, steps and edges. On the basis of DFT theoretical calculations, Nørskov et al. considered low-coordinated Au atoms on supported Au nanoparticles as the active sites and viewed the effect of support as structural and electronic promotion.14–16 Zanella et al. also attributed the decrease of the catalytic activity of Au nanoparticles supported on TiO2 with increasing calcination temperature to the particle shape change from the shape containing a large proportion of low-coordinated sites to truncated octahedra with smooth facets.11 The decrease in the size of Au nanoparticles could also change their electronic structure, in which the quantum size effect, i.e. the metal-to-nonmetal transition, is of particular interest. The critical thickness of supported Au nanoparticles for the metal-to-nonmetal transition was reported to be of two atom layer thickness.10,17 Goodman et al. correlated the size-dependent catalytic activity of supported Au nanoparticles on TiO2(110) to the quantum size effect and proposed that supported Au nanoparticles with two layers of gold were most catalytically active.10 Later they also demonstrated that the two-layered Au film vacuum-deposited on a thin layer of TiO2 on Mo(112) was the most active structure for low-temperature CO oxidation.18 Au clusters with a bi-layer structure were also proposed to be responsible for the high catalytic activity of coprecipitated Au/FeOOH dried at 393 K in low-temperature CO oxidation.19 However, in a highly active Au/Fe2O3 catalyst prepared with the colloidal deposition method, Liu et al. reported that supported Au nanoparticles had diameters larger than 1 nm and that bilayer structures and/or diameters of about 0.5 nm were not mandatory to achieve the high activity.20 Recent simultaneous reactivity measurements and grazing incidence small-angle X-ray scattering characterizations of the Au/TiO2(110) model catalyst revealed that supported Au nanoparticles with the optimal size (2.1 ± 0.3 nm) were with a height of about six atomic layers.13
Previous studies of the size-dependent catalytic activity of supported Au nanoparticles all employ “active” metal oxides or hydroxides as supports that can participate in low-temperature CO oxidation themselves,21,22 the observed catalytic activity thus depends not only on the structure of Au nanoparticles but also on the support and the Au–support interface, which makes it difficult to reach a consensus on the optimal size of supported Au nanoparticles and their structure. Meanwhile, it has not been explored much how the size of supported Au nanoparticles affects their ability to chemisorb and activate CO and O2. Shaikhutdinov et al. studied CO adsorption on Au clusters in a range of sizes on Fe3O4, FeO and Al2O3 thin films by means of temperature-programmed desorption.23 Their studies found that regardless of the substrate, Au nanoparticles of a threshold diameter (3.0 nm) desorbed CO at the same maximum temperature. Meier and Goodman employed infrared reflection absorption spectroscopy to study CO adsorption on Au clusters ranging in size from 1.8 to 3.1 nm supported on TiO2(110).24 The vibrational frequency of adsorbed CO blue-shifted slightly compared to that adsorbed on bulk Au, whereas the heats of adsorption increased sharply with decreasing cluster size and the Au cluster size corresponding to the highest heat of adsorption for CO was very close to that exhibiting the maximum catalytic activity for CO oxidation.
SiO2 is an “inert” support that does not contribute to the catalytic activity of supported Au nanocatalysts in low-temperature CO oxidation,22,25,26 therefore, Au/SiO2 catalysts are suitable for the study of the structure–activity relationship of supported Au nanoparticles. Employing Au/SiO2 catalysts, we have successfully demonstrated the Au–support interaction in supported Au catalysts27–30 and the roles of low-coordinated Au atoms31 and surface hydroxyls32 on the catalytic activity of supported Au nanoparticles in low-temperature CO oxidation. In this paper, we report comprehensive experimental results for the size–structure–chemisorption–catalytic activity relationship of Au nanoparticles supported on inert SiO2, not only solidly evidencing a volcano-shaped dependence of chemisorption and catalytic activity on the size but also clarifying the origin. Supported Au nanoparticles with both abundant low-coordinated Au atoms and bulk Au-like electronic structure are the catalytically active structure for catalyzing CO oxidation at RT without the involvement of oxide supports. The density of low-coordinated Au atoms increases with the decrease of their size, but their electronic structure eventually deviates from bulk Au-like electronic structure; therefore, the catalytic activity of SiO2-supported Au nanoparticles inevitably exhibits a volcano-shaped dependence on their size with the existence of an optimum size.
Experimental section
Au/SiO2 catalysts with a calculated Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : SiO2 weight ratio of 2% were prepared by the deposition–precipitation (DP) method employing HAuCl4 as the gold precursor.33,34 Typically, 0.9332 g HAuCl4·4H2O (Sinopharm Chemical Reagent Co., Ltd, Au content ≥47.8%) was dissolved in 50 mL distilled water to prepare a 0.0453 mol L–1 HAuCl4 aqueous solution. 8.97 mL HAuCl4 aqueous solution, 4.0 g SiO2 (40–120 mesh, specific surface area: 390 m2 g−1, Qingdao Haiyang Chemicals Co.) and 50 mL distilled water were co-added into a three-necked bottle and adequately mixed by stirring at 60 °C for 30 min. An appropriate amount of ammonium hydroxide was added to adjust the pH value of the system between 9 and 10, after which the system was stirred at 60 °C for 24 h. The solid was then filtered, washed with distilled water several times, dried at 60 °C for 12 h, and stored in darkness as the catalyst precursor. The catalyst precursor was then treated at desired temperatures and atmosphere (air or 10% H2 in Ar, flow rate: 20 mL min−1) for 4 hours to prepare various Au/SiO2 catalysts denoted Au/SiO2-A-T, where A and T stand for the treatment atmosphere and temperature, respectively.
: SiO2 weight ratio of 2% were prepared by the deposition–precipitation (DP) method employing HAuCl4 as the gold precursor.33,34 Typically, 0.9332 g HAuCl4·4H2O (Sinopharm Chemical Reagent Co., Ltd, Au content ≥47.8%) was dissolved in 50 mL distilled water to prepare a 0.0453 mol L–1 HAuCl4 aqueous solution. 8.97 mL HAuCl4 aqueous solution, 4.0 g SiO2 (40–120 mesh, specific surface area: 390 m2 g−1, Qingdao Haiyang Chemicals Co.) and 50 mL distilled water were co-added into a three-necked bottle and adequately mixed by stirring at 60 °C for 30 min. An appropriate amount of ammonium hydroxide was added to adjust the pH value of the system between 9 and 10, after which the system was stirred at 60 °C for 24 h. The solid was then filtered, washed with distilled water several times, dried at 60 °C for 12 h, and stored in darkness as the catalyst precursor. The catalyst precursor was then treated at desired temperatures and atmosphere (air or 10% H2 in Ar, flow rate: 20 mL min−1) for 4 hours to prepare various Au/SiO2 catalysts denoted Au/SiO2-A-T, where A and T stand for the treatment atmosphere and temperature, respectively.The compositions of catalysts were analyzed by an inductively coupled plasma atomic emission spectrometer (ICP-AES). XRD spectra were acquired on a Philips X'Pert PRO SUPER X-ray diffractometer with a Ni-filtered Cu Kα X-ray source operating at 40 kV and 50 mA. XPS measurements were performed on an ESCALAB 250 electron spectrometer using a monochromatized Al Kα excitation source (hν = 1486.6 eV). The binding energies in XPS spectra were referenced with respect to the Si 2p binding energy in SiO2 at 103.3 eV. Transmission electron microscopy (TEM) measurements were preformed on JEOL-2010 and JEOL-2100F high-resolution transmission electron microscopes.
Operando diffuse reflectance infrared spectroscopy (DRIFTS) measurements were performed on a Nicolet 6700 FTIR spectrometer equipped with an in situ DRIFTS reaction cell (Harrick Scientific Products, INC) at RT. A home-made pulse flow apparatus (volume: 0.28 mL) was connected to the reaction cell to realize the pulse adsorption process that can effectively suppress the interference from gas-phase CO. Prior to the measurements, 10 mg catalyst precursor was loaded on the sample stage of the reaction cell and treated at desired temperatures and atmosphere (air or 10% H2 in Ar, flow rate: 20 mL min−1) for 4 hours, then purged with Ar (flow rate: 20 mL min−1) and cooled down to RT to prepare various Au/SiO2 catalysts. The pulse gas consists of 1% CO in Ar and the time between two consecutive pulses was 20 s. The DRIFT spectra were measured in a series mode with 16 scans and a resolution of 4 cm−1 using a MCT/A detector. The DRIFTS spectrum of the freshly-prepared catalyst purged with Ar at RT was taken as the background spectrum.
In situ Au LIII-edge X-ray absorption (XAS) spectra were measured in a transmission mode with an energy step of 0.5 eV at the BL14W1 beamline of Shanghai Synchrotron Radiation Facility (SSRF). The catalyst precursor was mounted on the sample stage of an in situ reaction cell and treated at desired temperatures and atmosphere for 30 min, then purged with He (flow rate: 20 mL min−1) and cooled down to RT for the XAS measurement. The Au LIII-edge XAS spectra of the gold foil and HAuCl4 powder were also measured for comparisons. All XPS data were processed with the Ifeffit software (version 1.2.11) to acquire the Au LIII-edge X-ray absorption near-edge structure spectra (XANES).
The catalytic activity was evaluated on a fixed-bed flow reactor. Prior to the catalytic activity evaluation, 100 mg catalyst precursor in the catalytic reactor were treated at desired temperatures and atmosphere (air or 10% H2 in Ar, flow rate: 20 mL min−1) for 4 hours and then cooled to RT in Ar (flow rate: 20 mL min−1) to prepare various Au/SiO2 catalysts. Then the reaction gas consisting of 1% CO and 99% dry air was fed to the catalyst at a rate of 20 mL min−1. The catalyst was heated to desirable temperatures at a heating rate of 2 °C min−1, and the steady-state composition of the effluent gas was analyzed with an online GC-14C gas chromatograph equipped with a TDX-01 column (T = 80 °C, H2 as the carrier gas at 30 mL min−1) after the catalytic reaction proceeded for 30 min. The CO conversion was calculated from the change in CO concentrations in the inlet and outlet gases. The catalyst stability was examined by operating the catalytic reaction at desired reaction temperatures for 3 hours and the composition of the effluent gas was analyzed every 1 hour.
Results and discussion
ICP-AES analysis results show that the Au loadings in all Au/SiO2 catalysts are the same (1.8% weight ratio), which is reasonable because all catalysts were prepared employing the same catalyst precursor. No chlorine was detected by XPS in all Au/SiO2 catalysts. Fig. 1A shows the catalytic performance of various Au/SiO2 catalysts in CO oxidation up to 180 °C. Au/SiO2 catalysts prepared by calcination in air at 200 and 300 °C and higher temperatures are catalytically inactive while those prepared by reduction in H2 are active and their catalytic activities depend on the reduction temperature. Au/SiO2-H2-120 exhibits an observable activity in catalyzing CO oxidation at RT, and the catalytic activity of Au/SiO2-H2 at RT initially increases rapidly with the increase of reduction temperature, then slightly decreases when the reduction temperature reaches 600 °C. Au/SiO2-H2-300 achieves 77% and 100% CO conversions at RT and 60 °C, respectively. Under our catalytic reaction conditions, a 100% CO conversion corresponds to a reaction rate of 0.289 molCO gAu−1 h−1. With the increase of reaction temperature, the catalytic activity of Au/SiO2-H2 catalysts initially increases, then decreases. Such a dependence of the catalytic activity on the reaction temperature was previously observed in Au/ZnO/SiO2 and Au/CeO2/SiO2 catalysts28,30 which was interpreted by the temperature-dependent formation and competition between the surface reaction and desorption of a weakly chemisorbed surface intermediate involved in low-temperature CO oxidation. The coverage of this weakly chemisorbed species initially increases with increasing reaction temperature, resulting in an initial increase in the catalytic activity. But with further increases in reaction temperature, desorption of this weakly chemisorbed species occurs and eventually overwhelms, decreasing the coverage of the weakly chemisorbed species and thus the catalytic activity. Fig. 1B displays the stability of Au/SiO2-H2-200 in CO oxidation. Under the tested conditions, the CO conversion slightly decreases at RT but remains stable at elevated reaction temperatures.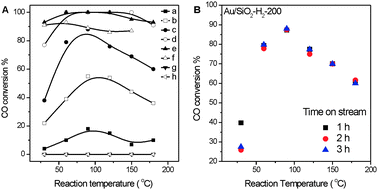 | ||
| Fig. 1 (A) Catalytic performance in CO oxidation of Au/SiO2-H2-120 (a), Au/SiO2-H2-150 (b), Au/SiO2-H2-200 (c), Au/SiO2-H2-300 (d), Au/SiO2-H2-400 (e), Au/SiO2-H2-600 (f), Au/SiO2-Air-200 (g) and Au/SiO2-Air-300 (h). (B) Stability of Au/SiO2-H2-200 in CO oxidation. | ||
It is of great interest that our Au/SiO2-H2 catalysts simply prepared by the conventional DP method employing HAuCl4 as the gold precursor followed by H2 reduction are active in catalyzing CO oxidation at low temperatures. On one hand, since SiO2 is an inert support, it is rather difficult to prepare Au/SiO2 catalysts active in low-temperature CO oxidation. Previous successful examples involve complicated methods or unusual gold precursors, including Au/SiO2 catalysts prepared by chemical vapor deposition,35 by using gold–inorganic–organic high-surface-area materials as precursors,36 by the deposition of fine Au sols followed by oxidation and reduction treatments,37 by the in situ formation of gold amine cation complexes,38 and by using a cationic gold complex cation ([Au(en)2]2+) as the gold precursor.39 Obviously, our method is simple and convenient. On the other hand, since SiO2 is an “inert” support,22,25,26 the observed different catalytic activities of various Au/SiO2 catalysts offer an appropriate system to investigate the structure–activity relationship of supported Au catalysts without the involvement of oxide supports. The structure of Au/SiO2-Air-200 has been previously reported;31,32,40 therefore, the structures of representative Au/SiO2-Air-300, Au/SiO2-H2-300, Au/SiO2-H2-200 and Au/SiO2-H2-120 catalysts were studied in detail.
Fig. 2 displays XRD patterns of the catalyst precursor, Au/SiO2-Air-300 and various Au/SiO2-H2 catalysts. The XRD pattern of the catalyst precursor does not show any diffraction peaks, and that of Au/SiO2-Air-300 clearly shows Au(111), Au(200) and Au(311) diffraction peaks. A weak Au(111) diffraction peak was firstly observed in the XRD pattern of Au/SiO2-H2 reduced at 90 °C and grows with the increase of reduction temperature, and Au(200) and Au(311) diffraction peaks appear for Au/SiO2-H2 reduced at 150 °C and then grow with the increase of reduction temperature.
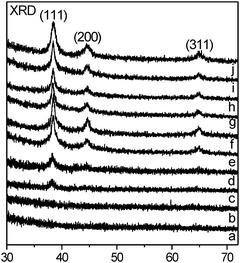 | ||
| Fig. 2 XRD patterns of the catalyst precursor (a), Au/SiO2-H2-60 (b), Au/SiO2-H2-90 (c), Au/SiO2-H2-120, (d) Au/SiO2-H2-150 (e), Au/SiO2-H2-200 (f), Au/SiO2-H2-300 (g), Au/SiO2-H2-400 (h), Au/SiO2-H2-600 (i) and Au/SiO2-Air-300 (j). | ||
Fig. 3 shows the representative transmission electron microscopy (TEM) images and the size distribution of Au nanoparticles by counting more than two hundred Au nanoparticles for each catalyst. Supported Au nanoparticles in these catalysts are all spheres but with a wide size distribution (Table 1). In Au/SiO2-Air-300, ∼95% Au nanoparticles are larger than 4.5 nm and their size distribution is very similar to that in Au/SiO2-Air-200 reported previously.31,32,40 In Au/SiO2-H2-300, ∼23% Au nanoparticles are larger than 4.5 nm, ∼70% Au nanoparticles are between 3 and 4.5 nm, and ∼7% Au nanoparticles are between 2 and 3 nm. In Au/SiO2-H2-200, ∼24% Au nanoparticles are larger than 4.5 nm, ∼54% Au nanoparticles are between 3 and 4.5 nm, and ∼22% Au nanoparticles are between 2 and 3 nm. In Au/SiO2-H2-120, ∼35.6% Au nanoparticles are larger than 4.5 nm, ∼39% Au nanoparticles are between 3 and 4.5 nm, ∼23% Au nanoparticles are between 2 and 3 nm, and ∼2.4% Au nanoparticles are finer than 2 nm. It is noteworthy that Au clusters finer than 2 nm could not be well resolved by the TEM facility employed in our study, therefore, it is likely that the number of Au nanoparticles finer than 2 nm in Au/SiO2-H2-120 is more than what we counted. However, as discussed below, this does not affect our data analysis and conclusions.
 | ||
| Fig. 3 TEM images and size distributions of Au/SiO2-Air-300 (a1,a2), Au/SiO2-H2-300 (b1,b2), Au/SiO2-H2-200 (c1,c2) and Au/SiO2-H2-120 (d1,d2). | ||
| Catalyst | Fraction (%) | |||
|---|---|---|---|---|
| >4.5 nm | 3–4.5 nm | 2–3 nm | < 2 nm | |
| Au/SiO2-Air-300 | ∼95% | |||
| Au/SiO2-H2-120 | ∼35.6% | ∼39% | ∼23% | ∼2.4% |
| Au/SiO2-H2-200 | ∼24% | ∼54% | ∼22% | |
| Au/SiO2-H2-300 | ∼23% | ∼70% | ∼7% | |
The above TEM results demonstrate that calcination of the catalyst precursor in air at 200 and 300 °C mainly leads to the formation of Au nanoparticles larger than 4.5 nm supported on SiO2 whereas reduction of the catalyst precursor in H2 can form Au nanoparticles finer than 4.5 nm whose size distributions depend on the reduction temperature. Moreover, we also found that, as shown in Fig. 3b2, c2 and d2, the contrast between Au nanoparticles finer than 3 nm and the SiO2 support obviously increases with the reduction temperature; meanwhile, most Au nanoparticles finer than 3 nm in Au/SiO2-H2-120 only exhibit one group of lattice fringe, but with the increase of the reduction temperature, more and more Au nanoparticles finer than 3 nm exhibit multi groups of lattice fringes. Therefore, the thickness of fine Au nanoparticles in Au/SiO2-H2 increases with the reduction temperature.
The size distribution and catalytic activity in CO oxidation of supported Au nanoparticles in various Au/SiO2 catalysts clearly reveals a volcano-shaped dependent catalytic activity of supported Au nanoparticles on their sizes since SiO2 does not participate in the catalytic reaction. Au/SiO2-Air-200 and Au/SiO2-Air-300 catalysts containing about 95% Au nanoparticles larger than 4.5 nm do not exhibit any catalytic activity, demonstrating that Au nanoparticles larger than 4.5 nm are inactive in catalyzing low-temperature CO oxidation. Au/SiO2-H2-120, Au/SiO2-H2-200 and Au/SiO2-H2-300 containing large fractions of Au nanoparticles finer than 4.5 nm (more than 65%) are active in catalyzing low-temperature CO oxidation but their catalytic activity obviously decreases with the increasing fraction of Au nanoparticles finer than 3 nm. The catalytic activity of Au/SiO2-H2 catalysts in low-temperature CO oxidation coincides best with the fraction of Au nanoparticles between 3 and 4.5 nm. Au/SiO2-H2-300 with the largest fraction of Au nanoparticles between 3 and 4.5 nm (70%) and the lowest fraction of Au nanoparticles finer than 3 nm (7%) is most active while Au/SiO2-H2-120 with the lowest fraction of Au nanoparticles between 3 and 4.5 nm (39%) and the largest fraction of Au nanoparticles finer than 3 nm (25.4%) is least active. It can be thus concluded that Au nanoparticles finer than 3 nm are less active in catalyzing low-temperature CO oxidation than those between 3 and 4.5 nm. Such a volcano-shaped dependence of the catalytic activity of supported Au nanoparticles on their size was previously observed,10–13 but all in TiO2-supported Au catalysts. Since the active TiO2 support participates in the catalytic CO oxidation itself, therefore, the observed volcano-shaped size-dependent catalytic activity of supported Au nanoparticles in Au/TiO2 catalysts cannot be solely correlated to the structure of supported Au nanoparticles. Therefore, our results for the first time demonstrate that, without the involvement of supports, supported Au nanoparticles intrinsically exhibit a volcano-shaped dependent catalytic activity on their sizes for low-temperature CO oxidation and the optimal size is between 3 and 4.5 nm.
Fig. 4 displays Au 4f XPS spectra of Au/SiO2-Air-300, Au/SiO2-H2-300, Au/SiO2-H2-200 and Au/SiO2-H2-120. The Au 4f XPS spectra of both Au/SiO2-Air-300 and Au/SiO2-H2-300 could be nicely fitted with one component centering at 84.0 eV with the same line shape (Lorentz weight = 46%) and full-width at half-maximum (FWHM = 1.19 eV). Such parameters were thus employed during the peak-fitting process of Au 4f XPS spectra of Au/SiO2-H2-120 and Au/SiO2-H2-200, whose results are summarized in Table 2.
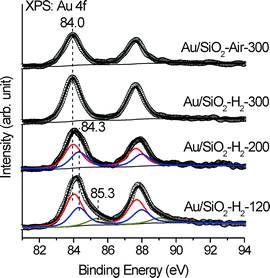 | ||
| Fig. 4 Au 4f XPS spectra of Au/SiO2-H2-120, Au/SiO2-H2-200, Au/SiO2-H2-300 and Au/SiO2-Air-300. Scattered circles and solid lines indicate the experimental data and fitting spectra, respectively. | ||
| Catalyst | Component I | Component II | Component III | |||
|---|---|---|---|---|---|---|
| BE (eV) | Fraction | BE (eV) | Fraction | BE (eV) | Fraction | |
| Au/SiO2-Air-300 | 84.0 | 100% | ||||
| Au/SiO2-H2-120 | 84.0 | 55.56% | 84.3 | 33.33% | 85.3 | 11.11% |
| Au/SiO2-H2-200 | 84.0 | 60% | 84.3 | 40% | ||
| Au/SiO2-H2-300 | 84.0 | 100% | ||||
The Au 4f7/2 XPS spectrum of Au/SiO2-H2-200 consists of two components centering at 84.0 (fraction: 60%) and 84.3 eV (fraction: 40%) while that of Au/SiO2-H2-120 consists of three components centering at 84.0 (fraction: 55.56%), 84.3 (fraction: 33.33%) and 85.3 eV (fraction: 11.11%). It has been well established that the Au 4f binding energy of Au nanoparticles shifts to a higher binding energy as their size decreases.41–43 The Au 4f7/2 binding energy of fine Au clusters on SiO2 was reported to reach 85.6 eV, 1.6 eV higher than that of supported large Au nanoparticles (84.0 eV).41 The Au 4f XPS spectrum of the catalyst precursor (not shown) demonstrates that the Au 4f7/2 binding energy of the supported Au(III) precursor lies at 86.9 eV. Meanwhile, the corresponding Au LIII-edge XANES spectra shown below suggest that few Au cations should exist in Au/SiO2-H2-120. Therefore, the observed different components in the Au 4f XPS spectra of Au/SiO2-H2-120 should result from metallic Au species with different size distributions. Comparing the Au 4f binding energy and size distribution of supported Au nanoparticles in various Au/SiO2 catalysts, we proposed that supported Au nanoparticles larger than 3 nm exhibit the Au 4f binding energy at 84.0 eV while those between 2 and 3 nm and finer than 2 nm exhibit the Au 4f binding energy at 84.3 and 85.3 eV, respectively. Bulk Au exhibits its Au 4f7/2 binding energy at 84.0 eV. These observations imply that Au nanoparticles supported on SiO2 larger than 3 nm exhibit the bulk Au-like electronic structure whereas those finer than 3 nm exhibit electronic structures deviating from that of bulk Au more or less.
TEM and XPS results demonstrate that treating the same catalyst precursor by calcinations in air and reduction in H2 leads to the formation of Au nanoparticles supported on SiO2 with different size distributions and electronic structures. In situ X-ray absorption spectroscopy was employed to understand the compositional and structural change during different treatment processes, whose results are shown in Fig. 5A and B. The Au LIII-edge XANES spectra for HAuCl4 and Au foil are also included in Fig. 5A for comparison. The white line peak of Au LIII-edge XANES spectrum at ∼11923 eV is associated with the 2p3/2 → 5d5/2,3/2 dipole transition probing the unoccupied densities of d states of gold species. Therefore, HAuCl4 exhibits a strong white line peak while the Au foil does not show an obvious white line peak, but exhibits a characteristic absorption peak at 11948 eV. The Au LIII-edge XANES spectra during the calcination process in air clearly vary very differently from those during the reduction process in H2. We defined the white line peak height as h1 and the peak height at 11948 eV as h2, and plotted h1 and h2 as the function of treatment temperature during different treatment processes (Fig. 5C and D). The catalyst precursor prepared by drying at 60 °C for 12 h mainly consists of SiO2 with deposited Au(III) hydroxides44–46 and exhibits a quite strong white line peak whose peak height is lower than that of HAuCl4, but no absorption peak at 11948 eV. Additional calcination of the catalyst precursor in air at 60 °C for 30 min only results in the further decrease of the white line peak but also without the appearance of the peak at 11948 eV. This observation infers that calcination of the catalyst precursor in air at 60 °C might reduce Au(III) to Au(I), but not to Au(0). Increasing the calcination temperature to 90 °C leads to the simultaneous quick decrease of h1 and increase of h2, demonstrating the simultaneous reduction of Au cations and formation of Au(0). The peak at 11948 eV further grows at the expense of the white line peak with the increase of the calcination temperature. Above 150 °C, the white line peak height reaches that of the Au foil, meanwhile, the peak height at 11948 eV still increases slowly.
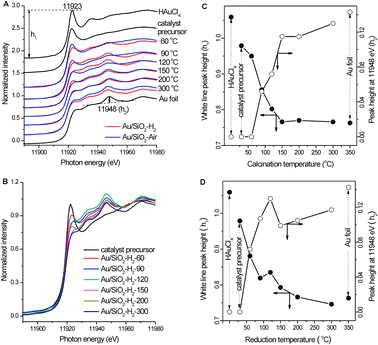 | ||
| Fig. 5 (A) In situ Au LIII-edge XANES spectra during the calcination of the catalyst precursor in air and during the reduction of the catalyst precursor in H2. (B) In situ Au LIII-edge XANES spectra during the reduction of the catalyst precursor in H2. (C) Variations of the white line peak height (h1) and the peak height at 11948 eV (h2) in the Au LIII-edge XANES spectra during the calcination of the catalyst precursor in air. (D) Variations of the white line peak height (h1) and the peak height at 11948 eV (h2) in the Au LIII-edge XANES spectra during the reduction of the catalyst precursor in H2. | ||
After the catalyst precursor was reduced in H2 at 60 °C, the white line peak attenuates rapidly, meanwhile, the peak at 11948 eV obviously appears, indicating the reduction of Au cations to Au(0). The peak at 11948 eV further grows at the expense of the white line peak when reduced at 90 °C. When the reduction temperature was increased to 120 °C, the peak at 11948 eV still grows, but interestingly, the white line peak height also increases. The fraction of Au cations surely decreases with the increase of the reduction temperature, and thus the weakening of the white line peak should be expected. Zhang and Sham47 reported that, resulting from the interplay of quantum-size and surface effect, the lattice contracts and the d charge at the Au atom site depletes relative to bulk Au as the size of the Au nanoparticle decreases, leading to enhanced white line peak intensity. Therefore, the observation of simultaneous intensity growth of the white line peak and the peak at 11948 eV indicates the formation of dominant Au clusters with depleted d charge (Aunδ+) when the catalyst precursor was reduced in H2 at 120 °C for 30 min. With the further increase of the reduction temperature, the white line peak intensity keeps decreasing; however, the peak height at 11948 eV drops much when the reduction temperature was increased from 120 to 150 °C, and then increases slowly.
The Au LIII-edge XANES spectrum obtained after the catalyst precursor was reduced in H2 at 120 °C for 30 min is worth further discussing. Its white line peak intensity is a local maximum, demonstrating the dominance of Au clusters with depleted d charge by the reduction of Au cations in the sample; meanwhile, its peak height at 11948 eV is also a local maximum and even much higher than that obtained after the catalyst precursor was reduced in H2 at 300 °C for 30 min. The great differences among these Au LIII-edge XANES spectra indicate that the electronic structure of dominant Au clusters formed after the reduction at 120 °C for 30 min is not in line with that formed after the reduction at higher temperatures. Reduction at high temperatures reasonably facilitates the formation of Au nanoparticles with metallic electronic structure; therefore, the dominant Au clusters formed by the reduction at 120 °C for 30 min are likely with non-metallic electronic structure resulting from the quantum-size effect. These Au clusters with the non-metallic electronic structure are likely to be Au nanoparticles finer than 2 nm with the Au 4f binding energy at 85.3 eV in Au/SiO2/H2-120. The population of Au clusters with non-metallic electronic structure is reasonably much less in Au/SiO2/H2-120 prepared by reducing the catalyst precursor in H2 at 120 °C for 4 h than in the sample prepared by reducing the catalyst precursor in H2 at 120 °C only for 30 min during the in situ XANES experiments. The change of the Au LIII-edge XANES spectrum when the reduction temperature was increased from 120 to 150 °C likely corresponds to the transition from non-metallic electronic structure to metallic electronic structure resulting from the size and thickness growth.
Therefore, when calcined in air, Au(III) hydroxides in the catalyst precursor seem to only decompose into Au(I) hydroxides or oxides at low temperatures, and then Au(I) hydroxides or oxides decompose to Au(0) at elevated temperatures, forming Au nanoparticles with metallic electronic structure. When reduced in H2, Au(III) hydroxides in the catalyst precursor seem to be facilely reduced to Au(0) at low temperatures, forming Au clusters likely with depleted d charge, then these Au clusters grow into Au nanoparticles with metallic electronic structure at elevated temperatures. These results explain well the different size distributions and electronic structures of supported Au nanoparticles in various Au/SiO2 catalysts.
Fig. 6 displays the operando diffuse reflectance infrared spectroscopy (DRIFTS) spectra of saturated pulse chemisorption of CO on various Au/SiO2 catalysts at RT. Au/SiO2-Air-300 does not chemisorb CO while Au/SiO2-H2 chemisorbs CO reversibly. The amount of CO chemisorbed on various Au/SiO2-H2 catalysts increases with the increase of reduction temperature but their vibrational frequencies do not vary, suggesting that the type of chemisorption site on various Au/SiO2-H2 catalysts is the same but its amount increases with the reduction temperature. The observed vibrational frequencies (2114 and 2117 cm−1) of chemisorbed CO agree well with those reported for CO chemisorbed on the top of low-coordinated Au atoms.24,40,48,49 Note that there is no IR band in the range of 2125–2250 cm−1 indicating the absence of cationic Au sites even in Au/SiO2-H2-120,50 agreeing well with the above XPS and XANES results. Therefore, Au nanoparticles in Au/SiO2-Air-300 expose few low-coordinated Au atoms, which could be reasonably associated with their large size and thus likely smooth surfaces. The experimental observation that the number of chemisorption sites in Au/SiO2-H2 increases with the reduction temperature is consistent with their fractions of supported Au nanoparticles between 3 and 4.5 nm, but contrasts the general assumption that finer Au nanoparticles expose more low-coordinated Au atoms because the population of fine Au nanoparticles in various Au/SiO2-H2 increases with the decrease of reduction temperature. This infers that Au nanoparticles finer than 3 nm in Au/SiO2-H2 should not actively chemisorb CO although they expose a high density of low-coordinated Au atoms, which could be reasonably associated with their electronic structure that deviates from bulk-Au electronic structure. These results indicate the important role of the electronic structure of Au nanoparticles on their ability to chemisorb CO. Thus, co-affected by the size-dependent density of low-coordinated Au atoms and the electronic structure of Au nanoparticles supported on SiO2, their ability to chemisorb CO at RT exhibits a volcano-shaped dependence on their size with the optimum size of 3–4.5 nm.
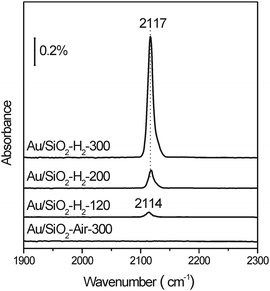 | ||
| Fig. 6 In situ DRIFTS spectra of saturated CO pulse adsorption on Au/SiO2-H2-120, Au/SiO2-H2-200, Au/SiO2-H2-300 and Au/SiO2-Air-300 at room temperature. | ||
Fig. 7A compares the integrated CO chemisorption peak area (CO(a) coverage) and the reaction rate at RT of Au/SiO2-H2 catalysts and Fig. 7B compares those normalized to the values of Au/SiO2-H2-120. Since chemisorbed CO on the Au surfaces participates in the catalytic reaction, the reaction rate of Au/SiO2-H2 catalysts at RT reasonably increases with the coverage of chemisorbed CO at RT, but it is clearly not proportional to the coverage of chemisorbed CO. This suggests the existence of factors other than the coverage of chemisorbed CO affecting the reaction rate, which is plausible because CO chemisorption is only one of the elementary steps in catalytic CO oxidation. Moreover, comparing with those of Au/SiO2-H2-120, the increasing factor of the reaction rate of Au/SiO2-H2-200 is more than twice than the increasing factor of CO(a) coverage whereas the increasing factor of the reaction rate of Au/SiO2-H2-300 is slightly lower than the increasing factor of CO(a) coverage. This infers that the influence of other factors of supported Au nanoparticles on their catalytic activity in CO oxidation at RT should also exhibit a volcano-shaped dependence on the size with the optimal size between 3 and 4.5 nm but the optimal size should be finer than that for CO chemisorption. This needs further investigations.
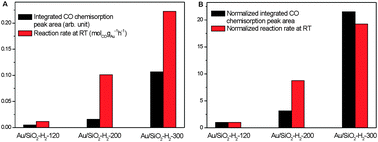 | ||
| Fig. 7 (A) Integrated CO chemisorption peak area and reaction rate at RT of Au/SiO2-H2-120, Au/SiO2-H2-200 and Au/SiO2-H2-300. (B) Integrated CO chemisorption peak area and reaction rate at RT of Au/SiO2-H2-200 and Au/SiO2-H2-300 normalized to those of Au/SiO2-H2-120. | ||
Our results provide unprecedented solid and comprehensive experimental evidence for the size–structure–chemisorption–catalytic activity relationship of supported Au nanoparticles without the involvement of oxide supports in low-temperature CO oxidation. As schematically illustrated in Fig. 8, both the ability to chemisorb CO and the activity in catalyzing CO oxidation at RT of supported Au nanoparticles clearly exhibit a volcano-shaped dependence on their size. Supported Au nanoparticles between 3 and 4.5 nm with the bulk Au-like electronic structure effectively chemisorb CO and catalyze CO oxidation at RT, which could be attributed to the existence of abundant low-coordination Au atoms; larger supported Au nanoparticles with few low-coordinated Au atoms do not chemisorb CO and catalyze CO oxidation at RT; however, finer supported Au nanoparticles with more abundant low-coordination Au atoms whose electronic structure deviates from that of bulk Au also do not chemisorb CO and catalyze CO oxidation at RT. Therefore, without the involvement of supports, the catalytically active structure of supported Au nanoparticles in low-temperature CO oxidation is low-coordinated Au atoms on Au nanoparticles with bulk Au-like electronic structure. With the decrease of the size of Au nanoparticles, the density of low-coordinated Au atoms generally increases while the electronic structure deviates from bulk Au-like electronic structure, therefore, the volcano-shaped dependence of the catalytic activity of Au nanoparticles on their size is an inevitable outcome resulting from the fact that their size-dependence of the geometric structure effect is opposite to their size dependence of the electronic structure effect.
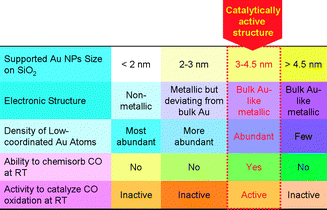 | ||
| Fig. 8 A schematic illustration of the size–structure–chemisorption–catalytic activity relationship and the catalytically active structure of supported Au nanoparticles in low-temperature CO oxidation without the involvement of oxide supports. | ||
The employed oxide support has been well recognized to significantly influence the catalytic performance of supported Au nanoparticles in low-temperature CO oxidation. On one hand, the oxide support can directly participate in the catalytic reaction; on the other hand, different oxide supports might exhibit different Au–support interactions so as to affect the electronic and geometric structures of supported Au nanoparticles in various aspects. Therefore, the size-dependent structures and properties of Au nanoparticles supported on “inert” SiO2 reported herein cannot be simply extended to Au nanoparticles supported on other oxide supports.
Conclusion
Employing Au/SiO2 catalysts with different size distributions prepared by treating Au(III) hydroxides supported on SiO2 under various conditions, we have elucidated the size–structure–chemisorption–catalytic activity relationship of supported Au nanoparticles without the involvement of oxide supports in low-temperature CO oxidation. Low-coordinated Au atoms on Au nanoparticles with bulk Au-like electronic structure are the catalytically active site of Au nanoparticles supported on SiO2 in low-temperature CO oxidation. With the decrease of the size of supported Au nanoparticles, the density of low-coordinated Au atoms generally increases while the electronic structure deviates from bulk Au-like electronic structure, therefore, supported Au nanoparticles exhibit a volcano-shaped dependence of their ability to chemisorb CO and catalyze CO oxidation at RT on their size with the optimal size between 3 and 4.5 nm.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (20973161, 11079033), National Basic Research Program of China (2013CB933104, 2010CB923301), the Fundamental Research Funds for the Central Universities, and the MPG-CAS partner group program. We gratefully acknowledge Dr Zhen Jiang, Prof. Yuyin Huang and Dr Shuo Zhang at Shanghai Synchrotron Radiation Facility for their assistance in the XAS experiments.Notes and references
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301 CrossRef CAS.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef.
- M. Haruta, Faraday Discuss., 2011, 152, 11 RSC.
- M. Haruta, Chem. Rec., 2003, 3, 75 CrossRef CAS.
- I. X. Green, W. Tang, M. Neurock and J. T. Yates Jr., Science, 2011, 333, 736 CrossRef CAS.
- G. C. Bond and D. T. Thompson, Gold Bull., 2000, 33, 41 CrossRef CAS.
- M. C. Kung, R. J. Davis and H. H. Kung, J. Phys. Chem. C, 2007, 111, 11767 CAS.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175 CrossRef CAS.
- G. R. Bamwenda, S. Tsubota, T. Nakamura and M. Haruta, Catal. Lett., 1997, 44, 83 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647 CrossRef CAS.
- R. Zanella, S. Giorgio, C.-H. Shin, C. R. Henry and C. Louis, J. Catal., 2004, 222, 357 CrossRef CAS.
- Y. Tai, W. Yamaguchi, K. Tajiri and H. Kageyama, Appl. Catal., A, 2009, 364, 143 CrossRef CAS.
- I. Laoufi, M.-C. Saint-Lager, R. Lazzari, J. Jupille, O. Robach, S. Garaudée, G. Cabailh, P. Dolle, H. Cruguel and A. Bailly, J. Phys. Chem. C, 2011, 115, 4673 CAS.
- N. Lopez, T. V. W. Janssens, B. S. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Nørskov, J. Catal., 2004, 223, 232 CrossRef CAS.
- T. V. W. Janssens, B. S. Clausen, B. Hvolbæk, H. Falsig, C. H. Christensen, T. Bligaard and J. K. Nørskov, Top. Catal., 2007, 44, 15 CrossRef CAS.
- H. Falsig, B. Hvolbæk, I. S. Kristensen, T. Jiang, T. Bligaard, C. H. Christensen and J. K. Nørskov, Angew. Chem., Int. Ed., 2008, 47, 4835 CrossRef CAS.
- Y. Maeda, M. Okumura, S. Tsubota, M. Kohyama and M. Haruta, Appl. Surf. Sci., 2004, 222, 409 CrossRef CAS.
- M. S. Chen and D. W. Goodman, Science, 2004, 306, 252 CrossRef CAS.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331 CrossRef CAS.
- Y. Liu, C.-J. Jia, J. Yamasaki, O. Terasaki and F. Schüth, Angew. Chem., Int. Ed., 2010, 49, 5771 CrossRef CAS.
- J. D. Grunwaldt, M. Maciejewski, O. S. Becker, P. Fabrizioli and A. Baiker, J. Catal., 1999, 186, 458 CrossRef CAS.
- M. M. Schubert, S. Hackenberg, A. C. van Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197, 113 CrossRef CAS.
- S. K. Shaikhutdinov, R. Meyer, M. Naschitski, M. Baumer and H.-J. Freund, Catal. Lett., 2003, 86, 211 CrossRef CAS.
- D. C. Meier and D. W. Goodman, J. Am. Chem. Soc., 2004, 126, 1892 CrossRef CAS.
- M. Daté, M. Okumura, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 2129 CrossRef.
- D. Gajan, K. Guillois, P. Delichère, J.-M. Basset, J.-P. Candy, V. Caps, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2009, 131, 14667 CrossRef CAS.
- K. Qian, W. X. Huang, Z. Q. Jiang and H. X. Sun, J. Catal., 2007, 248, 137 CrossRef CAS.
- K. Qian, W. X. Huang, J. Fang, S. S. Lv, B. He, Z. Q. Jiang and S. Q. Wei, J. Catal., 2008, 255, 269 CrossRef CAS.
- K. Qian, S. S. Lv, X. Y. Xiao, H. X. Sun, J. Q. Lu, M. F. Luo and W. X. Huang, J. Mol. Catal. A: Chem., 2009, 306, 40 CrossRef CAS.
- K. Qian, J. Fang, W. X. Huang, B. He, Z. Q. Jiang, Y. S. Ma and S. Q. Wei, J. Mol. Catal. A: Chem., 2010, 320, 97 CrossRef CAS.
- K. Qian, H. X. Sun, W. X. Huang, J. Fang, S. S. Lv, B. He, Z. Q. Jiang and S. Q. Wei, Chem.–Eur. J., 2008, 14, 10595 CrossRef CAS.
- K. Qian, W. H. Zhang, H. X. Sun, J. Fang, B. He, Y. S. Ma, Z. Q. Jiang, S. Q. Wei, J. L. Yang and W. X. Huang, J. Catal., 2011, 277, 95 CrossRef CAS.
- S. Ivanova, C. Petit and V. Pitchon, Appl. Catal., A, 2004, 267, 191 CrossRef CAS.
- S. Ivanova, V. Pitchon, Y. Zimmermann and C. Petit, Appl. Catal., A, 2006, 298, 57 CrossRef CAS.
- M. Okumura, S. Tsubota and M. Haruta, J. Mol. Catal. A: Chem., 2003, 199, 73 CrossRef CAS.
- G. Budroni and A. Corma, Angew. Chem., Int. Ed., 2006, 45, 3328 CrossRef CAS.
- G. Martra, L. Prati, C. Manfredotti, S. Biella, M. Rossi and S. Coluccia, J. Phys. Chem. B, 2003, 107, 5453 CrossRef CAS.
- F. Somodi, I. Borbáth, M. Hegedűs, A. Tompos, I. E. Sajó, Á. Szegedi, S. Rojas, J. L. G. Fierro and J. L. Margitfalvi, Appl. Catal., A, 2008, 347, 216 CrossRef CAS.
- H. G. Zhu, C. D. Liang, W. F. Yan, S. H. Overbury and S. Dai, J. Phys. Chem. B, 2006, 110, 10842 CrossRef CAS.
- K. Qian, L. F. Luo, C. B. Chen, S. F. Yang and W. X. Huang, ChemCatChem, 2011, 2, 161 CrossRef.
- C. C. Chusuei, X. Lai, K. Luo and D. W. Goodman, Top. Catal., 2001, 14, 71 CrossRef.
- R. Meyer, C. Lemire, S. K. Shaikhutdinov and H.-J. Freund, Gold Bull., 2004, 37, 72 CrossRef CAS.
- Z. Q. Jiang, W. H. Zhang, L. Jin, X. Yang, F. Q. Xu, J. F. Zhu and W. X. Huang, J. Phys. Chem. C, 2007, 111, 12434 CAS.
- J. H. Yang, J. D. Henao, C. Costello, M. C. Kung, H. H. Kung, J. T. Miller, A. J. Kropf, J. G. Kim, J. R. Regalbuto, M. T. Bore, H. N. Pham, A. K. Datye, J. D. Laeger and K. Kharas, Appl. Catal., A, 2005, 291, 73 CrossRef CAS.
- F. Moreau, G. C. Bond and A. O. Taylor, J. Catal., 2005, 231, 105 CrossRef CAS.
- S. Wang, K. Qian, X. Z. Bi and W. X. Huang, J. Phys. Chem. C, 2009, 113, 6505 CAS.
- P. Zhang and T. K. Sham, Phys. Rev. Lett., 2003, 90, 245502 CrossRef.
- M. L. Kottke, R. G. Greenler and H. G. Tompkins, Surf. Sci., 1972, 32, 231 CrossRef CAS.
- C. Ruggiero and P. Hollins, J. Chem. Soc., Faraday Trans., 1996, 92, 4829 RSC.
- E. Roze, E. Quinet, V. Caps and D. Bianchi, J. Phys. Chem. C, 2009, 113, 8194 CAS.
| This journal is © The Royal Society of Chemistry 2013 |