The use of enzymes in organic synthesis and the life sciences: perspectives from the Swiss Industrial Biocatalysis Consortium (SIBC)
Hans-Peter Meyer*a, Eric Eichhornb, Steven Hanlonc, Stephan Lützd, Martin Schürmanne, Roland Wohlgemuthf and Raffaella Coppolecchiag
aLonza AG, 3930 Visp, Switzerland. E-mail: hans-peter.meyer@lonza.com; Fax: +41 27 947 51 78; Tel: +41 27 948 51 78
bGivaudan Schweiz AG, Ueberlandstrasse 138, 8600 Dübendorf, Switzerland
cF. Hoffmann-La Roche AG, Grenzacherstrasse 124, 4070 Basel, Switzerland
dNovartis Pharma, Postfach, 4002 Basel, Switzerland
eDSM Innovative Synthesis BV, Urmonderbaan 22, NL-6167RD Geleen, The Netherlands
fSigma-Aldrich, Industriestrasse 25, 9470 Buchs, Switzerland
gCerbios Pharma SA, Via Pian Scairolo 6, 6917 Barbengo-Lugano, Switzerland
First published on 10th August 2012
Abstract
The potential of biotechnology by means of biocatalysis or biosynthesis in organic synthesis is far from being fully exploited. For this reason a group of life science companies active in pharmaceuticals, flavour and fragrance, vitamin and fine chemicals businesses describe some examples of the use of enzymes in industrial organic synthesis and discuss why enzymes are still the exception rather than the rule in organic synthesis.
1. Introduction
“It takes 20 years to become an overnight success”. This sentence seems appropriate to describe the situation of biotechnology in organic chemistry in general and the use of biocatalysis in industrial organic synthesis in particular. The potential of biotechnology for sustainable manufacturing is far from being fully realized in many industries. 80 years after the industrial applications of enzymes in the bioconversion of steroids, the use of enzymes is still under expectation in chemistry.There is however progress, the most recent examples being the ketoreductases (alcohol dehydrogenases, KRED or ADH respectively) and the transaminases which are starting to establish themselves alongside the widely-used hydrolases. Test kits and several enzyme variants are commercially available, which allows inclusion of these enzymes in the planning of synthetic strategies by the organic chemist. Bornscheuer et al.1 described very recently how biocatalysis reached its present industrially proven level through three technological waves of research and innovation leading to enzymatic steps in blockbusters such as atorvastatin (Lipitor®) or sitagliptin (Januvia®). However, despite these and other applications of enzymatic reactions also run at an industrial scale, e.g. the acrylamide process with nitrile hydratase, the production of beta-lactam antibiotics or the recent production of the sitagliptin API with a transaminase, the potential of biocatalysis is not yet fully realised.
These few achievements should not conceal the fact that although biocatalysis has been more frequently used in areas such as fine chemicals and pharmaceuticals, it still often represents a second generation process. As a matter of fact it is exactly this area which requires better and commercially available enzymes as well as the rapid development of synthetic routes and sustainable manufacturing processes. It is of critical importance to further expand the toolbox with commercially available biocatalysts of all six different enzyme classes to meet the ever increasing degree of sophistication of the molecules (small and large) to be produced, modified and functionalised and to offer green processes in a timely manner. Because of the challenges and difficulties in the life sciences and the pharmaceutical industry in particular (time pressure, heterogeneous chiral molecules, increasing molecular complexity), these industries remain the most important drivers for innovation in biocatalysis.
Another important breakthrough which we put forward is the application of directed or targeted evolution for biological protein engineering.2 Directed evolution has established itself as an industrial tool and several companies offering this service have been founded in recent years.
However, this technological breakthrough will not make biotransformation an overnight success in organic chemical synthesis. It is also most important to reach an agreement between chemists and biologists on one side and between industry and academia on the other. All parties involved should agree on a roadmap and on priorities, i.e. on where to focus development and where to reduce priorities. Such a roadmap is proposed in this paper. Another point, which is put forward, is to critically question the way biocatalysis and chemical synthesis work together, and the need to reinvent the interface between them. Innovation is also needed in contracting and business models. We describe a bottom-up solution from a group of companies with a common interest in using and promoting biocatalysis in its various aspects as a rapid, efficient and economic technology for the production of a variety of industrially significant chemicals.
In a 2007 publication which received much attention, Constable et al.3 described the needs of the pharmaceutical industry with respect to organic chemical synthesis. The present contribution goes beyond that in two directions: we specifically identify the issues with respect to the enzyme toolbox and we also go beyond the pharmaceutical industry by including the view of other industrial partners as well.
2. Biocatalysis and biotransformation
Switzerland has become one of the global leaders in red biotechnology, the biotechnology of large parenteral molecules, mainly therapeutic proteins. This success story started from the already established small molecule pharmaceuticals within the Swiss chemical and pharmaceutical industry at that time. The application of enzymes for the preparation of small molecule pharmaceuticals was neglected while red biotechnology became a huge success story over the last 20 years: Swiss companies are global leaders in the large scale manufacturing of monoclonal antibodies for example.The principles of biocatalysis and biotransformation can be of use for many different markets with incredibly diverse products theoretically representing a large global commercial potential. However, biocatalysis has begun to establish itself more firmly in the pharmaceutical industry only, although the process remains a slow uphill battle with classic organic chemical synthesis remaining the standard approach of choice. Table 1 summarizes some facts with regard to recent developments in biocatalysis and its industrial application.
| Ranking of the industries using biotechnology in terms of value added: 1. Pharma, 2. Food industry, 3. Agrochemicals, 4. Cosmetics, 5. Polymers, 6. Others |
| 7 of 40 small molecule pharmaceuticals produced using biocatalysis, are blockbusters |
| The market value of the blockbusters represents over 30 billion US$. This tendency is increasing |
| Enzyme classes relevant to the production of the mentioned products are Oxidoreductases EC 1: 50%, Hydrolases EC 3: 35%, Transferases EC 2: >5%, Lyases EC 4: >5%, Isomerases EC 5: >1% |
| Biotransformation still often represents second generation processes. The diversity of the enzyme toolbox remains a problem that must be solved |
| Biotechnology and biocatalysis are key technologies for a sustainable (green) chemistry |
Theoretically, almost any organic chemical reaction can be carried out by enzymes directing in the right thermodynamic energy path. The reason why the potential of biocatalysis is not fully exploited for manufacturing in the life sciences and chemical industries is that the industry is generally lacking a broad panel of enzyme catalyzed reaction platforms including the respective enzyme libraries for its various applications and the often long lead times. The compounds include natural and non-natural intermediates as well as final Active Pharmaceutical Ingredients (APIs), flavour and fragrance compounds, natural products, fine chemicals, specialty chemicals, metabolites and derivatives thereof.
Creative solutions are not only needed in the laboratory or the pilot plant: they must be applicable at a large scale as well. The Swiss Industrial Biocatalysis Consortium (SIBC) was founded in 2004 to help overcome the limitations, which apply in many industries, namely the penury of commercially available strains and enzymes, which is common to all SIBC members.4 In 2004 the SIBC formulated that the priorities should be to find new strains, enzymes and reaction types. Since then the consortium has regularly assessed the situation; the assessments of 2004 and 2012 are summarized in Table 2.
| a FMO: Flavin Monooxygenase, ThDP: Thiaminediphosphate, CYP450: Cytochrome P450 monooxygenases. |
|---|
| Oxidoreductases |
| Dehydrogenases |
| 1. NAD(P)H-dependent dehydrogenases for the asymmetric reduction of ketones, ketoacids, olefins etc.… |
| 2. Oxidation of alcohols with dehydrogenases |
| Oxygenases |
| 1. Monohydroxylations, especially for the hydroxylation of non-activated centers and of non-natural substrates. Improve practicability, robustness and substrate scope of the in vitro CYP450s systems, develop an FMO-based alternative system |
| 2. Peroxidases |
| 3. Transformation of ribonucleotides, stereospecific epoxidations, oxidation of ketones to esters and lactones (Baeyer Villiger) |
| Lyases |
| 1. Synthetically useful enzymes for C–C bond formation (preferably asymmetric) using aldolases, hydroxynitrile lyases and ThDP-dependent lyases |
| 2. C–N (aminolyases) and C–O (hydratases) bond formations. Identification of lyases with a broad substrate acceptance |
In 2004 the SIBC members (Ciba, Givaudan, Hoffmann-La Roche, Lonza, Novartis, Sigma-Aldrich) also identified a few areas with lower R&D needs. Hydrolases (lipases, esterases, proteases, acylases) were identified as being of lower priority as the technology had been implemented in industry and their improvement and adaptation are routinely carried out by companies with expertise in the field. Epoxide hydrolases, amidases, nitrilases, nitrile hydratases and glycosidases are other types of hydrolytic enzymes of some industrial interest, which often perform below expectations but for which modern biological engineering techniques are nowadays offered by several companies. With respect to transferases, transaminases may have the highest impact followed by glycosyltransferases and sulfotransferases (both for the generation of drug candidates and metabolites), but also methyltransferases have found increased interest. The importance of hydroxylation stressed in Table 2 applies for the generation of drug metabolites as well as for synthetic purposes of pharmaceuticals, flavours and fragrances or other compounds. Despite some processes at a large scale, racemases and isomerases have limited industrial applications probably mainly due to their narrow substrate specificities.
The current consortium members (Cerbios-Pharma, DSM, Givaudan, Hoffmann-La Roche, Lonza, Novartis, Sigma-Aldrich) conclude from regular assessments that the key issues essentially remain the same. However a few new aspects listed in Table 3 have gained importance since then.
| a An internal discussion and review among consortium members in 2012 confirmed that the challenge remains the myriad of yet unknown substrates and products for biocatalysis, and which requires a corresponding toolbox. |
|---|
| 1. Sugar chemistry, glycosylations and deglycosylations |
| 2. Nucleotide and peptide chemistry by enzymatic coupling reactions |
| 3. Halogenations, especially fluorinations |
| 4. Fast and cost efficient directed evolution and rapid scale-up |
Biotransformation processes are far more diverse than therapeutic protein production processes. The substrates and/or products of biotransformation processes may inhibit the reaction, may be poorly soluble or unstable novel small molecules. Timeline compressions in the development cycle of pharmaceuticals, in combination with the absence of suitable biocatalysts result in the fact that bioprocesses typically represent the second generation process choice in the manufacturing of small molecule pharmaceuticals or intermediates.
In order to answer the question of the research focus on biocatalysis all abstracts submitted to the “International Symposium on Biocatalysis and Biotransformations” (the Biotrans conference series) for the years 2003 to 2011 have been analysed and the result is summarized in Table 4.
| Enzyme class | 2011 | 2009 | 2007 | 2005 | 2003 |
|---|---|---|---|---|---|
| a As an example: 34% of all presentations in 2007 dealt with oxidoreductases. The sum must not add up to 100% since some of the presented papers did not deal with biotransformations. | |||||
| Oxidoreductases | 39 | 32 | 34 | 24 | 28 |
| Transferases | 13 | 10 | 8 | 6 | 3 |
| Hydrolases | 35 | 46 | 41 | 55 | 58 |
| Lyases | 9 | 9 | 12 | 12 | 10 |
| Isomerases | 3 | 3 | 2 | 2 | 1 |
| Ligases | 1 | 0 | 1 | 1 | 0 |
Hydrolytic enzymes remain the most widely presented enzyme group among the lectures and posters presented at the 2011 conference. A trend seen over the last 10 years is that oxidoreductases and transferases are the only two enzyme classes which show a significant increase in interest. Table 4 shows also that there are still unknown “biotransformation” territories to be conquered and that considerable efforts have to be made for the discovery and introduction of novel enzyme panels/reactions types. In our opinion both aspects can be best dealt with by coordinated activities in an academic environment where “risky” and more long-term projects can be performed better than in industry. However, hydrolytic enzymes no longer really represent a prime academic topic, in particular as there are small companies specialised in improving and adapting these biocatalysts to industrial needs.
3. Selected biotransformation applications at an industrial scale by SIBC member companies
Some examples of biotransformations applied at an industrial scale in SIBC member companies are given in the following.3.1. Oxidoreductases EC 1
The advantage of catalysing selective oxidations with safe and environmentally-friendly oxidants in non-flammable solvents is of major importance for process improvements from a safety, health and environmental perspective.5 Since inherently chiral oxidoreductases in addition often catalyze reactions in an enantioselective way, the added benefit by the application of biocatalysts is enormous.Laccase–mediator systems are relevant to fragrance chemistry for the oxidation of terpene molecules. Niku-Paavola and Viikari10 investigated the oxidation of several fragrance precursors using a laccase from Trametes hirsuta in three laccase–mediator systems. The mediators appeared to differ from each other under optimal reaction conditions and in specificity towards a given target compound.
In the flavour and fragrance industry there is a constant demand for compounds possessing unique olfactory properties within the floral and woody odour notes. Such compounds extend the perfumer's and flavourist's palettes and result in greater product diversity for consumers. Using a laccase-mediator system technology Givaudan created a novel class of octahydroazulene derivatives, which possess very strong and pleasant floral and woody odour as well as organoleptic characteristics.11 These molecules can be used as such, but they are particularly suitable when present in a mixture resulting from a laccase-catalyzed transformation of an olefinic mixture enriched in α-guaiene resulting, e.g., from Patchouli (Pogostemon cablin) oil distillation (Fig. 1). These ingredients are intended for use in alcoholic perfumes as well as other fragranced consumer products, and perhaps also in flavoured consumer goods.
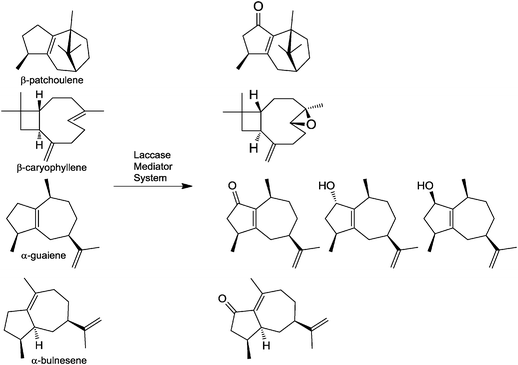 | ||
| Fig. 1 Oxidation of an α-guaiene rich olefinic fraction with a laccase-mediator system. The olefinic fraction is treated with the Denilite® II S laccase (Novozymes) resulting in the oxidation of oil-resident olefins to the corresponding alcohols, ketones or epoxide. | ||
There is an ongoing trend to build on green chemistry and biocatalysis reactions in the flavour and fragrance industry. Lack of specific and efficient enzyme systems is still limiting the number of successful applications. Laccase–mediator systems are quite unique with respect to the wide spectrum of substrates that can be transformed based on the specific selection of enzyme and the best suited mediator. The availability of a laccase–mediator toolbox allows the rapid screening of substrates to identify a laccase–mediator system that generates products meeting certain criteria and specifications with a fair chance to upscale the reaction with minimal technical hurdles and at an attractive cost.
In the following, a recent example for the efficient preparation of drug metabolites with biocatalysts displaying high stereoselectivity is shown. It concerns the preparation of exo and endo monohydroxylated metabolites of an amino-benzathiazole derivative 1 which had been under development as an adenosine 2a receptor antagonist and potentially useful as a treatment for neurological disorders.13 The major metabolite 2 found in plasma and urine samples from clinical studies arising from exo-hydroxylation at the oxa-bicyclohexyl moiety was not readily accessible by organic synthesis and without biocatalysis would have required a major synthetic effort. As this metabolite was not found in tox species it was necessary to provide sufficient material for pharmacological activity testing and for analytical reference material. A second metabolite 4 formed by hydroxylation and N-demethylation was also required.
As recombinant CYP450s did not produce 2, a screening was carried out with 1100 bacterial and fungal strains in a low-well format. After incubation for 3 days the samples were analyzed by HPLC-DAD and those potentially containing metabolites by HPLC-MS/MS. Only two strains (both Actinoplanes sp.) were found which produced 2 in significant amounts and one of these, CBS 614.71, was selected for scaleup as the retention time and MS/MS spectrum were identical with that of 2 in human samples. 2 × 8 L biotransformations were carried out containing a total of 3.2 g 1. The substrate was completely converted after 48 h and 2 was subsequently purified by prep-HPLC and subsequent crystallization affording 2 in 56% overall yield (1.8 g) with a purity of 99%.
For the preparation of 4, the N-demethylated substrate 3 had to be employed14 as no strain was capable of the double hydroxylation of 1. On incubation of 3 with the two Actinoplanes strains it was found that ATCC 5371 reactions contained a product with identical retention time to 4 in human samples. Two 8 L biotransformations were carried out containing a total of 0.8 g 3. After 1 day biotransformation, 4 was isolated by extraction, chromatography and crystallization, affording 85 mg 4 in 10% overall yield and 98% purity.
Confirmation of identity by NMR analysis of the pure 2 and 4 revealed the configuration of the hydroxyl group was exo and endo, respectively. This result nicely demonstrated the great potential of microbial biotransformations in stereoselective preparation of authentic drug metabolites (Fig. 2).
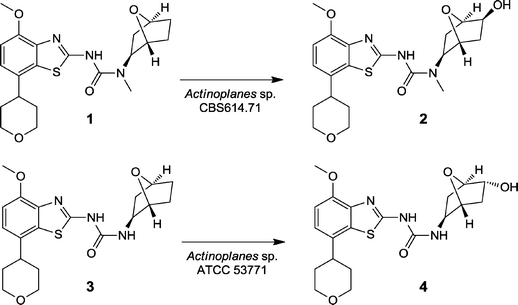 | ||
| Fig. 2 Stereoselective hydroxylation of an A2a receptor antagonist 1 and N-desmethyl metabolite 2 by two Actinoplanes strains. | ||
3.2. Transferases (EC 2)
In the context of production of reduced folates, active ingredients widely used in oncology applications, Cerbios was interested in evaluating a diastereo-selective, enzymatic alternative to the chemical tetrahydrofolic acid formylation, with the purpose of obtaining the active ingredient (6S)-5-formyltetrahydrofolate. Therefore attention was paid to the enzyme glutamate formyl transferase.| Tetrahydrofolate + N-formyl-L-glutamate ⇌ 5-formyltetrahydrofolate + L-glutamate |
This reaction was assumed earlier to be catalyzed by a different enzyme (EC 2.2.2.6) and has only been recently ascribed to GFT. As frequently observed among transferases, pyridoxal phosphate is required as a cofactor for the transfer of the formyl group to THF by GFT. In nature GFT is found both in prokaryotes and eukaryotes as catalyst involved in histidine utilization, as well as in the biochemical pathway involving folic acid utilization. The reaction of interest is only one of the possible transfer reactions catalyzed by GFT; also possible is the reversible transfer of one-carbon group (formyl-, formidoyl-) from 5-formimidoyl-/or formyl-tetrahydrofolate to L-glutamate. GFT has been studied for its role as THF-dependent glutamate formiminotransferase involved in histidine utilization interconverting L-glutamate and N-formimino-L-glutamate. GFT is generally a bifunctional enzyme catalyzing also the cyclodeaminase reaction on N-formimino-THF leading to 5,10-methenyl-THF and ammonia. The crystal structure of the enzyme has been studied in the context of its catalytic mechanism.15 Quite a large number of eukaryotic and prokaryotic gene sequences encoding for GFT are deposited at the National Center for Biotechnology Information (NCBI) gene bank. The gene coding for the microbial GFT of Streptococcus pyogenes MGAS5005 (GeneID: 3571098) was chosen because of its monofunctional glutamate formyltransferase activity, in contrast to the homologous eukaryotic enzyme, which exhibits a formimidoyltetrahydrofolate cyclodeaminase (EC 4.3.1.4) activity as well. In this view, the use of the S. pyogenes GFT offers the advantage of obtaining (6S)-5-formyltetrahydrofolate as a homogenous product starting from tetrahydrofolate and N-formyl-L-glutamate. The GFT protein used here in (6S)-5-formyltetrahydrofolate production was prepared by overexpression of the corresponding gft gene of S. pyogenes ATCC 19615 in the lon−E. coli strain BL21(DE3) (Fig. 3).
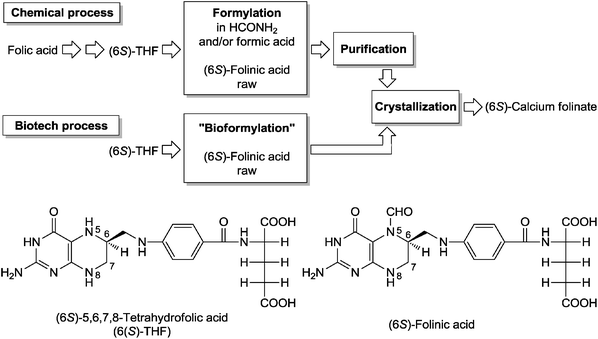 | ||
| Fig. 3 Chemical and biotech processes for the formylation of (6S)-5,6,7,8-tetrahydrofolic acid (THF) in the synthesis of (6S)-calcium folinate. The use and disposal of the relatively toxic compounds formamide and formic acid can be avoided in the biotech process where N-formylglutamic acid is used as a formyl-group donor. | ||
3.3. Hydrolases (EC 3)
While biocatalysis certainly has a lot to offer in terms of asymmetric synthesis (e.g. transaminases or ketoreductases), classical resolution reactions, although limited to a maximum of 50% yield of one enantiomer, are still useful tools and frequently used in drug discovery chemistry. At early stages, time and availability of material is sometimes more important than a highly efficient (i.e. high yielding) process. Moreover, resolution reactions by definition allow access to both optical antipodes. This is particularly useful, when establishing a comprehensive set of chiral building blocks.It is therefore not surprising that hydrolytic enzymes, which have been a preferred research topic in the last 20 years, are frequently used in different industries. They do not require cofactors, are easily available at a large scale and a broad range of hydrolases has been well characterized with predictable results. Thus they also still play a role in the synthesis of chiral building blocks at the Bioreactions group within the Novartis Institutes for BioMedical Research and at Sigma-Aldrich for example. The groups' task is to use biocatalysis in support of the medicinal chemistry effort to produce drug metabolites and chiral building blocks with isolated enzymes and microbial systems.
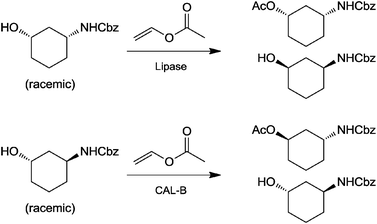 | ||
| Fig. 4 Resolution of 1,3-aminocyclohexanols. | ||
The resolution of the cis-racemate (Fig. 4, upper half) was conducted with three different enzymes, the lipase B from Candida antarctica (CAL-B) and the lipases from Pseudomonas fluorescens and Thermomyces lanuginosus. The highest selectivity was obtained using the latter enzyme, with an E value of more than 1000. Due to his high discrimination between the enantiomers, the reaction stopped at 50.4% of conversion and the alcohol could be obtained in very high optical purity of 99.9% ee. CAL-B was only modestly selective (E ca. 100), which resulted in an ee of 95.4% for the alcohol. The lipase from P. fluorescens had medium selectivity (E ca. 300) and yielded the alcohol with 98.6% ee. Using the trans-racemate as substrate (Fig. 4, lower half), the alcohol was obtained with 37% yield and 99.8% optical purity. In this case, only CAL-B was used. The reaction was carried out on a 660 g scale, yielding 244 g of the chiral building block.
In this way, all four stereoisomers are accessible via the lipase resolution. This illustrates the usefulness of kinetic resolutions in the supply of chiral building blocks for drug discovery chemistry. In addition to hydrolase-catalyzed kinetic resolutions of racemic mixtures, the synthesis of chiral building blocks from symmetric starting materials has been expanded over the last 20 years at Sigma-Aldrich.
Hydrolase-catalyzed desymmetrization reactions are particularly interesting because of theoretically complete substrate-to-product conversion. Although the development of both enantiotope-selective hydrolyses of prochiral acylated diols and dicarboxylic esters in aqueous buffers as well as the enantiotope-selective acylation of prochiral diols has been developed decades ago, short synthetic routes to valuable chiral compounds from easily accessible starting materials are still of industrial interest.
If pure prochiral starting compounds are used, chiral products of high chemical and enantiomeric purity can be obtained in high yield. Esterases have proven to be useful biocatalysts in the desymmetrization of a variety of prochiral glutaric esters, e.g. in the desymmetrization of dimethyl 3-hydroxy-3-methylglutarate as a key step in the synthesis of (R)-mevalonolactone or in the desymmetrization of dimethyl cis-1,2,3,6-tetrahydrophthalate to (1S,2R)-1-methyl-cis-1,2,3,6-tetra-hydrophthalate. Diols and their derivatives can be prepared by lipase-catalyzed hydrolysis (Fig. 5) or by epoxide hydrolase-catalyzed ring-opening reactions of the corresponding epoxides.
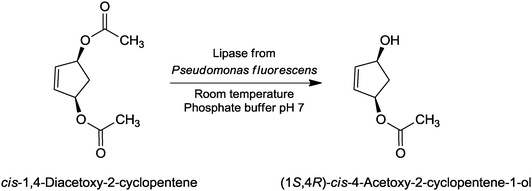 | ||
| Fig. 5 Synthesis of (1S,4R)-cis-4-acetoxy-2-cyclopentene-1-ol by lipase-catalyzed hydrolytic desymmetrization. | ||
Especially large scale peptide manufacturing of quantities greater than 100 kg is still a highly cost-intensive endeavor, especially for long chain peptides.18 Peptides are not a minor market with annual sales in the order of about 10 billion US$. In order to minimize costs Lonza has successfully combined chemistry and biotechnology. Below two examples employing a combination of chemical synthesis, biocatalysis and recombinant fermentation are given.19,20
Chymotrypsin was used for an enzymatic fragment condensation for the synthesis of dicarba analogs of calcitonin. Elcatonin is a 32 amino acid peptide hormone of the calcitonin family. In elcatonin, the aminosuberic acid containing analog of eel calcitonin, the substitution of the intrachain disulfide loop of natural hormone with an ethylene bridge in 1–7 positions, gives better stability and higher activity than elcatonin making it resistant to proteolysis. Elcatonin stimulates osteoblasts and is used in the treatment of osteoporosis, Paget's disease and musculoskeletal pain. It is formulated both as injectable and as nasal spray. The semisynthesis of eel[L-α-aminosuberic acid]calcitonin or elcatonin was accomplished by a α-chymotrypsin-catalyzed coupling of two peptide segments at low temperature in a single reaction without protection of any functional group. One of the peptide segments, the amino terminal cyclic desamino 9 amino acid peptide fragment was synthesized chemically. The other 23 amino acid carboxyterminal fragment was produced recombinantly in E. coli.
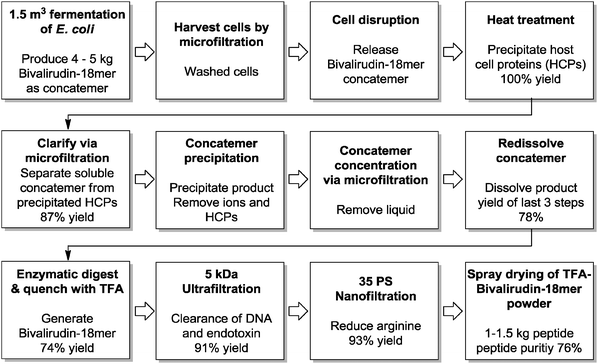 | ||
| Fig. 6 Process flow scheme of the semi-synthesis of bivalirudin. As this peptide would be recognized as foreign structures by microbial host cells, it must be concealed and protected from the cellular proteolytic machinery. One way to do that is to artificially enlarge the peptide by expressing as a long concatemer chain of 18 amino acid peptides and digest the chain into the appropriate peptides. The bivalirudin-18 mer process is run at a 1.5 m3 piloting scale. TFA: trifluoroacetic acid. | ||
3.4. Lyases (EC 4)
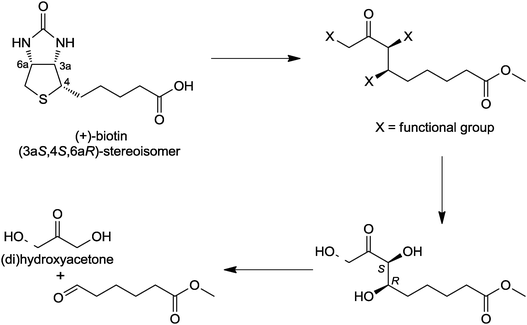 | ||
| Fig. 7 Retrosynthetic scheme from DHA and HA to (+)-biotin. | ||
From a chemical point of view, the following general problems associated with efficient routes to this vitamin have to be solved in an economically and ecologically satisfactory manner: introduction of nitrogen and sulfur functionalities to form the highly functionalized bi-heterocycle, introduction of the C5 side chain, and generation of the three stereogenic centers of the all-cis-thiophane ring.26,27
A general new route by using aldolase reactions is depicted in Fig. 7. Acetone or its functionalized derivatives hydroxyacetone or dihydroxyacetone are reacted with acceptor substrate (electrophile) adipic acid semialdehyde methylester (AASM). Remarkably, the full carbon skeleton is built up, and chirality is introduced in one reaction step. The follow-up chemistry applies functional group transformations for introduction of the sulfur and nitrogen substituents.
For the synthesis of the key intermediates, three variations of this route employing the three donor substrates (nucleophiles) dihydroxyacetone (DHA), (mono-)hydroxyacetone (HA) and acetone were investigated with the acceptor substrate (electrophile) adipic acid semialdehyde methylester (AASM, Fig. 8).
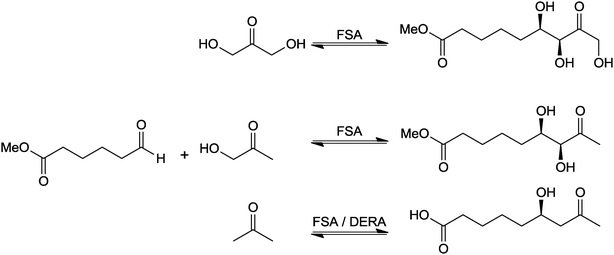 | ||
| Fig. 8 Investigated aldolase reaction with 3 different donor substrates and two aldolases (variants). FSA: D-fructose 6-phosphate aldolase, DERA: 2-deoxy-D-ribose 5-phosphate aldolase. | ||
Two different aldolases were applied to achieve the target conversion: wild-type and a variant 2-deoxy-D-ribose 5-phosphate aldolase (DERA) which had been used for the industrial production of statin intermediates28,29 as well as wild-type and variant D-fructose 6-phosphate aldolases (FSAs) were tested. FSAs have been reported as a new type of aldolases capable of using the cheap and readily available donor substrates hydroxyacetone and dihydroxyacetone30,31 instead of the expensive and unstable dihydroxyacetone-phosphate required by D-fructose 1,6-bisphosphate aldolases such as RAMA.32 To the best of our knowledge the efficient conversion of the donor substrate of acetone by an aldolase had not been described. Wong and co-workers as pioneers in aldolase chemistry report only minimal activity for acetone (∼0%) compared to its physiological substrate acetaldehyde33 although a 14% conversion of acetone and 3-hydroxy-2-azido-propanal was described.34
The two FSAs from Escherichia coli represent unusually temperature stable enzymes for their mesophilic origin, a property which could be attributed to its tightly packed tertiary and quaternary structure consisting of a donut shaped dimer of pentamers.35 To test these enzymes in the target reactions the genes fsaA and fsaB encoding the corresponding aldolases FsaA and FsaB were amplified from genomic DNA of E. coli W3110 and cloned into and pBAD-derived expression vector. For the more efficient conversion of dihydroxyacetone Ala129Ser mutations36 were introduced in both fsa genes by site-directed mutagenesis. The enzymes were recombinantly expressed in E. coli TOP10 at the shake-flask level and tested in the target reactions. Wild-type FsaB was produced in a batch fermentation on a 10 litre scale.
The aldol reaction of the acceptor AASM with acetone was found to be catalyzed by wild-type DERA, variant DERA and both wild-type FSAs (in ascending order of efficiency). This acceptor substrate was generally converted faster and to higher yields by FsaB than FsaA. As anticipated the wild-type FSAs were more active with AASM and hydroxyacetone than with dihydroxyacetone. On the other hand the FSA variants containing the Ala129Ser exchange had higher activities with dihydroxyacetone and AASM compared to hydroxyacetone (data not shown).
The reaction of wild-type FsaB with HA and AASM was scaled-up to 0.5 litre with substrate concentrations of 2.5 and 2.0 mol l−1, respectively. Almost quantitative conversion of AASM was obtained after overnight stirring at room temperature. High volumetric activities and specific activities close to the natural reaction were observed for this reaction (Table 5). HPLC and NMR analysis proved excellent enantioselectivity and diastereoselectivity of the enzymes for these substrates of >99%, respectively, for the 3S,4R-configured product. The other stereo-isomers were all below the detection level.
| Hydroxyacetone | Dihydroxyacetone | |
|---|---|---|
| Substrate concentration | 2.0 mol l−1 | 0.5 mol l−1 |
| Product concentration | 40% (w/v) | 10% (w/v) |
| Volumetric productivity | 170 g/(l*h) | 80 g/(l*h) |
| Specific activity | 13 U mg−1 | 6 U mg−1 |
FsaB from the same fermentation was used in the reaction of DHA and AASM at 0.5 mol l−1 each. The specific activity and volumetric productivity for this reaction was lower than with HA, but still in a high range compared to the physiological substrates (Table 5). Also this reaction occurred with apparently full stereo-selectivity as no other stereo-isomer could be detected.
Most of the subsequent steps towards (+)-biotin were proven to be feasible and efficient as well (to be published elsewhere or reported before). In summary a novel and efficient route to (+)-biotin was developed based on three aldolase options making use of cheap, readily available and stable donor substrates. Exceptionally high activities and productivities were achieved with D-fructose 6-phosphate aldolases and the donor substrates hydroxyacetone and dihydroxyacetone.
3.5. Isomerases (EC 5)
Typically the substrate scope of isomerases is rather narrow and restricted to their natural substrates, e.g. sugars and amino acids. Nevertheless one of the biggest industrial processes with an enzyme is the manufacturing of High Fructose Corn Syrup (HFCS) from glucose with thermostable Xylose isomerase. This process runs on a multi-million ton scale, which underlines its economic significance.37 The use of isomerases in the synthesis of fine chemicals is rather limited, as most often the target molecule is chiral, and the stereoselective synthesis is often a reason to choose a biocatalytic route, so to plan a route with a biocatalytic racemisation reaction might seem counter-intuitive. Recently, several examples for beneficial use of racemases have been given. Racemases can be combined with physicochemical enantioseparation processes, e.g. chromatography or preferential crystallisation and lead to higher yields for the desired enantiomer by recycling the other half of the starting material. One example is the chromatographic separation of racemic methionine combined with a coupled enzymatic racemisation.38,39 Another example is the combination of preferential crystallisation with enzymatic in situ racemisation, as exemplified for L-asparagine40 and allo-threonine.413.6. Ligases (EC 6)
These enzymes are frequently used in laboratory applications, particularly in the field of molecular biology, but large-scale use in biocatalysis is still missing because costly cofactors such as ATP are required. Thus these enzymes, if used for preparative purposes, will only be useful for high value products.4. Process engineering
The advantage of many biocatalytic processes, especially those using hydrolases, is the fact that they can be carried out in normal chemical stirred tank reactors and do not need special costly equipment (Fig. 9). The process is often adapted to the properties of the biological system, although the opposite is possible and the preferred way.42 For most process-related problems more or less practicable solutions are available today. | ||
| Fig. 9 The success of scale-up procedures for a large variety of hydrolase-catalyzed processes is based on robust, scalable and simple process designs without cofactor regeneration as shown here for a standard multi-purpose reactor for hydrolase-catalyzed desymmetrizations and resolutions using standard process control for the reaction and further processing and product isolation being performed at the bottom of the reactor in the lower level (Copyright @ Sigma-Aldrich). | ||
In order to perform bioreactions, the biocatalyst can be operated under various process modes and conditions which include batch, fed-batch and continuous operation43 in one pot single stage or cascade reactors. Artificial cell production using prokaryotic as well as eukaryotic machineries is an emerging option. Down Stream Processing (DSP) is normally the least problematic aspect of biocatalysis, as scalable chemical process technology for small molecules is available. Poor solubility of reactants may require non-aqueous reaction media in one single or two separate phases for which enzymes have to be adapted. However, the problem of product inhibition still awaits a scalable technical multi-product solution for removal of the inhibiting product from a biocatalysis broth e.g. by in situ Product Removal (ISPR) with resins based solutions, electrodialysis or membrane based solutions. In the case of substrate as well as product inhibition, the in situ Substrate Feed and Product Recovery (SFPR) methodology has proven useful for a number of reaction types and processes. Product recovery needs to be considered in the context of the whole bioprocess in order to isolate and purify the product of interest within the windows of stability in terms of pH, temperature and solvent.
For economic reasons whole-cell biocatalysis is used wherever possible. Immobilization of the cells is one efficient method to further improve the productivity and process economics. This is true for biocatalysis as well as for the production of an enzyme by fermentation. Immobilization of the biomass can be done by adsorption (Fig. 10), crosslinking, covalent fixation, entrapment or cell separation cell.44,45
 | ||
| Fig. 10 Construction installed in a 15 m3 fermenter with the aim to hold 400 m2 of polyurethane tubes as a matrix for the immobilization of microbial cells by adsorption for the production of an oxidoreductase. One can see the Rushton impellers, flow breakers on the wall and the polyurethane tubes being mounted on a scaffold in the reactor. For scale: note the engineer installing the tubes in the upper right corner of the picture, standing on the top impeller (copyright @ Lonza s.r.o. Czech Republic). | ||
5. Outlook
The huge potential of enzymes as nature's privileged catalysts for making possible the diverse metabolic pathways of living cells points us to an intensified global exploration of the organic chemistry, biology and engineering fundamentals of enzyme-catalyzed transformations. The translation of many of these enzyme-catalyzed transformations into industrially viable processes and technologies will help in the value creation and establish biocatalysis as a key area within catalysis science and technology. The scientific and technological exchange between the metal and organometallic catalysis, the bio- and organocatalysis areas is therefore not only important for the cross-fertilization, but also for the development of novel routes and multi-step conversions by the combination of different catalysts in a one-pot reaction.Biocatalysis and biotransformation have given the life sciences and chemical industries solid ways and means to help out on a large scale manufacturing processes relying on well established chemistries with reactions catalyzed by lipases, proteases and alcohol dehydrogenases. A further set of enzymatic chemistries encompassing reactions catalyzed by nitrilases, amidases and hydroxynitrilases, transaminases, can be termed as expanding since over recent years a trend was noted that these enzymes are finding their applications on a large scale. A number of important chemical reaction types are, however, still not accessible to large scale chemical synthesis. Among these are especially oxidation reactions catalyzed by a variety of oxygenases, C–C bond formation reactions as catalyzed by e.g. decarboxylases, aldolases, or reactions catalyzed by enoate or ene reductases. These enzyme classes deserve major investigative efforts in order to establish robust biocatalysts applicable at a large scale.
Massive pathway engineering and synthetic biology will be increasingly implemented into manufacturing processes. For the application of biotechnology for chemical synthetic purposes one should not differentiate between biocatalysis and biosynthesis (fermentation) as both are complementary. However, biocatalysis might become more and more a niche technology for difficult and highly specific synthetic purposes.
Classical biocatalysis (one step reactions catalyzed with an isolated enzyme) will continue to have its rightful place in the synthetic repertoire. However, to fully exploit the potential of biocatalysis it is important that the enzyme toolbox is filled with new and improved enzymes to enable new types of chemistry and to remain competitive with synthetic organic chemistry. The “toolbox” character cannot be stressed enough for an enzyme to be attractive for industrial use. Ideally, we would have CAL-B type enzymes (in terms of substrate scope, versatility, availability and process robustness) also for e.g. reductive amination, hydroxylation or bond formations (Table 2 and 3). It will certainly not be realistic to expect this in one single enzyme molecule, but a set of e.g. 10–20 robust peroxidases, with known sequences and recombinantly overexpressed, covering a substrate range from small aliphatics to multiple ring (hetero)aromatics would certainly be useful and ensure future use of biocatalysis.
However, to realize the full potential of this niche technology the following approaches are suggested:
• The development of robust biocatalysts in the classes which have not yet proven to be applicable at a large scale because of being more challenging.
• A selection of ready to use and stable enzymes from all EC classes (not only hydrolases), similar to ketoreductases/alcohol dehydrogenases for synthetic purposes.
• The development of meaningful test kits of biocatalysts in the different classes, which have not yet proven to be applicable at a large scale (outside hydrolases and alcohol dehydrogenases/ketoreductases).
• Increased availability of enzyme sequences from genomics and their functional characterization. Ideally enzyme sequence are known and available (in case the vendor goes out of business to ensure supply), they are preferably overexpressed in aerobic GRAS organisms. Enzyme variants should be available covering a decent substrate scope, as well as both (R)- and (S)-selective variants to ensure enantiocomplementarity of route design.
• Technologies for rapid scale up and large scale production.
• New models of (intensified) collaboration/networking to get facile and economic access to specific biocatalysts and technologies.
References
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. More and K. Robins, Nature, 2012, 485, 185 CrossRef CAS.
- S. Lütz, L. Giver and J. Lalonde, Biotechnol. Bioeng., 2008, 101, 657 Search PubMed.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, Jr., R. J. Lindermann, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- B. Wirz, M. Kittelmann, H.-P. Meyer and R. Wohlgemuth, Chimia, 2011, 64, 780 CrossRef.
- Modern Biocatalysis, ed. W. D. Fessner and T. Anthonsen, Wiley-VCH, Weinheim, 2009 Search PubMed.
- S. Riva, Trends Biotechnol., 2006, 24, 219 CrossRef CAS.
- A. I. Cañas and S. Camarero, Biotechnol. Adv., 2010, 28, 694 CrossRef.
- S. Witayakran and A. J. Ragauskas, Adv. Synth. Catal., 2009, 351, 1187 CrossRef CAS.
- P. Baiocco, A. M. Barrecca, M. Fabbrini, C. Galli and P. Gentili, Org. Biomol. Chem., 2002, 1, 191 RSC.
- M. L. Niku-Paavola and L. Viikari, J. Mol. Catal. B: Enzym., 2000, 10, 435 CrossRef CAS.
- B. Schilling, T. Granier and E. Locher, Patent application, WO 2012/001018/A1, 2012 Search PubMed.
- M. El Yacoubi, C. Ledent, M. Parmentier, R. Bertorelli, E. Ongini, J. Costentin and J.-M. Vaugeois, Br. J. Pharmacol., 2001, 134, 68 CrossRef CAS.
- B. Wirz, P. Spurr and C. Pfleger, Tetrahedron: Asymmetry, 2010, 21, 159 CrossRef CAS.
- preparation: Christian Jenny, Chemical Synthesis, Roche, 2012, unpublished.
- D. Kohls, T. Sulea, E. O. Purisima, R. E. MacKenzie and A. Vrielink, Structure, 2000, 8, 35 CrossRef CAS.
- C. E. Brocklehurst, K. Laumen, L. La Vecchia, D. Shaw and M. Vögtle, Org. Process Res. Dev., 2011, 15, 294 CrossRef CAS.
- H.-P. Meyer and N. Turner, Mini-Rev. Org. Chem., 2009, 6, 300 CrossRef CAS.
- H.-P. Meyer and O. Werbitzky Oleg, in Biocatalysis for Green Chemistry and Chemical Process Development, ed. A. Tao and R. Kazlauskas, John Wiley & Sons, Hoboken New Jersey USA, 2012, pp. 23–45 Search PubMed.
- J. Brass, J. Frank, F. Wagner, H. Stocker and E. Wünsch, Future aspects in peptide chemistry, 1999, vol. 1, p. 165 Search PubMed.
- J. M. Brass and W. B. Huizinga, Chem. Today, 2000, 10, 165 Search PubMed.
- J. Richards, J. Brass, H. de Witt, B. Dubuis, R. Gloeckler, J. Gueck, F. Hoeks and A. Tinschert, Poster presented at the Tides Peptide Meeting, 2000, Las Vegas, USA Search PubMed.
- J. Wenger and J. Brass, Lonza AG, 2005, unpublished.
- J. Wenger, Lonza AG, 2006, unpublished.
- M. Casutt, T. Koppe and M. Schwarz, in Ullmann's Enzyclopedia of Industrial Chemistry, VCH, Weinheim, 1996, vol. A 27, pp. 566–575, 609–611 Search PubMed.
- M. Seki, Med. Res. Rev., 2006, 26, 434 CrossRef CAS.
- P. J. De Clercq, Chem. Rev., 1997, 97, 1755 CrossRef CAS.
- W. Bonrath, R. Karge, Th. Netscher, F. Roessler and F. Spindler, Chimia, 2009, 63, 265 CrossRef CAS.
- S. Jennewein, M. Schürmann, M. Wolberg, I. Hilker, R. Luiten, M. Wubbolts and D. Mink, Biotechnol. J., 2006, 1, 537 CrossRef CAS.
- M. Schürmann, S. Panke, H. Kierkels and M. Wolberg, in Green Chemistry in the Pharmaceutical Industry, ed. P. Dunn, A. Wells and M. T. Williams, WILEY-VCH Verlag, Weinheim, 2006 Search PubMed.
- M. Schürmann and G. A. Sprenger, J. Biol. Chem., 2001, 276, 11055 CrossRef.
- M. Schürmann, M. Schürmann and G. A. Sprenger, J. Mol. Catal. B: Enzym., 2002, 19–20, 247 CrossRef.
- M. Schümperli, R. Pellaux and S. Panke, Appl. Microbiol. Biotechnol., 2007, 75, 33 CrossRef.
- L. Chen, D. P. Dumas and C. H. Wong, J. Am. Chem. Soc., 1992, 114, 741 CrossRef CAS.
- J. Liu and C. H. Wong, Angew. Chem., Int. Ed., 2002, 41, 1404 CrossRef CAS.
- S. Thorell, M. Schürmann, G. A. Sprenger and G. Schneider, J. Mol. Biol., 2002, 319, 161 CrossRef CAS.
- J. A. Castillo, C. Guérard-Hélaine, M. Gutiérrez, X. Garrabou, M. Sancelme, M. Schürmann, T. Inoue, V. Hélaine, F. Charmantray, T. Gefflaut, L. Hecquet, J. Joglar, P. Clapés, G. A. Sprenger and M. Lemaire, Adv. Synth. Catal., 2010, 352, 1039 CrossRef CAS.
- A. Liese, K. Seelbach and C. Wandrey, Industrial Biotransformations, Wiley-VCH Verlag, Weinheim, 2nd edn, 2006 Search PubMed.
- M. Bechtold, S. Makart, R. Reiss, P. Alder and S. Panke, Biotechnol. Bioeng., 2007, 98, 812 CrossRef CAS.
- K. Würges, K. Petruševska, S. Serci, S. Wilhelm, C. Wandrey, A. Seidel-Morgenstern, M. P. Elsner and S. Lütz, J. Mol. Catal. B: Enzym., 2009, 58, 10 CrossRef.
- K. Würges, K. Petruševska-Seebach, M. P. Elsner and S. Lütz, Biotechnol. Bioeng., 2009, 104, 1235 CrossRef.
- K. Würges, U. Mackfeld, M. Pohl, S. Lütz, S. Wilhelm, W. Wiechert and T. Kubitzki, Adv. Synth. Catal., 2011, 353, 2431 CrossRef.
- O. Ghisalba, H.-P. Meyer and R. Wohlgemuth, in Encyclopedia of Industrial Biotechnology: Bioprocess, Bioseparation, and Cell Technology, ed. M. C. Flickinger, John Wiley and Sons Inc., 2010, vol. 3, pp. 1–18 Search PubMed.
- K. Würges, D. Minör, N. Rao and S. Lütz, Org. Process Res. Dev., 2009, 13, 607 CrossRef.
- K. T. Robins and J. Gordon, in Biocatalysis for Green Chemistry and chemical process development, ed. J. Tao and R. Kazlauskas, 2011, John Wiley & Sons, 2011 Search PubMed.
- F. W. J. M. M. Hoeks, H. Kulla and H.-P. Meyer, J. Biotechnol., 1992, 22, 117 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |