Recent advances in the chemistry of heterometallacycles of group 4 metallocenes†
Torsten Beweries*, Martin Haehnel and Uwe Rosenthal*
Leibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: torsten.beweries@catalysis.de; uwe.rosenthal@catalysis.de; Fax: +49 381 1281 51104; Fax: +49 381 1281 51176; Tel: +49 381 1281 104 Tel: +49 381 1281 176
First published on 3rd October 2012
Abstract
This perspective gives an overview of recent developments in the chemistry of heterometallacycles of group 4 metallocenes. This class of compounds has been known for several decades now, however, in recent years significant efforts were made in order to further explore the structural scope of these complexes and their potential in catalytic applications. This overview shows some special heterometallacycles which could be of great interest for theoretical and synthetic chemists, owing to their unusual structural and bonding parameters as well as their applicability in a wide range of synthetic and catalytic transformations.
 Torsten Beweries | Torsten Beweries, born 1982, studied chemistry at the University of Rostock (2001–2006). He obtained his PhD under the supervision of Uwe Rosenthal (Leibniz Institute for Catalysis, Rostock) in 2008 for studies on the organometallic chemistry of hafnocene complexes. After a postdoctoral stay with Robin N. Perutz at the University of York in 2009, working on halogen bonding and transition metal fluoride complexes, he returned to Rostock, where he is now head of the group “Organometallic Water Splitting”. His current research is focussed towards the investigation of unusual group 4 metallacycles as well as photocatalytic water splitting and hydrogen storage. |
 Martin Haehnel | Martin Haehnel, born 1985, studied chemistry at the Friedrich Schiller University Jena (2005–2010) and graduated (2010) in the group of Matthias Westerhausen in the chemistry of redox behaviour of zinc 2-pyridylmethylamide complexes. He then moved to Rostock for his PhD studies in the group of Uwe Rosenthal (Leibniz Institute for Catalysis, Rostock) on the field of organometallic chemistry of group 4 metallocenes. His scientific interests include highly strained heterometallacycles of titanium, zirconium and hafnium as well as catalytic water splitting. |
 Uwe Rosenthal | Uwe Rosenthal studied chemistry (1968–1972), received his PhD under E. Kurras (1976), and completed his habilitation (1991) at the University of Rostock. After some time at the Nesmeyanov Institute of Organoelement Compounds in Moscow with M. E. Vol'pin and V. B. Shur (1988) and at the Max Planck Institute of Kohlenforschung in Mülheim/Ruhr with G. Wilke and K. Pörschke (1990–1991) he headed the Max Planck Research group “Complex Catalysis” (1992–1996) and became Professor of Inorganic Chemistry at the University of Rostock (1993). Today he is Deputy Director of the Leibniz Institute for Catalysis in Rostock with scientific interests in organometallic chemistry and complex catalysis. He has published around 300 papers and patents for this subject. |
Introduction
Metallacycles are an important class of organometallic complexes and have recently emerged from lab curiosities to become widely used and often described intermediates or compounds for numerous synthetic and catalytic applications. These include olefin oligomerisation reactions,1 cycloadditions2 as well as a vast number of different functionalisation reactions.3However, the isolation of well-defined metallacycles is often problematic, mostly due to the high ring-strain, which has to be taken into account when discussing the stability of some of such molecules. An elegant approach for the minimisation of ring strain is the incorporation of metal centres which can be further modified via their ligand environment into the cyclic units. A reasonable quantitative measure of the stabilisation effect of this metal-incorporation into unsaturated cyclic structures was found to be the hydrogenation enthalpy.4 The chemistry of such metallacycles of group 4 metals containing exclusively carbon atoms in the ring was studied and reviewed extensively in the past.5 Additionally, an enhancement of stability is gained when substituting carbon atoms in the ring by heteroatoms. Examples for this include the six-membered cyclic alkyne 1,2,3,4-tetrasilahex-5-yne6 and cyclic heterocycles such as 3,4-didehydrothiophenes7 and pyrroles (Fig. 1).8
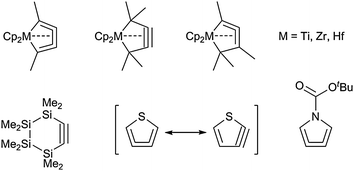 | ||
| Fig. 1 Stabilisation of highly strained cyclic structures by incorporation of metals or heteroatoms. | ||
A combination of both approaches (i.e. incorporation of metal and heteroatoms) to yield the corresponding heterometallacycles provides an elegant way for the stabilisation of unusual organometallic structures. Moreover, interesting synthetic applications such as functionalisations of the heteroatom substrate as well as activation of the latter for coupling reactions could be facilitated by coordination to a metal centre. For example, a wide range of zirconocene-mediated organic transformations to generate heterocyclic compounds is known to date with most of the reaction products being available in a straight-forward way instead of from less convenient multistep reaction protocols.9 Pioneering contributions demonstrating the role of heterozirconacycles in catalytic transformations were made by Nugent et al. for the hydroamination of terminal olefins, which involved metallaaziridines as key-intermediates in the catalytic cycle.10 Additionally, Bergman and co-workers investigated the zirconocene imido complex-catalysed hydroamination of internal alkynes and found that four-membered azazirconacyclobutenes are present during the course of the catalytic reactions.11
The concept of heterometallacycles as a direct extension of the well-known all-carbon-metallacycles was described by us based on 1-metalla-2,5-heterocyclopent-3-yne complexes in a recent review article.5b In this perspective, we summarise a selection of recent advances in the field of heterometallacycles of group 4 metallocenes, focussing on synthetic and catalytic applications as well as on the unusual structural and bonding characteristics of these organometallic compounds.
Main part
Some sources for group 4 metallocene starting materials
The herein described heterometallacycles feature a metallocene core as the metal component, which is essential for the stabilisation of the unusual cyclic structures. In organometallic chemistry, different ways of generating reactive group 4 metallocene fragments are known. Some of the most common examples are listed below (Scheme 1).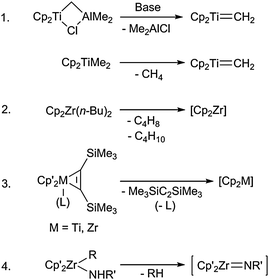 | ||
| Scheme 1 Common metallocene sources. | ||
1. Tebbe's reagent:12 Cp2TiCl2 (Cp = η5-cyclopentadienyl) is reacted with AlMe3 to yield the dimetallic Cp2Ti-μ-Cl, μ-CH2-AlMe2, which with base forms the reactive carbene species [Cp2Ti![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2]. As a variation of this approach, the more air-stable Petasis' reagent Cp2TiMe2 also forms the carbene fragment under reaction conditions (with concomitant release of methane). A very similar species was described by Beckhaus et al., who used the decamethyltitanocene vinylidene complex [Cp*2TiCCH2] in coupling reactions with a variety of unsaturated substrates.13
CH2]. As a variation of this approach, the more air-stable Petasis' reagent Cp2TiMe2 also forms the carbene fragment under reaction conditions (with concomitant release of methane). A very similar species was described by Beckhaus et al., who used the decamethyltitanocene vinylidene complex [Cp*2TiCCH2] in coupling reactions with a variety of unsaturated substrates.13
2. Negishi's reagent:14 Cp2ZrCl2 is reacted with n-butyllithium at low temperatures to give Cp2Zr(n-Bu)2. Warming of this species generates the reactive zirconocene [Cp2Zr] under release of butene and butane. It should be noted that addition of the substrate at low temperatures is essential due to the instability of the reagent under ambient conditions. In an analogous way, the complex Cp2ZrEt2 is capable of generating the ethene complex Cp2Zr(ethene), which can then react with unsaturated substrates to yield metallacycles.3b
3. Metallocene bis(trimethylsilyl)acetylene complexes (Rosenthal):15 These complexes are formed by reduction of Cp′2MCl2 (Cp′ = substituted or unsubstituted η5-cyclopentadienyl, M = Ti, Zr) in the presence of bis(trimethylsilyl)acetylene. The latter acts as a spectator ligand and can easily be released under reaction conditions to generate the reactive 14-electron fragment [Cp′2M]. These complexes are long-term stable at room temperature.
4. Zirconocene imido complexes (Bergman):16 Elimination of RH or RNH2 from Cp′2Zr(R)NHR′ or Cp′2Zr(NHR)2, respectively, yields the electronically and coordinatively unsaturated fragment [Cp′2ZrNR], which can react with unsaturated substrates to give azazirconacycles.
Three-membered heterometallacycles
Probably some of the first examples for heterometallacycles of group 4 metallocenes were reported in the context of dinitrogen binding and activation at titanocenes.17 The exact nature and coordination mode of dinitrogen to the metal centre was not known for some time until Pez and co-workers described the crystal structure of one of the first titanocene dinitrogen complexes, which displayed a side-on, end-on “μ3-N2” ligand, thus formally forming a three-membered TiN2-metallacyclic unit.18 Numerous other examples for side-on coordinated dinitrogen at group 4 metallocene fragments were reported later, some of them showing very interesting and unprecedented reactivities with other small molecules such as isocyanates, CO and CO2.19In our group, we have studied the ligand exchange reactions of azobenzene with group 4 metallocene bis(trimethylsilyl)acetylene complexes. The nature of the reaction products was found to be strongly dependent on the metal centre as well as on the cyclopentadienyl substituents. Whereas in the case of the bridging ligand rac-(ebthi) [ebthi = 1,2-ethylene-1,1′-bis(η5-tetrahydroindenyl)] η2-coordination of azobenzene to give three-membered heterometallacycles takes place for M = Ti, Zr, for the [Cp*2Ti] moiety a mixture of products is formed including the aforementioned η2-coordination mode and two complexes formed by cleavage of the azobenzene N–N double bond (Scheme 2).20
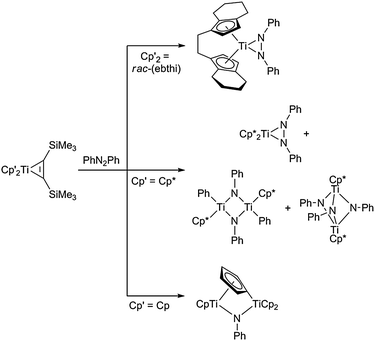 | ||
| Scheme 2 Selected reactions of group 4 metallocenes with azobenzene. | ||
Reaction of the Cp substituted complex Cp2Ti(η2-Me3SiC2SiMe3) with azobenzene yields a dinuclear complex, which was formed by C–H and N–N bond activation steps.21 In contrast, the same reaction with [Cp2Zr] results in simple ligand exchange to give the three-membered diazazirconacycle,20,21 a type of complex which was also described before by Bergman and co-workers.22 A very similar η2-coordination was observed by Beckhaus et al. when reacting titanocene bis(trimethylsilyl)acetylene complexes with trans-4,4′-azobispyridine: in this case a tetranuclear molecular square formed with two azobispyridine units serving as the bridging functionalities via the pyridine and diazo nitrogen atoms.23
Structurally similar side-on coordinated dipnictene complexes of the heavier homologues arsenic, antimony and bismuth are less common, however, examples were reported for the latter two elements: Breunig and co-workers found that reaction of the titanocene source Cp2Ti(η2-Me3SiC2SiMe3) with the organostibane RSbH2 (R = 2,6-Mes-C6H3) yields a coordinated distibene, which was formed in situ by coupling of two stibane molecules.24 Following a similar approach, Robinson and Schleyer et al. published the synthesis of a zirconocene dibismuthene complex from sodium reduction of zirconocene dichloride in the presence of a dichlorobismuthane RBiCl2 (R = 2,6-Mes-C6H3).25
Group 4 metallocene olefin complexes are known for decades and the ethene complexes [Cp2Ti(PMe3)(η2-C2H4)] and Cp*2Ti(η2-C2H4) were found to be versatile starting materials for organometallic chemists.26 Examples for similar compounds of the heavier homologues were described recently by Marschner et al. A full set of isostructural disilene complexes of the type Cp2M[η2-(Me3Si)2Si2(SiMe3)2] was prepared by salt metathesis from 1,2-dipotassiotetrakis(trimethylsilyl)disilane and the respective metallocene dichloride for all group 4 metallocenes. Addition of the Lewis base PMe3 to the Zr and Hf species resulted in the formation of a metallacyclic MSi2 unit (Scheme 3).27 The same approach was used for the synthesis of zirconocene and hafnocene digermene and silagermene complexes (Scheme 3). The corresponding distannene and diplumbene complexes are not known to date. The reason for this can be found in the comparatively weaker E–E bond (E = Sn, Pb) in these species, thus yielding the respective metallocene diylene complexes.28
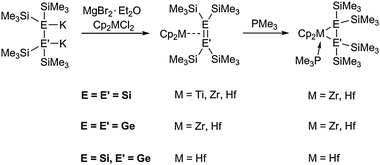 | ||
| Scheme 3 Group 4 metallocene disilene-, digermene- and silagermene complexes. | ||
Four-membered heterometallacycles
The aforementioned method of salt elimination from a metalated organosilicon precursor and a metallocene dihalide is also applicable to four-membered heterometallacycles. Marschner and co-workers showed that reaction of the α,ω-silyl dianion K2{Me2Si[Si(SiMe3)2]2} with Cp2ZrCl2 results in clean formation of the expected four-membered ring Cp2Zr[κ2-Si,Si-Si(SiMe3)2Si(Me)2Si(SiMe3)2].29 Similar zirconocene and hafnocene complexes with four-membered rings consisting of three stannylene groups [Sn(SiMe3)2] were obtained from reactions of the corresponding metallocene dichlorides with 1,2-dipotassiotetrakis(trimethylsilyl)distannane. Formation of three-membered stannene complexes (vide supra) does not take place since in this case the tetrylene–ditetrene equilibrium is shifted towards the –ylene species, thus resulting in a coupling of three stannylenes to give the four-membered heterometallacycles27In an attempt to synthesise the well-known diphenyldiphosphene from reduction of phenyldichlorophosphine with the bis(trimethylsilyl)acetylene complex Cp2Ti(η2-Me3SiC2SiMe3), formation of the previously described30 four-membered cyclotitanatriphosphine complex Cp2Ti(κ2-P,P-PPh-PPh-PPh) was observed.31 It should be noted that this reaction pattern was not observed for sterically demanding dichlorophosphines such as Mes*PCl2 (Mes* = 2,4,6-tri-tert-butylphenyl). In these cases, due to the increased bulk at phosphorus atoms, formation of the desired diphosphenes took place and titanocene monochloride [Cp2TiCl]2 was found to be the only organometallic product.
Very recently, we have studied the coordination chemistry of unsaturated heteroallene substrates at group 4 metallocene fragments. Reaction of the titanocene and zirconocene alkyne complexes with the sulfurdiimide Me3SiNSNSiMe3 resulted in ligand exchange to yield mononuclear unsaturated metallocene(IV) compounds with an N–S–N scaffold (Scheme 4).32 Besides the MII bis-π complex, a Cp2MIII sulfurdiimide species and finally a Cp2MIV metallacycle may be formulated as a result of the transfer of one and two electrons from the metallocene fragment to the sulfurdiimide ligand, respectively. Theoretical calculations of the titanocene compound suggest a σ-complex with cyclic delocalisation of electrons in the four-membered ring to be the best description.
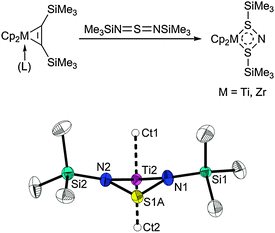 | ||
| Scheme 4 Formation of group 4 metallocene sulfurdiimide complexes (top) and butterfly structure of the heterometallacycle (bottom). | ||
A similar type of heterometallacyclic complex, Cp2M(κ2-P,P-Ph2P-N-PPh2), was obtained from reactions of the bis(diphenyl)phosphinoamide fragment [N(PPh2)2] with various titanocene and zirconocene sources (Scheme 5). The titanocene compound can be prepared by reaction of the corresponding phosphine complex with n-butyllithium as well as from the reaction of the titanocene alkyne complex Cp2Ti(η2-Me3SiC2SiMe3) with the amine (Ph2P)2NH. The synthesis of the zirconocene analogue is possible by reduction of the monochloride complex Cp2Zr(Cl)(κ2-N,P-Ph2P-N-PPh2).33 Similarly as described before for the sulfurdiimide complexes cyclic delocalisation of electrons in the ring is an important factor for the stabilisation of the heterometallacycles. It should be noted that the [N(PPh2)2] fragment is well documented as a ligand, mainly to stabilise late transition metals. In combination with early transition metal fragments it is an interesting ligand for oligomerisation and polymerisation catalysis since it provides the possibility of fine tuning at both P donor atoms as well as at the N atom.
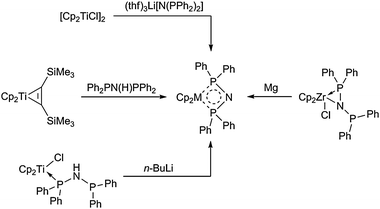 | ||
| Scheme 5 Formation of four-membered titanocene and zirconocene bis(diphenylphosphine)amido complexes. | ||
A different approach of [2 + 2] addition at metallocene imido complexes was used by Clot, Jones, Mountford and co-workers in the synthesis of an unsymmetrical titanocene heterometallacycle Cp2Ti[κ2-C,N-C(tBu)PN(NPh2)]. Reaction of the imido complex Cp2Ti(py)NNPh2 (py = pyridine) with the phosphaalkyne tBuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P allows for the generation of an unusual ligand fragment following a straightforward functionalisation reaction (Scheme 6).34 Structurally similar four-membered ring systems containing phosphorus as well as sulfur, selenium and tellurium, respectively, were described by Nixon et al., who reacted the corresponding decamethylzirconocene complexes Cp*2Zr(py)E (E = S, Se, Te) with the same phosphaalkyne tBuCP. However, in this case, a slightly different regiochemistry of the cycloaddition was observed, thus yielding both regioisomers for the sulfur case (S,C and S,P coordination) and exclusively a Se,P bound ligand in Cp*2Zr[κ2-Se,P-SeC(tBu)P], respectively. In contrast, for the Zr–Te complex, a connectivity similar to the above-mentioned titanocene complex was observed (Scheme 6). A possible explanation for the addition of the phosphaalkyne fragment in the opposite way to that found for the ZrS and ZrSe systems may be found in the longer Zr–Te bond distance compared to the Zr–S and Zr–Se bond lengths, thus leading to the expansion of the four-membered metallacycle, facilitating the approach of the tBu groups in the α position.35
P allows for the generation of an unusual ligand fragment following a straightforward functionalisation reaction (Scheme 6).34 Structurally similar four-membered ring systems containing phosphorus as well as sulfur, selenium and tellurium, respectively, were described by Nixon et al., who reacted the corresponding decamethylzirconocene complexes Cp*2Zr(py)E (E = S, Se, Te) with the same phosphaalkyne tBuCP. However, in this case, a slightly different regiochemistry of the cycloaddition was observed, thus yielding both regioisomers for the sulfur case (S,C and S,P coordination) and exclusively a Se,P bound ligand in Cp*2Zr[κ2-Se,P-SeC(tBu)P], respectively. In contrast, for the Zr–Te complex, a connectivity similar to the above-mentioned titanocene complex was observed (Scheme 6). A possible explanation for the addition of the phosphaalkyne fragment in the opposite way to that found for the ZrS and ZrSe systems may be found in the longer Zr–Te bond distance compared to the Zr–S and Zr–Se bond lengths, thus leading to the expansion of the four-membered metallacycle, facilitating the approach of the tBu groups in the α position.35
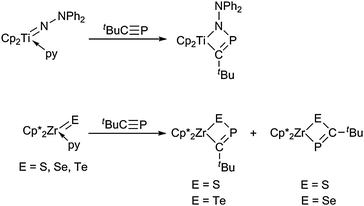 | ||
| Scheme 6 Formation of four-membered heterometallacycles by cycloaddition reactions with tBuCP (py = pyridine). | ||
In an attempt to trap an elusive stibidine species [CpCp*HfSbR], Waterman and Tilley isolated a hafnastibacyclobutene from the reaction of a hafnocene methyl triflate complex with the lithium salt LiSbHdmp (dmp = 2,6-dimesitylphenyl) and but-2-yne.36 This reactivity is very similar to the cycloaddition chemistry observed with other group 4 metallocene compounds with ME double bonds (E = N, P).
Four-membered metallocene amidinate complexes are well-precedented in the literature, however, only some examples are known for formamidinate complexes. Floriani et al. first prepared corresponding zirconocene(IV) compounds of the type Cp2Zr(Cl)(κ2-N,N-N(R)C(H)NR) (R = Ph, Cy) by hydrozirconation of the corresponding carbodiimides RNCNR.37a The methyl complex Cp2Zr(Me)(κ2-N,N-N(Ph)C(H)NPh) was obtained from Cp2ZrMe2 and the PhN(H)–CHNPh reagent.36 Later, Erker and co-workers published an alternative synthesis of the former zirconocene chloro species from Cp2ZrCl2 and the THF-stabilised formamidinyl lithium dimer precursors.38
Amidinate complexes with aromatic and aliphatic substituents at the central C atom can in principle be obtained from the salt metathesis reaction of the metallocene dichlorides and the metallated amidinate ligands. Recent examples for this method were reported by Guo and Liu et al.39 As an extension of this concept, parent group 4 metallocene guanidinate compounds are accessible following the same synthetic pathway (Scheme 7).40 Interestingly, one of these complexes was used as a monomeric volatile Ti(III) gas-phase precursor for chemical deposition techniques such as ALD (atomic layer deposition).
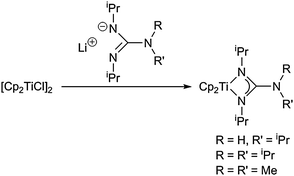 | ||
| Scheme 7 Synthesis of titanocene guanidinate complexes. | ||
A less directed way of preparing dinuclear titanocene amidinate complexes was described by our group very recently (Scheme 8): in the reaction of the titanocene alkyne complex Cp2Ti(η2-Me3SiC2SiMe3) with carbodiimides RNCNR′ (R = R′ = Cy;41a R = R′ = iPr;40b R = Et, R′ = tBu40b) diamidinate complexes are formed.
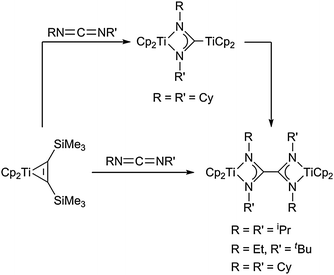 | ||
| Scheme 8 Formation of four-membered dinuclear titanocene amidinate complexes. | ||
A likely way to explain the formation of these complexes includes the generation of mononuclear four-membered diazatitanacycles, which dimerise to give the diamidinate species. This mechanism is corroborated by the successful isolation of the dinuclear carbene-like complex Cp2Ti[κ2-N,N-(NCy)2C–TiCp2], which with time eliminates the C-coordinated titanocene and thus dimerises to yield the corresponding diamidinate products.40a
Further studies have shown that stabilisation of the four-membered diazatitanacycle is also possible by substitution of the central carbon atom, e.g. with organic fragments such as in the guanidinate complex Cp2Ti{κ2-N,N-N(SiMe3)C[=N(SiMe3)2]N(SiMe3)}.40b DFT calculations implied that introduction of sterically demanding substituents at the carbodiimide substrate should facilitate internal complexation to the metal centre, which would be essential for the stability of the mononuclear four-membered diazametallacycles. However, C–N bond activation processes involve dominant reaction motifs when reacting bulky carbodiimides RCNR (R = mesityl, 2,6-(diisopropyl)phenyl) with the titanocene source Cp2Ti(η2-Me3SiC2SiMe3).40
A four-membered phosphazirconacycle was prepared by Chirik et al. from a zirconocene dihydride complex and white phosphorus. In the reaction, elimination of dihydrogen takes place to yield a dinuclear [P4H2]4− bridged dinuclear zirconocene complex with two fused ZrP3 rings.42 Direct and most preferably catalytic functionalisation of P4 by insertion into metal–carbon or metal–hydogen bonds would be an elegant way to synthesise more complex organophosphorus structures.
Five-membered heterometallacycles
Formal addition of another atom into the metallacyclic unit should result in an increase in the stability of the systems, which should allow for the incorporation of double and triple bonds into the systems. The general feasibility of this principle was demonstrated before on several occasions for all-carbon metallacycles.5Bergman and co-workers have developed a multifaceted coupling chemistry employing zirconocene imido complexes,16 thus resulting in the formation of a wide range of heterozirconacycles. For example, a [2 + 2] cycloaddition reaction of a THF stabilised zirconocene imido complex Cp2Zr(thf)NAr (Ar = 2,6-Me-C6H3) with substituted allenes results in the formation of azametallacyclobutenes. The latter species readily rearrange and undergo ring expansion to yield the corresponding isomeric five-membered zirconocene monoazadiene (MAD) complexes (Scheme 9).43 As evidenced by data obtained from X-ray analysis, these compounds are best described as metallacyclopentenes, rather than as an η4-monoazadiene complex.
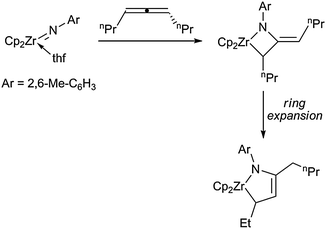 | ||
| Scheme 9 Example for the formation of MAD complexes from four-membered azazirconacyclobutanes. | ||
Reactions of the imido complex Cp2Zr(thf)NtBu with epoxides resulted in the ring opening of the highly strained heterocycles, giving azaoxazirconacyclopentanes. It should be noted that the absence of β-hydrogen atoms at the epoxide substrate is essential for the formation of metallacyclic products. In contrast, epoxides with accessible β-hydrogens form allylic alkoxy amides as the dominant products. The reason for this reactivity was found to be the nucleophilicity of the imido nitrogen atom of the zirconocene starting material, which facilitates hydrogen abstraction from the β position of the substrate.44 A similar ring opening reaction with N-tosyl-2-phenylaziridine gave rise to a diazazirconacyclopentane complex.43 This type of saturated metallacyclic structure is also accessible by simple salt elimination from the dilithiated amine ligand and a zirconium chloro species as was shown by Jordan et al. for an ansa-zirconocene complex.45
Five-membered metallacycles containing exclusively Si atoms and the metal centre were extensively studied by Marschner and co-workers. Similarly as for the preparation of smaller ring systems (vide supra), simple salt metathesis from oligosilyl-α,ω-dipotassium compounds and metallocene dichlorides yields the corresponding zircona- and hafnacyclopentasilanes Cp2M[κ2-Si,Si-Si(SiMe3)2(SiMe2)2Si(SiMe3)2] (Scheme 10).46
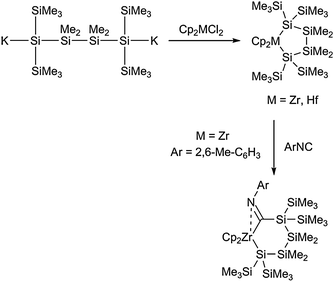 | ||
| Scheme 10 Generation of metallacyclopentasilanes and insertion of an isocyanide into the M–Si bond. | ||
Insertion of an isonitrile into one of the M–Si bonds of the zirconocene complex results in the formation of a six-membered unsymmetrical metallacycle (Scheme 10). Additionally, this insertion reaction was also studied for the four-membered zirconacyclotetrasilanes, thus yielding the respective five-membered ring with an exocyclic CN double bond and a dative Zr–N interaction.28 This interaction was found to be responsible for the exclusive formation of mono-insertion-products; reaction at the second Zr–Si bond was not observed.
Later, Marschner, Baumgartner, Müller and co-workers also studied five-membered silametallacycles in the oxidation state +3. These were obtained either from reactions of the oligosilyl-α,ω-dipotassium compounds with the M(III) source Cp2MCl2·K(18crown6) (M = Ti, Zr, Hf) or by reduction of the M(IV) metallacyclopentasilanes Cp2M[κ2-Si,Si-Si(SiMe3)2(SiMe2)2Si(SiMe3)2], respectively.47 In this context, it should be noted that such group 4 metal oligosilyl complexes form attractive model complexes for catalytic reactions involving silanes. Examples include the dehydrogenative coupling polymerisation of polysilanes, an atom-economic alternative to Wurtz type reactions, which involve large amounts of salts as by-products.48
A different way of obtaining silametallacycles was described by our group recently: Si–H activation and subsequent coupling of alkynylsilanes at hafnocene centres results in the formation of five-membered hafnasilacyclopentenes (Scheme 11).49 Most likely, generation of these unusual complexes takes place via initial activation of the Si–H bond at the metal centre, followed by formation of a silyl hydride and a hafnasilacyclopropane. The latter could then insert a second molecule of the alkynylsilane and yield the product complex. DFT calculations confirmed the general plausibility of the suggested intermediates and some of the elementary steps in the proposed mechanism of their formation.
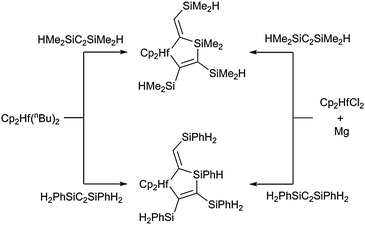 | ||
| Scheme 11 Generation of five-membered hafnasilacyclopentenes by coupling of alkynylsilanes. | ||
Another hafnasilacyclopentene complex was obtained as a by-product in the formation of the decamethylhafnocene alkyne complex Cp*2Hf(η2-Me3SiC2SiMe3). In a second reaction pathway (besides η2-coordination of the alkyne to the hafnium centre), tandem Si–C and C–H bond activation take place to generate a tucked-in hafnocene alkenyl complex, which further rearranges to yield a metallacyclic structure as the thermodynamically more stable product (Scheme 12).50 These examples demonstrate the special reactivity of hafnocene complexes compared to similar titanocene and zirconocene complexes, which should be taken into account when employing such compounds for stoichiometric and catalytic applications.
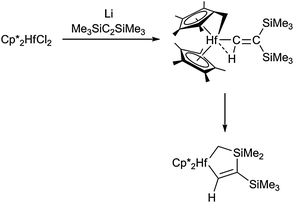 | ||
| Scheme 12 Si–C and C–H activations at a decamethylhafnocene centre. | ||
A more strained five-membered silametallacycle, 1-zircona-2,5-disilacyclopent-3-yne, was prepared by reaction of Cp2ZrCl2 and the alkyne ClMe2SiC2SiMe2Cl in the presence of magnesium as the reducing agent (Scheme 13).51 This approach is very similar to that used before for the generation of unusual all-carbon metallacyclopent-3-ynes.52 Structural and spectral parameters as well as DFT analysis of the structure and bonding revealed the cyclic 1-zircona-2,5-disilacyclopent-3-yne structure with a weak metal-triple-bond interaction to be the best description for this complex. It should be noted that similar titanocene and hafnocene complexes of this type could not be prepared.
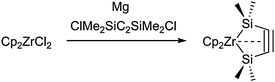 | ||
| Scheme 13 Preparation of a 1-zircona-2,5-disilacyclopent-3-yne. | ||
In attempts to generate highly strained four-membered heterometallacycloallene complexes, titanocene and zirconocene bis(trimethylsilyl)acetylene complexes were reacted with a variety of alkyl and aryl substituted carbodiimides. As a result, the expected formation of mono- or dinuclear metallocene carbodiimide species was not observed for some cases. Instead, Si–C bond activation at the bis(trimethylsilyl)acetylene ligand and coupling with the carbodiimide takes place to yield five-membered heterometallacycloallenes with one nitrogen atom incorporated into the ring (Scheme 14).53 X-ray analysis revealed that both, metallacycle and bis-π-complex, contribute to the structure of the complexes, whereas theoretical studies implied that the heterometallacycloallene form should be the main contributor to the resonance hybrid of the complex. Similarly as observed before for five-membered all-carbon metallacycles (i.e. 1-metallacyclopenta-2,3,4-trienes and 1-metallacyclopent-3-ynes) a significant interaction of the central double bond with the metal centre was found to be present in these complexes, thus contributing to the stability of these unusual complexes.
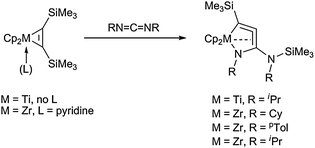 | ||
| Scheme 14 Synthesis of five-membered heterometallacycloallenes by insertion of carbodiimides. | ||
Very recently, two more directed approaches for the synthesis of these five-membered heterometallacycloallene complexes were presented in a joint publication by the groups of Erker and Suzuki. Firstly, reaction of a (η2-iminoacyl)zirconocene chloride complex, which was initially formed from Cp2Zr(H)Cl and an isocyanide, with trimethylsilylethynyl lithium yielded the corresponding metallacyclic product (Scheme 15a).54 Moreover, the authors showed that generation of this species necessarily takes place via initial formation of a hetero(enyne) complex. The second described methodology uses Negishi's zirconocene reagent [Cp2Zr] and alkynyl imines as the heterosubstrates (Scheme 15b). This approach is very similar to that used before by the same group for the synthesis of parent all-carbon zirconacyclopenta-3,4-dienes55 and gives rise to a wide range of structurally similar five-membered heterometallacycloallene complexes.
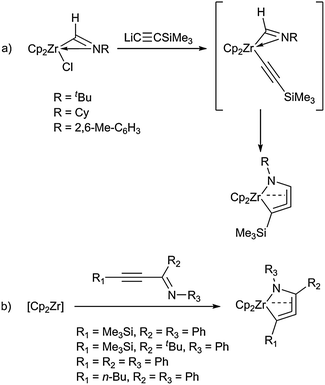 | ||
| Scheme 15 Synthesis of five-membered heterometallacycloallenes by coupling at a zirconocene fragment. | ||
A different type of product was obtained in the reaction of the titanocene bis(trimethylsilyl)acetylene complex with 1,3-di-p-tolylcarbodiimide, namely a dinuclear titanocene complex with two carbodiimide fragments serving as a diamidate bridging unit.52 This type of complex was described before by Floriani et al., who reacted the titanocene source Cp2Ti(CO)2 with the same carbodiimide substrate as a model for CO2 coupling and functionalisation.56
Group 4 metallocene diazadiene (DAD) complexes can be readily obtained by reaction of the corresponding metallocene precursor with the appropriate ligand fragment. Scholz and co-workers reported on the synthesis of a series of titanocene DAD complexes starting from Cp2TiCl2 and the diazadiene in the presence of magnesium as the reducing agent (Scheme 16). Moreover, the preparation of a hafnocene complex was accomplished by reaction of the disodium salt Na2[PhN–C(Ph)C(Ph)NPh] with Cp2HfCl2.57 A structurally similar zirconocene complex was later reported by Gardiner et al., who isolated this diazametallacycle from the reaction of Cp2ZrCl2 with the dilithio compound.58
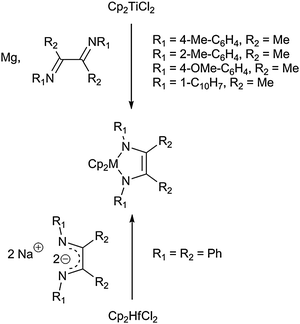 | ||
| Scheme 16 Formation of group 4 metallocene DAD complexes. | ||
More unusual, fused metallocene diazadiene complexes are accessible from insertion of isocyanides into metal–carbon bonds of metallacycles. Examples include the reactions of the unusual five-membered 1-zirconacyclopent-3-ynes with tert-butylisocyanide, thus yielding 1-zircona-2,5-diazacyclopent-3-enes with a backbone consisting of a four-membered cyclobutene with exocyclic double bonds (which originated in the coordinated butatriene fragment of the 1-zirconacyclopent-3-yne starting material).59 A similar insertion reaction of an isocyanide into the metal–carbon contact of a zirconocene η2-cyclobutadiene complex gave a highly unusual 1-zircona-2,5-diazacyclopent-3-ene with four anellated ring fragments (Scheme 17).60
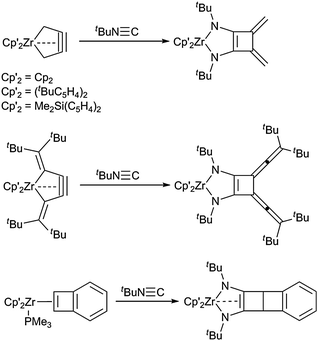 | ||
| Scheme 17 Isocyanide insertions into 1-zirconacyclopent-3-ynes yield diazazirconacyclopentenes. | ||
A macromolecular system with three metallacyclic units was described by Beckhaus and co-workers. Reaction of the titanocene source Cp2Ti(η2-Me3SiC2SiMe3) with quinoxaline results in the formation of a hexaazatrinaphthylene (HATN) ligand, which coordinates three titanocene fragments (Scheme 18).61 Initial C–H bond activation induces spontaneous coupling of three quinoxaline molecules to yield a complex heterocyclic structure with potential applications in polymer-based hydrogen storage materials or molecular magnets – most remarkably within only one synthetic step.
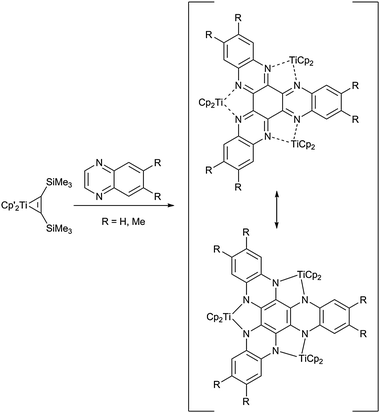 | ||
| Scheme 18 Coupling of quinoxalines at a titanocene centre. | ||
Larger heterometallacycles and cluster compounds
Larger heterometallacycles, especially those containing zirconocene fragments, were studied intensively by Xi et al. Six- or even seven-membered ring systems can be obtained from reactions of the metallocene reagent [Cp2Zr]–Si-tethered diyne with a variety of unsaturated organic heterosubstrates such as nitriles and isocyanides (Scheme 19). The bicyclic zirconocene starting material displays a highly unusual, seemingly stable, but highly reactive zirconacyclobutene–silacyclobutene ring-fused structure.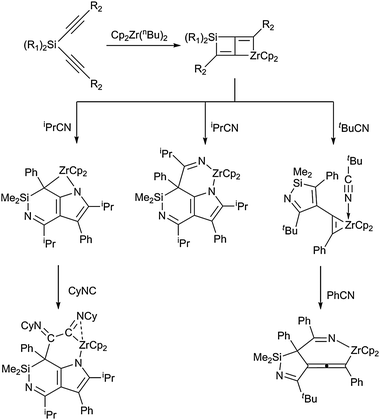 | ||
| Scheme 19 Selected examples for larger azazirconacycles generated from zirconocene–Si-tethered diyne and organic nitriles and isocyanides. | ||
Upon hydrolysis, these cyclic species yield highly functionalised organic molecules, which are potentially interesting for further synthetic applications.62 However, it should be noted in this context that it is difficult to predict general reactivity patterns, most likely due to the structure of the zirconocene–Si-tethered diyne adduct, which can be described in at least four different resonance forms, thus leading to a multifaceted coupling chemistry.63
Another example for the formation of seven-membered heterometallacycles was reported by Erker and co-workers.64 Reaction of the five-membered zirconacycloallenoid Cp2Zr[CH2-C(Me)CC(tBu)] with acetonitrile gave a zirconacyclic allene complex, which was most likely formed by insertion of the nitrile into the metal–carbon bond followed by tautomerisation to yield the metallocene enamido product complex (Scheme 20).
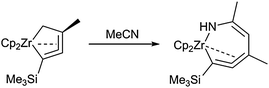 | ||
| Scheme 20 Formation of a seven-membered zirconocene allene complex. | ||
Moreover, examples for heterometallacyclic complexes with cluster structures are well precedented in the literature. Breunig and co-workers described the synthesis of two polynuclear titanocene organoantimony cluster compounds by reaction of the titanocene alkyne complex Cp2Ti(η2-Me3SiC2SiMe3) with organostibanes R2Sb2R2 (R = Me3SiCH2, 2-(Me2NCH2)C6H4).65 Zirconocene polyphosphorus compounds were prepared by Scheer et al., who reacted the tetraphosphabicyclobutane complex Cp′2Zr(P4) (Cp′ = η5-1,3-tBu2C5H3) with the phosphaalkyne PCtBu to obtain two zirconocene complexes as well as an organophosphorus cage species.66 Both of the organometallic products formally display fused heterometallacyclic units.
Structure and bonding in heterometallacycles
The principles of structure and bonding in three- and five-membered group 4 metallacycles containing exclusively the metal centre and carbon ring atoms were described by our group before.67 For highly strained and low valent complexes featuring unsaturated motifs, internal complexation was found to be one of the preferred stabilising factors. Additionally, external complexation via additional ligands or interactions with metal centres in proximity is a viable option for the realisation of unusual metallacycles.For the parent heterometallacycles, similar interactions can be considered as responsible factors for stabilisation, thus resulting in exotic structural motifs such as strong deviations from linearity as found in the five-membered zirconacycloallene Cp2Zr{N(Cy)C[N(Cy)(SiMe3)]CC(SiMe3)} with an angle for the coordinated allene unit of 154°.52 Additionally, due to free electron pairs at hetero-ring atoms, cyclic delocalisation of electrons has to be taken into account when interpreting the structures and bonding of these species.
As for the rationalisation of the bonding situations, in many cases more than one mesomeric structure can be formulated, thus yielding resonance hybrids that describe the overall structures of the metallacycles. An example for this is shown in Scheme 21: in the aforementioned azazirconacycloallenes formally both mesomeric structures (metallacycle and bis-π-complex) contribute to the molecular structure, however the presence of two σ bonds was found to be favourable in this case.52
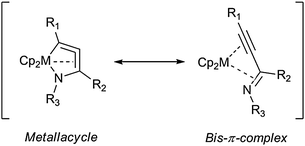 | ||
| Scheme 21 Resonance forms of an azametallacycloallene complex. | ||
Evidence for an unusual and hitherto unknown bonding feature was found in mononuclear four-membered diazatitanacycles (Scheme 22).41b The presence of an interannular titanium–carbon interaction (b) was found to be essential for the stabilisation of such ring systems, thus reducing the biradicaloid nature due to unpaired electrons at both, the metal centre and the central carbon atom (a). However, to date, isolation of such a complex was not successful. Instead, stabilisation by external complexation (i.e. dimerisation or substitution at the central carbon atom) was present.
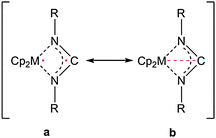 | ||
| Scheme 22 Electronic situation in a four-membered diazatitanacycloallene complex. | ||
Conclusions and outlook
The herein described examples for heterometallacycles with group 4 metallocene fragments demonstrate that unusual and highly strained metallacyclic structures can be generated by well-established and straightforward synthetic methodologies. These include simple coordination of the ligand precursor at the metallocene, coupling reactions of unsaturated organic substrates with a metallocene reagent as well as salt metathesis reactions, e.g. from readily available metallocene halide complexes. Some of the ligand frameworks generated in the coordination sphere of the metallocenes are highly unusual and are only accessible following significantly more complex organic transformations.As for applications in catalysis, heterometallacycles of group 4 metallocenes are discussed as intermediates in various catalytic processes, e.g. in hydroamination reactions, [2 + 2 + 2] cycloaddition reactions as well as in the dehydrogenative coupling polymerisation of silanes. Mechanistic studies to gain further insights into these transformations were performed and should be the subject of future investigations.
Structure and bonding in heterometallacycles is a topic of current interest, especially in the context of exploring the frontiers of unusual structural motifs and geometries. Combinations of classical preparative and computational chemistry revealed that internal or external complexations are essential for the stabilisation of most of the exotic unsaturated metallacyclic complexes. Further studies to broaden the scope of strained and unusual heterometallacycles and ligand architectures are promising and should hold expected outcomes as well as surprises for both, synthetic and computational chemists.
Acknowledgements
We would like to thank all former and present co-workers who contributed to this work in our group. Financial support by the DFG (project codes RO 1269/7-2 and RO 1268/8-1) is gratefully acknowledged.Notes and references
- (a) M. J. Overett, K. Blann, A. Bollmann, J. T. Dixon, D. Haasbroek, E. Killian, H. Maumela, D. S. McGuinness and D. H. Morgan, J. Am. Chem. Soc., 2005, 127, 10723 CrossRef CAS; (b) S. Peitz, B. R. Aluri, N. Peulecke, B. H. Müller, A. Wöhl, W. Müller, M. H. Al-Hazmi, F. M. Mosa and U. Rosenthal, Chem.–Eur. J., 2010, 16, 7670 CrossRef CAS.
- B. Heller and M. Hapke, Chem. Soc. Rev., 2007, 36, 1025 RSC.
- (a) E. Negishi and S. Huo, in Titanium and Zirconium in Organic Synthesis, ed. I. Marek, Wiley-VCH, Weinheim, 2002 Search PubMed; (b) T. Takahashi and Y. Li, in Titanium and Zirconium in Organic Synthesis, ed. I. Marek, Wiley-VCH, Weinheim, 2002 Search PubMed.
- E. D. Jemmis, A. K. Phukan, H. Jiao and U. Rosenthal, Organometallics, 2003, 22, 4958 CrossRef CAS.
- (a) U. Rosenthal, V. V. Burlakov, P. Arndt, W. Baumann and A. Spannenberg, Organometallics, 2005, 24, 456 CrossRef CAS; (b) U. Rosenthal, V. V. Burlakov, M. A. Bach and T. Beweries, Chem. Soc. Rev., 2007, 36, 719 RSC; N. Suzuki and D. Hashizume, Coord. Chem. Rev., 2010, 254, 1307–1326 Search PubMed.
- W. Ando, F. Hojo, S. Sekigawa, N. Nakaayama and T. Shimizu, Organometallics, 1992, 11, 1009 CrossRef CAS.
- X.-S. Ye and H. N. C. Wong, J. Org. Chem., 1997, 62, 1940 CrossRef CAS.
- J.-H. Liu, H.-W. Chan, F. Xue, Q.-G. Wang, T. C. Mak and H. N. C. Wong, J. Org. Chem., 1999, 64, 1630 CrossRef CAS.
- Selected examples: (a) P. J. Fagan and W. A. Nugent, J. Am. Chem. Soc., 1988, 110, 2310 CrossRef CAS; (b) B. Jiang and T. D. Tilley, J. Am. Chem. Soc., 1999, 121, 9744 CrossRef CAS; (c) C. Fan, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2009, 48, 2955 CrossRef CAS.
- W. A. Nugent, D. W. Ovenall and S. J. Holmes, Organometallics, 1983, 2, 161 CrossRef CAS.
- P. J. Walsh, A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1992, 114, 1708 CrossRef CAS.
- F. N. Tebbe, G. W. Parshall and G. S. Reddy, J. Am. Chem. Soc., 1978, 100, 3611 CrossRef CAS.
- R. Beckhaus, I. Strauß, T. Wagner and P. Kiprof, Angew. Chem., Int. Ed. Engl., 1993, 32, 264 CrossRef.
- E. Negishi, F. E. Cederbaum and T. Takahashi, Tetrahedron Lett., 1986, 27, 2829 CrossRef CAS.
- (a) U. Rosenthal, V. V. Burlakov, P. Arndt, W. Baumann and A. Spannenberg, Organometallics, 2003, 22, 884 CrossRef CAS; (b) T. Beweries and U. Rosenthal, Science of Synthesis Knowledge Updates 2011/4, ed. D. G. Hall, K. Ishihara, J. J. Li, I. Marek, M. North, E. Schaumann, S. M. Weinreb and M. Yus, Stuttgart, 2011 Search PubMed.
- P. J. Walsh, F. J. Hollander and R. G. Bergman, Organometallics, 1993, 12, 3705 CrossRef CAS.
- P. J. Chirik, Organometallics, 2010, 29, 1500 CrossRef CAS , and references cited therein.
- G. P. Pez, P. Apgar and R. K. Crissey, J. Am. Chem. Soc., 1982, 104, 482 CrossRef CAS.
- Selected recent examples: (a) W. H. Bernskoetter, A. V. Olmos, J. A. Pool, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 10696 CrossRef CAS; (b) W. H. Bernskoetter, E. Lobkovksy and P. J. Chirik, Angew. Chem., Int. Ed., 2007, 46, 2858 CrossRef CAS; (c) D. J. Knobloch, E. Lobkovsky and P. J. Chirik, Nat. Chem., 2010, 2, 30 CrossRef CAS; (d) D. J. Knobloch, S. P. Semproni, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2012, 134, 3377 CrossRef CAS.
- K. Kaleta, P. Arndt, T. Beweries, A. Spannenberg, O. Theilmann and U. Rosenthal, Organometallics, 2010, 29, 2604 CrossRef CAS.
- K. Kaleta, P. Arndt, A. Spannenberg and U. Rosenthal, Inorg. Chim. Acta, 2011, 370, 187 CrossRef CAS.
- P. J. Walsh, F. J. Hollander and R. G. Bergman, J. Organomet. Chem., 1992, 428, 13 CrossRef CAS.
- O. Theilmann, W. Saak, D. Haase and R. Beckhaus, Organometallics, 2009, 28, 2799 CrossRef CAS.
- H. J. Breunig, T. Borrmann, E. Lork, C. I. Rat and U. Rosenthal, Organometallics, 2007, 26, 5364 CrossRef CAS.
- Y. Wang, B. Quillian, X.-J. Yang, P. Wei, Z. Chen, C. S. Wannere, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2005, 127, 7672 CrossRef CAS.
- (a) S. L. Buchwald and R. B. Nielsen, Chem. Rev., 1988, 88, 1047 CrossRef CAS , and references cited therein; (b) H. G. Alt, K.-H. Schwind, M. D. Rausch and U. Thewalt, J. Organomet. Chem., 1988, 349, C7 CrossRef CAS; (c) P. Binger, F. Langhauser, P. Wedemann, B. Gabor, R. Mynott and C. Krüger, Chem. Ber., 1994, 127, 39 CrossRef CAS.
- (a) R. Fischer, M. Zirngast, M. Flock, J. Baumgartner and C. Marschner, J. Am. Chem. Soc., 2005, 127, 70 CrossRef CAS; (b) M. Zirngast, M. Flock, J. Baumgartner and C. Marschner, J. Am. Chem. Soc., 2009, 131, 15952 CrossRef CAS.
- H. Arp, J. Baumgartner, C. Marschner, P. Zark and T. Müller, J. Am. Chem. Soc., 2012, 134, 10864 CrossRef CAS.
- R. Fischer, D. Frank, W. Gaderbauer, C. Kayser, C. Mechtler, J. Baumgartner and C. Marschner, Organometallics, 2003, 22, 3723 CrossRef CAS.
- H. Köpf and R. Voigtländer, Chem. Ber., 1981, 114, 2731 CrossRef.
- M. Schaffrath, A. Villinger, D. Michalik, U. Rosenthal and A. Schulz, Organometallics, 2008, 27, 1393 CrossRef CAS.
- K. Kaleta, M. Ruhmann, O. Theilmann, S. Roy, T. Beweries, P. Arndt, A. Villinger, E. D. Jemmis, A. Schulz and U. Rosenthal, Eur. J. Inorg. Chem., 2012, 611 CrossRef CAS.
- M. Haehnel, S. Hansen, A. Spannenberg, P. Arndt, T. Beweries and U. Rosenthal, Chem.–Eur. J., 2012, 18, 10546 CrossRef CAS.
- J. D. Selby, C. Schulten, A. D. Schwarz, A. Stasch, E. Clot, C. Jones and P. Mountford, Chem. Commun., 2008, 5101 RSC.
- S. E. d'Arbeloff, P. B. Hitchcock, J. F. Nixon, H. Kawaguchi and K. Tatsumi, J. Organomet. Chem., 2003, 672, 1 CrossRef.
- R. Waterman and T. D. Tilley, Chem. Commun., 2006, 4030 RSC.
- (a) S. Gambrotta, S. Strologo, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Am. Chem. Soc., 1985, 107, 627 Search PubMed; (b) F. Benetollo, G. Cavinato, L. Crosara, F. Milani, G. Rossetto, C. Scelza and P. Zanella, J. Organomet. Chem., 1998, 555, 177 CrossRef CAS.
- C. Cornelißen, G. Erker, G. Kehr and R. Fröhlich, Z. Naturforsch., 2004, 59b, 1246 Search PubMed.
- S.-D. Bai, H.-B. Tong, J.-P. Guo, M.-S. Zhou and D.-S. Liu, Inorg. Chim. Acta, 2009, 362, 1143 CrossRef CAS.
- (a) S. E. Potts, C. J. Carmalt, C. S. Blackman, F. Abou-Chahine, D. Pugh and H. O. Davies, Organometallics, 2009, 28, 1838 CrossRef CAS; (b) Y. A. Wasslen, E. Tois, S. Haukka, K. A. Kreisel, G. P. A. Yap, M. D. Halls and S. T. Barry, Inorg. Chem., 2010, 49, 1976 CrossRef CAS.
- (a) O. Theilmann, M. Ruhmann, A. Villinger, A. Schulz, W. W. Seidel, K. Kaleta, T. Beweries, P. Arndt and U. Rosenthal, Angew. Chem., Int. Ed., 2010, 49, 9282 CrossRef CAS; (b) M. Haehnel, M. Ruhmann, S. Roy, T. Beweries, P. Arndt, A. Spannenberg, A. Villinger, E. D. Jemmis, A. Schulz and U. Rosenthal, J. Am. Chem. Soc., 2012, 134, 15979 CrossRef CAS.
- P. J. Chirik, J. A. Pool and E. Lobkovsky, Angew. Chem., Int. Ed., 2002, 41, 3463 CrossRef CAS.
- F. E. Michael, A. P. Duncan, Z. K. Sweeney and R. G. Bergman, J. Am. Chem. Soc., 2005, 127, 1752 CrossRef CAS.
- (a) S. A. Blum, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 2003, 125, 14276 CrossRef CAS; (b) S. A. Blum, V. A. Rivera, R. T. Ruck, F. E. Michael and R. G. Bergman, Organometallics, 2005, 24, 1647 CrossRef CAS.
- M. D. LoCoco, X. Zhang and R. F. Jordan, J. Am. Chem. Soc., 2004, 126, 15231 CrossRef CAS.
- C. Kayser, G. Kickelbick and C. Marschner, Angew. Chem., Int. Ed., 2002, 41, 989 CrossRef CAS.
- H. Arp, M. Zirngast, C. Marschner, J. Baumgartner, K. Rasmussen, P. Zark and T. Müller, Organometallics, 2012, 31, 4309 CrossRef CAS.
- R. D. Miller and J. Michl, Chem. Rev., 1989, 89, 1359 CrossRef CAS.
- M. Lamac, A. Spannenberg, W. Baumann, H. Jiao, C. Fischer, S. Hansen, P. Arndt and U. Rosenthal, J. Am. Chem. Soc., 2010, 132, 4369 CrossRef CAS.
- T. Beweries, V. V. Burlakov, M. A. Bach, S. Peitz, P. Arndt, W. Baumann, A. Spannenberg, U. Rosenthal, B. Pathak and E. D. Jemmis, Angew. Chem., Int. Ed., 2007, 46, 6907 CrossRef CAS.
- M. Lamac, A. Spannenberg, H. Jiao, S. Hansen, W. Baumann, P. Arndt and U. Rosenthal, Angew. Chem., Int. Ed., 2010, 49, 2937 CrossRef CAS.
- N. Suzuki, N. Aihara, H. Takahara, T. Watanabe, M. Iwasaki, M. Saburi, D. Hashizume and T. Chihara, J. Am. Chem. Soc., 2004, 126, 60 CrossRef CAS.
- K. Kaleta, M. Ruhmann, O. Theilmann, T. Beweries, S. Roy, P. Arndt, A. Villinger, E. D. Jemmis, A. Schulz and U. Rosenthal, J. Am. Chem. Soc., 2011, 133, 5463 CrossRef CAS.
- S. K. Podiyanachari, R. Fröhlich, C. G. Daniliuc, J. L. Petersen, G. Kehr, G. Erker, N. Suzuki, S. Yuasa, K. Hagimori, S. Inoue, T. Asada, T. Takemoto and Y. Masuyama, Dalton Trans., 2012, 41, 10811 RSC.
- (a) J. Ugolotti, G. Kehr, R. Fröhlich, S. Grimme and G. Erker, J. Am. Chem. Soc., 2009, 131, 1996 CrossRef CAS; (b) N. Suzuki, T. Shimura, Y. Sakaguchi and Y. Masuyama, Pure Appl. Chem., 2011, 83, 1781 CrossRef CAS.
- M. Pasquali, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Am. Chem. Soc., 1979, 101, 4740 CrossRef CAS.
- J. Scholz, G. A. Hadi, K.-H. Thiele, H. Görls, R. Weimann, H. Schumann and J. Sieler, J. Organomet. Chem., 2001, 626, 243 CrossRef CAS.
- A. C. Benjamin, A. S. P. Frey, M. G. Gardiner, C. L. Raston, B. W. Skelton and A. H. White, J. Organomet. Chem., 2008, 693, 776 CrossRef CAS.
- (a) M. A. Bach, T. Beweries, V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg and U. Rosenthal, Organometallics, 2007, 26, 4592 CrossRef CAS; (b) N. Suzuki, D. Hashizume, H. Yoshida, M. Tezuka, K. Ida, S. Nagashima and T. Chihara, J. Am. Chem. Soc., 2009, 131, 2050 CrossRef CAS.
- T. V. V. Ramakrishna, S. Lushnikova and P. R. Sharp, Organometallics, 2002, 21, 5685 CrossRef CAS.
- I. Piglosiewicz, R. Beckhaus, W. Saak and D. Haase, J. Am. Chem. Soc., 2005, 127, 14190 CrossRef CAS.
- Selected recent examples: (a) S. Zhang, W.-X. Zhang and Z. Xi, Chem.–Eur. J., 2010, 16, 8419 CrossRef CAS; (b) S. Zhang, W.-X. Zhang, J. Zhao and Z. Xi, J. Am. Chem. Soc., 2010, 132, 14042 CrossRef CAS; (c) S. Zhang, J. Zhao, W.-X. Zhang and Z. Xi, Org. Lett., 2011, 13, 1626 CrossRef CAS; (d) S. Zhang, W.-X. Zhang, J. Zhao and Z. Xi, Chem.–Eur. J., 2011, 17, 2442 CAS.
- W.-X. Zhang, S. Zhang and Z. Xi, Acc. Chem. Res., 2011, 44, 541 CrossRef CAS.
- G. Bender, G. Kehr, R. Fröhlich, J. L. Petersen and G. Erker, Chem. Sci., 2012 10.1039/C2SC20745A.
- H. J. Breunig, E. Lork, O. Moldovan, C. I. Rat, U. Rosenthal and C. Silvestru, Dalton Trans., 2009, 5065 RSC.
- U. Vogel, M. Eberl, M. Eckhardt, A. Seitz, E.-M. Rummel, A. Y. Timoshkin, E. V. Peresypkina and M. Scheer, Angew. Chem., Int. Ed., 2011, 50, 8982 CrossRef CAS.
- (a) E. D. Jemmis, S. Roy, V. V. Burlakov, H. Jiao, M. Klahn, S. Hansen and U. Rosenthal, Organometallics, 2010, 29, 76 CrossRef CAS; (b) S. Roy, E. D. Jemmis, M. Ruhmann, A. Schulz, K. Kaleta, T. Beweries and U. Rosenthal, Organometallics, 2011, 30, 2670 CrossRef CAS.
Footnote |
| † This perspective covers developments from 2000 onwards. We are aware that this does not include all examples from the literature, however, we would like to make this limitation in the interest of the volume of the article. |
| This journal is © The Royal Society of Chemistry 2013 |