Remarkable Lewis acid catalytic performance of the scandium trimesate metal organic framework MIL-100(Sc) for C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) N bond-forming reactions†
N bond-forming reactions†
Laura
Mitchell
a,
Berenice
Gonzalez-Santiago
a,
John P. S.
Mowat
a,
Mary E.
Gunn
a,
Patrick
Williamson
a,
Nadia
Acerbi
b,
Matthew L.
Clarke
*a and
Paul A.
Wright
*a
aEaStCHEM School of Chemistry, University of St Andrews, Purdie Building, North Haugh, St Andrews, Fife, KY16 9ST, UK. E-mail: mc28@st-andrews.ac.uk; paw2@st-andrews.ac.uk; Fax: +44 (0)1334 463808
bJohnson Matthey Technology Centre, Blount's Court Road, Sonning Common, Reading, RG4 9NH, UK
First published on 11th October 2012
Abstract
The porous metal organic frameworks scandium trimesate MIL-100(Sc), scandium terephthalates MIL-101(Sc), MIL-88B(Sc) and MIL-68(Sc), scandium 4,4′-biphenyl-dicarboxylate MIL-88D(Sc) and the scandium 3,3′,5,5-azobenzene-tetracarboxylate socMOF(Sc) have been compared as Lewis acid catalysts against Sc3+-exchanged zeolite Beta, MIL-100(Cr), MIL-101(Cr), MIL-100(Fe) and the divalent MOFs HKUST-1(Cu), CPO-27(Ni) and STA-12(Ni), each of which can be prepared with coordinatively unsaturated metal sites. The performance of these MOFs has been investigated in several Lewis acid-catalysed reactions that are of importance in organic synthesis but have rarely been studied using MOF catalysts. These reactions were (i) the intermolecular carbonyl ene reaction of nucleophilic alkenes and electron-poor aldehydes, (ii) a Friedel–Crafts type Michael addition between electron-rich heterocycles and electron-deficient alkenes and (iii) ketimine and aldimine formation. In each of these, MIL-100(Sc) is both active and selective and significantly outperforms the other catalysts. Filtration and recycle tests indicate that catalysis over MIL-100(Sc) is heterogeneous. The study of Michael addition reactions carried out over scandium-bearing MOFs with different window sizes on indole-based substrates of varying molecular dimensions indicates that most of the catalysis that involves molecules small enough to enter the pores occurs within the internal pore space. These results indicate MIL-100(Sc) is an exceptional Lewis acidic MOF catalyst, and suggest that MIL-100(Sc) and new derivatives of it could find application as recyclable solid catalysts in synthetic chemistry.
1. Introduction
In recent years there has been tremendous interest in the synthesis and structural characterisation of porous metal organic frameworks (MOFs).1 While much of this activity has been driven by possible applications in gas storage and/or separation2,3 and bio-medical applications4 there is an increasing emphasis on research into the catalytic applications of MOFs.5–7 Like zeolites, porous crystalline MOFs offer high surface areas that can host active sites accessible via well-defined pore openings, and therefore have potential in heterogeneous catalysis with possible shape selectivity. Furthermore, MOFs have the advantages over zeolites of both greater chemical variability and also porosity that can extend into the mesoporous range, the latter allowing adsorption of larger molecules than can be taken up by zeolites. However, MOFs have lower thermal and chemical stability than zeolites, so they are better suited to the catalytic preparation of fine chemicals rather than bulk chemical synthesis, where zeolites play the leading role.Both the framework structure and the local environments around the metal cations in MOFs can be tuned by altering the ligands that bridge between the metal centres, even after the frameworks are synthesised.8 This versatility, reminiscent of homogeneous organometallic catalysts, suggests that it should be possible to control selectivity in the synthesis of fine chemicals and target synthesis over a MOF-based catalyst. Recyclable heterogeneous MOF catalysts with the desired molecular features to exert control of regiochemistry, chemoselectivity, molecular selection from mixtures and enantioselectivity are therefore a realistic prospect. Furthermore, as true single site heterogeneous catalysts of the type described by Thomas,9 MOFs might have better-defined active site environments than heterogenised molecular catalysts supported on silica or polymers.
One approach to using porous MOFs in catalysis is to functionalise a molecular catalyst (for example with carboxylate groups) such that it can become incorporated as a linker within the MOF.10 In this approach, the majority of metal ions in the MOF are inert vertices and the catalytic activity originates from the molecular catalyst that is cross-linking the MOF architecture. An alternative approach is to design MOFs where the metal cations that are integral to the framework can be made catalytically-active by removal of their solvent ligands to leave a vacant co-ordination site as a Lewis acid so long as the MOF remains porous. One of the most widely studied MOFs for Lewis acid catalysis is HKUST-1 (Cu3BTC2, BTC = 1,3,5-benzenetricarboxylate),11 which possesses framework Cu2+ cations from which water can be removed to leave square planar coordinated Cu2+. IR studies of small molecule adsorption identify these as Lewis acid sites.12 HKUST-1 has been widely studied as a catalyst13 but few studies compare it with other Lewis-acidic MOFs under relevant catalytic reactions. Among MOFs with coordinatively unsaturated metal sites active as Lewis acid catalysts, examples include copper, chromium, iron and zirconium carboxylates14–18 and a cobalt phosphonate:19 in some cases associated bifunctionality18,20 and/or enantioselectivity14 has been introduced.
In the study reported here, our starting point was the knowledge that homogeneous scandium triflate is a very powerful Lewis acid catalyst, the discovery of which21 led to major advances in the application of Lewis acid catalysis in organic synthesis, even in the presence of water. Although it was not obvious that scandium coordinated by carboxylate ligands in a porous solid would be as powerful a Lewis acid as the triflate, previous studies have shown that non-porous scandium carboxylate coordination polymers possess activity in relatively facile Lewis acid catalysed reactions, via surface catalysis.22 Furthermore, we have previously reported23 the synthesis of a range of porous scandium carboxylate MOFs with structures analogous to the structures MIL-88B,24 MIL-10025 and MIL-10126 containing other trivalent metals (such as Cr and Fe) that have been described previously. MIL-88B(Sc) and related materials have also been reported elsewhere27 and a further report of MIL-100(Sc) and MIL-101(Sc) has appeared subsequently.28 The scandium in [Sc3O(O2CR)6(OH)(OH2)2] trimers in these solids can lose coordinated water to leave five-fold coordinated scandium cations that we reasoned would be strong Lewis acid catalysts. Indeed, recent spectroscopic studies have shown that they interact strongly as Lewis acidic sites with molecules such as CO and even H2.29 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency of CO chemisorbed on dehydrated MIL-100(Sc), for example, is 2182 cm−1, which is shifted to higher wavenumbers than when adsorbed on the coordinatively unsaturated metal cation sites of other large pore MOFs, including Cu2BTC3 (HKUST-1),12a the Ni(II) 1,4-dioxido-2,5-benzenedicarboxylate of the CPO-27/MOF-74 family30,31 and the Ni(II) N,N′-piperazine-bismethylenephosphonate of the STA-12 family.32,33
O stretching frequency of CO chemisorbed on dehydrated MIL-100(Sc), for example, is 2182 cm−1, which is shifted to higher wavenumbers than when adsorbed on the coordinatively unsaturated metal cation sites of other large pore MOFs, including Cu2BTC3 (HKUST-1),12a the Ni(II) 1,4-dioxido-2,5-benzenedicarboxylate of the CPO-27/MOF-74 family30,31 and the Ni(II) N,N′-piperazine-bismethylenephosphonate of the STA-12 family.32,33
We therefore aimed to prepare porous scandium-bearing MOFs and to compare their activity with that of the well-known divalent and trivalent large pore Lewis acidic MOFs HKUST-1(Cu), CPO-27(Ni), STA-12(Ni), MIL-100(Fe), MIL-100(Cr) and MIL-101(Cr) in archetypal Lewis acid-catalysed reactions. Here we show that MIL-100(Sc) significantly outperforms the other heterogeneous Lewis acid catalysts in a range of synthetically important Lewis acid-catalysed reactions.
2. Results and discussion
Table 1 summarises the compositions and properties of the MOFs examined as catalysts. Pore volumes are those measured either by N2 adsorption at −196 °C or, for MIL-68(Sc), CO2 adsorption at −77 °C (ESI†). The window sizes are measured assuming solvent O atoms are coordinated to the metal cations. If these were removed, the effective ring diameters would be increased.| MOF [Formula] | Ligand | Structure | Pore volume/cm3 g−1 | Pore sizea/Å |
|---|---|---|---|---|
| a Window sizes estimated for materials with solvent O atoms attached to metal site that can become coordinatively unsaturated. | ||||
| STA-12(Ni) [Ni2L] |

|

|
0.2 | Channels 10 |
|
CPO-27(Ni)
[Ni2(DHTP)] |

|

|
0.47 | Channels 11 |
|
MIL-100 (Sc,Cr,Fe)
[M3O(OH)(BTC)2] |

|

|
0.93 (Sc)
0.80 (Cr) 0.90 (Fe) |
Cages 25 & 30
Windows 5 & 9 |
|
MIL-101 (Sc,Cr)
[M3O(OH)(BTC)2] |

|

|
0.33 (Sc)
1.46 (Cr) |
Cages 29 & 34
Windows 12 & 16 |
|
HKUST-1 (Cu)
[Cu3(BTC)2] |

|

|
0.71 |
Cages 25
Windows 6 |
|
MIL-88B (Sc)
[Sc3O(OH)(BDC)3] |

|

|
<0.02 |
Cage 7
Channel <2 |
|
MIL-88D (Sc)
[Sc3O(OH)(BPDC)3] |

|

|
0.47 |
Cage 16
Channel 13 |
|
MIL-68(Sc)
[Sc(OH)(BDC)] |

|

|
0.63 | Channel 16 |
|
socMOF(Sc)
[Sc3O(ABTC)1.5NO3−] |

|

|
0.56 | 3D network of 5 Å channels |
2.1 Synthesis and characterization of known scandium carboxylates
The scandium terephthalates MIL-88B(Sc) and MIL-101(Sc) were prepared following our published syntheses23 or via their slight modification. Both possess scandium ‘trimer’ units as the metal-based building units (Fig. 1 and 2). MIL-88B(Sc) (Fig. 1) exhibits marked ‘breathing’ behaviour, so that although it has little porosity for N2 when in the fully desolvated state, it adsorbs polar solvents such as methanol to give a greatly expanded framework with a three dimensionally-connected pore system.23 In the case of the MIL-101 structure26 (Fig. 2) the framework is rigid, and has the same tetrahedral connectivity as the zeolite topology type MTN where super-tetrahedral building units are at the tetrahedral nodes. In MIL-101(Sc) the scandium trimers are at the vertices of these supertetrahedra and terephthalate groups link along their edges (Fig. 2). The MIL-101 structure contains two kinds of supercage, the larger of which at 34 Å, are themselves tetrahedrally-connected via windows of 16 Å in free diameter whereas the smaller ones (29 Å) are connected to other supercages through windows of 12 Å. This should lead to very high permanent porosities (a BET surface areas of 2800 m2 g−1 has been measured for MIL-101(Cr) following activation in vacuum34 and similar values reported for MIL-101(Sc) activated at low temperatures)28 but the highest value we have obtained via N2 adsorption for MIL-101(Sc) after evacuation under heating is ca. 640 m2 g−1, even though the crystallinity is retained (ESI†). Although not directly relevant to catalytic performance in solutions, this low surface area following evacuation suggests that the surface structure may recrystallize and result in pore blockage upon thermal activation. Furthermore, MIL-101(Sc) was observed to lose crystallinity upon extended exposure to moist air or heating above 100 °C, as previously observed.28 | ||
| Fig. 1 Views parallel to the c-axes (top), of cages (left, below) and approximately perpendicular to the c-axis of the isoreticular terephthalate MIL-88B (left) and 4,4′-biphenyldicarboxylate MIL-88D (right), with the dicarboxylate linkers joining metal trimer units, where ScO6 units are represented as octahedra. MIL-88B is shown in its closed state, adopted in apolar solvents, whereas the framework MIL-88D is represented in the state of opening measured for MIL-88D(Sc) in this work. In the material studied this work, it is likely that the structure is interpenetrated, but the second interpenetrating framework is not shown. | ||
 | ||
| Fig. 2 (Left) Connectivity of Sc3O(OH–, H2O)3(O2C–)6 trimers and terephthalate and trimesate linkers to give supertetrahedra that give MIL-100 and MIL-101 structures, respectively, with the connectivity of the tetrahedra shown in the stick diagram that represents the MTN topology. (Right, above) The 6- and 5-membered ring windows into smaller and larger cages in the MIL-100 structure. (Right, below) The tetrahedral connectivity of the larger supercages (represented in light and dark blue) via windows that include six trimer units. | ||
The large pore scandium trimesate MIL-100(Sc) was prepared solvothermally using DMF at 150 °C. MIL-100 contains two types of supercage, the larger of which (diameter 30 Å) are themselves tetrahedrally-connected via windows of 9 Å whereas the smaller ones (diameter 25 Å) are connected to other supercages only through windows of ca. 5 Å (Fig. 2). The measured porosity of MIL-100(Sc) to N2 (1.45 cm3 g−1) was close to its theoretical maximum and MIL-100(Sc) presents the largest internal surface area of the large pore scandium MOFs reported here (typical preparations have BET surface areas of 1000–1400 m2 g−1). For comparison, MIL-100(Cr) (BET surface area 1430 m2 g−1), MIL-100(Fe) (BET surface area 1363 m2 g−1) and MIL-101(Cr) (BET surface area 3250 m2 g−1) were also prepared.
Scandium 3,3′,5,5-azobenzene-tetracarboxylate, socMOF(Sc),23,35 was also prepared for comparison. It also contains scandium trimers, and is highly porous, but the internal pore space is only accessible via narrow 5 Å channels, so that catalysis would only be expected to occur on the external surfaces. It therefore acts as a control for surface activity of a scandium trimer-containing solid.
 | ||
| Fig. 3 The framework structure of as-prepared MIL-68(Sc), Sc(OH)(O2CC6H4CO2), (left) showing the chains of corner-sharing ScO4(OH)2 octahedra and (right) viewed down its channel axis, with the position of an extraframework DMF molecule shown, as determined in this work by constrained Rietveld refinement of PXRD data. | ||
Another scandium dicarboxylate, initially assigned as the scandium 4,4′-biphenyldicarboxylate MIL-88D(Sc), was prepared for the first time (MIL-88D(Fe) has been reported previously).24 MIL-88D is a larger pore, isoreticular version of MIL-88B, also containing scandium trimers. Supporting evidence for the structural assignment was obtained by comparison of the unit cell symmetry and size determined from indexing the X-ray pattern with that expected for the MIL-88D framework, and also by comparing simulated and observed powder X-ray patterns (see ESI†). Unlike MIL-88B(Sc), MIL-88D(Sc) shows little structural flexibility upon evacuation or upon immersion in solvents. The structural framework model consistent with the unit cell determined from powder X-ray diffraction (Fig. 1) has 3-dimensional connectivity via large pores, but N2 adsorption at 77 K indicates that the observed pore volume (0.47 cm3 g−1) is much less than that expected from the crystal structure. This fact, taken together with the framework rigidity of the structure, suggests that there could be a high degree of interpenetration of the framework structure by a second framework. Indeed, recent reports of interpenetrated (and therefore rigid and permanently porous) versions of MIL-88D(Fe)40 and MIL-88D(In)41 shows that this is able to occur, and our measured PXRD is consistent with that. Although the quality of the PXRD is insufficient to determine the degree of interpenetration, this would explain the low pore volume and framework inflexibility. As well as reducing the pore volume, interpenetration will reduce the window size and therefore the accessibility of the internal pore volume to organic molecules.
Activation of the scandium-containing MOFs prior to use in catalysis was carried out by stirring the material in methanol for 24 h and drying under vacuum for 5 h at 150 °C, but simple washing with methanol is also sufficient to activate MIL-100(Sc) or remove water or products between catalytic reactions that involve this solid.
For comparison, selected divalent MOFs that can be activated to give coordinatively unsaturated metal sites were prepared. These were Cu3BTC2 (HKUST-1) and CPO-27(Ni) and STA-12(Ni). These MOFs cannot be activated by simple washing and were therefore activated by heating (100–220 °C) under vacuum for 5 hours. In addition, MIL-100(Cr), MIL-100(Fe) and MIL-101(Cr) were compared as catalysts with their scandium-bearing counterparts. We also investigated the catalytic activity of Sc3+-exchanged zeolite Beta, to examine whether extra framework cations in zeolites could act as catalytically active Lewis acid centres. The large pore (8 Å) zeolite Beta42 was chosen because all extra-framework cations are accessible to adsorbed species. Aqueous cation exchange using scandium nitrate resulted in incorporation of Sc3+, with the Sc/Al ratio (0.6) suggesting over-exchange, i.e. the scandium is included with associated oxy,hydroxy-species. Sc-Beta was heated at 350 °C prior to use in catalysis to remove physisorbed water.
2.2 Lewis acid catalysis
| Entry | Catalyst | Reactanta (%) | Producta (%) | Hydratea (%) | Othera (%) |
|---|---|---|---|---|---|
| a Determined by 19F NMR. b Reactions repeated using 1-fluoronaphthalene as an internal standard gave the same results. c Reaction using as-prepared MIL-100(Sc) washed with methanol rather than activated by heating under vacuum. | |||||
| 1 | No catalystc | 85 | 12 | 2 | 1 |
| 2 | HKUST-1(Cu)b | 54 | 31 | 9 | 6 |
| 3 | STA-12(Ni) | 44 | 48 | 7 | 1 |
| 4 | CPO-27(Ni)b | 42 | 47 | 9 | 2 |
| 5 | MIL-100(Sc)b | 0 | 99 | 0 | 1 |
| 6 | MIL-100(Sc)c | 0 | 99 | 0 | 1 |
| 7 | MIL-100(Cr)b | 29 | 65 | 4 | 2 |
| 8 | MIL-100(Fe)b | 56 | 29 | 8 | 4 |
| 9 | MIL-88B(Sc) | 55 | 26 | 17 | 2 |
| 10 | MIL-88D(Sc)b | 46 | 45 | 8 | 1 |
| 11 | MIL-101(Sc) | 53 | 24 | 13 | 10 |
| 12 | MIL-101(Cr) | 36 | 58 | 5 | 1 |
| 13 | MIL-68(Sc) | 55 | 14 | 14 | 17 |
| 14 | socMOF(Sc) | 83 | 4 | 8 | 5 |
| 15 | Zeolite H-β | 22 | 32 | 21 | 25 |
| 16 | Zeolite Sc-β | 16 | 20 | 45 | 19 |
Cu3BTC2 (HKUST-1), which has been reported previously to possess appreciable Lewis acidity upon dehydration because of the generation of coordinatively unsaturated Cu2+ cations, might on the basis of the many reports of its catalytic activity be regarded as a benchmark MOF catalyst. In this ene reaction and under the chosen conditions it gives 31% conversion to the desired product. Slightly better performances (47%, 48%) are obtained with the Ni2+ forms of the large pore carboxylate CPO-27 and the phosphonate STA-12, each of which has metal sites that are rendered 5-coordinate (and therefore Lewis acidic) by removal of water from the as-prepared materials.
By comparison with these divalent cation MOFs, the performance of MIL-100(Sc) is very promising, because a single use of the catalyst results in almost stoichiometric conversion of reactants to product (99%), with only a very minor amount of hydrate by-product and no isomerisation products. Even without activation by heating under vacuum, full conversion is achieved (Table 2, entry 6). This very high activity and selectivity is not matched by the other scandium carboxylates or the scandium-exchanged zeolites. In addition, conversions over MIL-100(Cr) and MIL-101(Cr) while still high (65% and 58% product, respectively) are not close to that of the scandium form, and MIL-100(Fe) is significantly less active.
Whereas the MIL-88B and −88D solids do give conversion (as might be expected from the presence of the same scandium trimer species in their framework as in MIL-100) their activities are lower (Table 2, entries 9 and 10). Powder diffraction of MIL-88B(Sc) indicates that the structure remains in a ‘closed pore’ configuration when immersed in toluene, so that only surface scandium trimer species would be involved. The moderate activity of MIL-88D(Sc) (45% product) is consistent with reduced access of reactant molecules to active sites due to interpenetration of the structure. MIL-101(Sc) shows low activity, probably associated with its low stability and the difficulties experienced in its activation to give high porosity. For MIL-68(Sc), the low activity can be explained both by pore blocking (the porosity is much lower than expected from the crystal structure) and also by the lack of coordinatively unsaturated Sc3+sites, whereas for the scandium socMOF the low activity, despite the high porosity of this material and the presence of scandium trimers, results from the pores being too small to admit substrate, and also from low surface activity.
Finally, the large pore zeolite Beta containing with Sc3+ was also found to be an unselective catalyst, giving high yields of unwanted products. It is likely this results from the presence of Bronsted acidity associated with hydrolysis on the Sc3+ site, since a similar pattern of reactivity is observed over the protonic form. The local environment of Sc3+ within the carboxylate MOF framework is therefore essential for its selective catalytic performance.
The MIL-100(Sc) catalyst therefore possesses Lewis acid sites accessible to reactant molecules (Sc3+ within the trimers) that are able to activate the carbonyl-containing reactant to addition and selectively catalyse the intermolecular ene reaction. The ease of activation of MIL-100(Sc) to give a highly porous solid is of significant practical importance in giving a useful catalyst, and simply washing the hydrated solid with methanol is an easier activation process than for the other catalysts examined. Furthermore, the 30 Å supercages that are tetrahedrally-connected via 9 Å windows (Fig. 2) are large enough to allow free passage of reactant and product molecules. Three-dimensional connectivity of supercages will greatly increase the ease of molecular transport through the crystals. (There are other cages within the MIL-100 structure that are large enough to host the reactant molecules but these are only accessible via windows 5 Å in diameter, too small to take in reactant molecules.)
To confirm that the catalysts can be re-used, recycling reactions were performed for both STA-12(Ni) and MIL-100(Sc), each time slightly adjusting the amount of substrate and solution according to the amount of catalyst recovered to keep the ratio the same in each case. This was necessary since these are small scale reactions. For STA-12(Ni) no change in either the crystallinity (as measured by PXRD on samples recovered, see ESI†) or the catalytic performance was measured over three catalytic tests (Table 3). Furthermore, if either catalyst is removed during the reaction, the conversion of substrate is stopped. The catalytic reaction was proved to be fully heterogeneous in this way. Further recycling tests were carried out using MIL-100(Sc) that also did not show any significant loss of crystallinity. A reduced catalyst loading (1 mol%) was used to give conversions that could readily be followed by our experimental method. Comparison of reaction over as-prepared (methanol-washed) and once-used catalysts showed there was no significant reduction in performance (see ESI† and Table 2, entries 4 and 5).
| Entry | Catalyst | Cycle | Producta (%) |
|---|---|---|---|
| α-Methyl styrene (1.2 mmol) and ethyltrifluoropyruvate (1 mmol) added to a suspension of activated MOF (2.5 mol%) in toluene (5 ml) and stirred at 20 °C for 8 h (MIL-100(Sc)) or 16 h (STA-12(Ni)). The amount of reactant used was adjusted in each recycle to account for small losses in catalyst mass.a Determined by 19F NMR.b Conversion of reaction after 3 h using 1 mol% catalyst, see ESI for graphs of conversion vs. time. 1-Fluoronaphthalene used as internal standard. | |||
| 1 | STA-12(Ni) | 1 | 64 |
| 2 | STA-12(Ni) | 2 | 63 |
| 3 | STA-12(Ni) | 3 | 63 |
| 4 | MIL-100(Sc)b | 1 | 55 |
| 5 | MIL-100(Sc)b | 2 | 54 |
| 6 | MIL-100(Sc) | 1 | 99 |
| 7 | MIL-100(Sc) | 2 | 96 |
| 8 | MIL-100(Sc) | 3 | 95 |
The strong performance of the MIL-100(Sc) catalyst in this ene reaction of an activated carbonyl group encouraged us to investigate it in reactions of other carbonyl compounds (Table 4). Depolymerised ethylglyoxylate (distilled at ∼150 °C) reacted with α-methylstyrene, methylene cyclohexane and methylene cyclopentane in the presence of MIL-100(Sc) to give conversions of 97–99%, considerably higher than over HKUST-1 in each case. For example, using the highly activated methylenecyclohexane HKUST-1 gave 57% conversion, only 9% higher than the control reaction.
| Entry | Catalyst | Alkene | Product | Product (%) |
|---|---|---|---|---|
| Alkene (2.7 mmol) and enophile (2.7 mmol) added to a suspension of activated MOF (5 mol%) in toluene (5 ml) and stirred at 90 °C for 8 h. Conversion determined by 1H NMR.a 2.5 mol% of catalyst/substrate. | ||||
| 1 | No catalyst |

|

|
0 |
| 2 | MIL-100(Sc) | ” | ” | 84 |
| 3 | HKUST-1(Cu) | ” | ” | 6 |
| 4 | No catalyst |

|

|
48 |
| 5 | MIL-100(Sc) | ” | ” | 99 |
| 6 | MIL-100(Sc)a | ” | 97 | |
| 7 | HKUST-1(Cu) | ” | ” | 57 |
| 8 | No catalyst |

|

|
55 |
| 9 | MIL-100(Sc) | ” | ” | 99 |
| 10 | HKUST-1(Cu) | ” | ” | 75 |
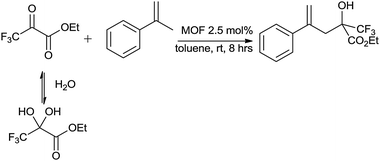
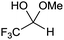
Tandem deprotection carbonyl ene reactions. In the course of these studies we noticed that the hydrate by-product sometimes converted into the desired product. The ability to promote dehydration and other related processes is significant because all electron-poor aldehydes are either supplied in a protected form (polymerised, hemiacetal, hydrate) or readily decompose to a hydrate or polymer. We therefore investigated three different tandem processes (Table 5): (i) the dehydration-ene reaction of ethyl trifluoropyruvate hydrate, (ii) the depolymerisation-carbonyl ene reaction of ethyl glyoxylate and (iii) the deprotection-carbonyl ene reaction of trifluoroacetaldehyde methyl hemiacetal.
| Entry | Catalyst | Reactant | Product | Product (%) |
|---|---|---|---|---|
| α-Methyl styrene (2.7 mmol) and enophile (2.7 mmol) were added to a solution of activated MOF (5 mol%) in toluene (5 ml) and the solution was stirred at 20 °C for 16 h (entries 1 and 2) or 90 °C for 8 h (entries 3–6). Conversion determined by 1H NMR. | ||||
| 1 | No catalyst |

|

|
0 |
| 2 | MIL-100(Sc) | ” | ” | 99 |
| 3 | No catalyst |

|

|
0 |
| 4 | MIL-100(Sc) | ” | ” | 99 |
| 5 | No catalyst |
|

|
0 |
| 6 | MIL-100(Sc) | ” | ” | 99 |
The MIL-100(Sc) catalyst is able to fully convert the hydrated form of the ethyltrifluoropyruvate with >99% selectivity so it can promote a tandem dehydration carbonyl ene reaction. Since ethyltrifluoropyruvate forms its hydrate on exposure to air, this is a useful feature. MIL-100(Cr) also performs well in the reaction, but does not equal MIL-100(Sc). The Ni-containing catalysts are not active.
The use of ethyl glyoxylate as a substrate in this reaction is complicated by its occurrence as a polymer that needs to be cracked at high temperature before use. To our knowledge, there are only two reports on particularly active homogeneous catalysts that use ethyl glyoxylate polymer directly.47 In the case of MIL-100(Sc) pure product was obtained directly from the ene reaction with using the polymeric form of ethyl glyoxylate, by simple filtration and removal of solvent after the reaction (Table 5, entry 4); in the case of this reaction without a catalyst there is no conversion. Pure product is also obtained over MIL-100(Sc) if methylenecyclohexane is used as a reactant.
The reaction was also carried out with trifluoroacetaldehyde methylhemiacetal; in order to carry out this carbonyl ene reaction the removal of methanol to form trifluoroacetaldehyde is required before reaction with methylstyrene can occur. MIL-100(Sc) catalysed the removal of methanol and subsequent ene reaction with >99% selectivity (Table 5, entry 6).

| Entry | Catalyst | Solvent | Producta (%) |
|---|---|---|---|
| a From 1H NMR. Remaining mass balance is reactant. b Conversion after 2 h using 5 mol% catalyst, see ESI for graphs of conversion vs. time. c Reactions repeated using 1-methylnaphthalene used as internal standard and gave same results. | |||
| 1 | No catalyst | CH2Cl2 | 0 |
| 2 | Sc(OTf)3 | CH2Cl2 | 99(99) |
| 3 | Sc(OTf)3 | CH3CN | 89 |
| 4 | Sc(OTf)3 | Methanol | 90 |
| 5 | MIL-100(Sc)c | CH2Cl2 | 99(99) |
| 6 | MIL-100(Sc) | Toluene | 93 |
| 7 | MIL-100(Sc) | Methanol | 15 |
| 8 | MIL-100(Sc) | CH3CN | 39 |
| 9 | MIL-100(Sc)b | CH2Cl2 | 72 |
| 10 | MIL-100(Sc)b | CH2Cl2 | 73 |
| 11 | MIL-100(Cr)c | CH2Cl2 | 79(75) |
| 12 | MIL-100(Fe) | CH2Cl2 | 40 |
| 13 | STA-12(Ni) | CH2Cl2 | 10(9) |
| 14 | CPO-27(Ni)c | CH2Cl2 | 68(68) |
| 15 | HKUST-1(Cu)c | CH2Cl2 | 11(10) |
| 16 | MIL-101(Sc) | CH2Cl2 | 51(50) |
| 17 | MIL-101(Cr) | CH2Cl2 | 56 |
| 18 | MIL-88B(Sc) | CH2Cl2 | 12 |
| 19 | MIL-88D(Sc)c | CH2Cl2 | 66(65) |
| 20 | MIL-88D(Sc) | Toluene | 61 |
| 21 | MIL-68 | CH2Cl2 | 7 |
| 22 | socMOF(Sc)c | CH2Cl2 | 0 |
| Entry | Catalyst | Indole | Ketone | Productb (%) |
|---|---|---|---|---|
| a Indole (0.84 mmol) and methyl vinyl ketone (0.84 mmol, 0.07 ml) added to suspension of MOF in CH2Cl2 (5 ml) and was stirred for 6 h at room temperature under N2. b Determined by 1H NMR. Mass balance is reactant. c Reaction repeated using 1-methylnaphthalene used as internal standard and gave same result. | ||||
| 1 | No catalyst |

|

|
0 |
| 2 | MIL-100(Sc)c | ” | 88(88) | |
| 3 | MIL-100(Cr) | ” | 36(35) | |
| 4 | MIL-100(Fe) | 25 | ||
| 5 | Sc(OTf)3 |

|
” | 90(90) |
| 6 | MIL-100(Sc) | ” | ” | 89(87) |
| 7 | MIL-88B(Sc) | ” | ” | 15 |
| 8 | MIL-100(Sc) |

|

|
91(90) |
Using dichloromethane as a solvent, MIL-100(Sc) is again the most active of the MOF catalysts (99% conversion). Conversions over MIL-100(Cr), MIL-100(Fe), MIL-101(Cr), CPO-27(Ni), MIL-88D(Sc) and MIL-101(Sc) are lower but the solids are still active catalysts. MIL-88B(Sc), which has similar metal cation sites to MIL-100(Sc) but remains in the closed form in CH2Cl2, socMOF(Sc), MIL-68(Sc) and STA-12(Ni) show very low activity. The low activity of MIL-88B(Sc) and socMOF(Sc) indicates (by comparison) that most of the Lewis acidic scandium trimer sites responsible for the catalysis in the active MIL-100(Sc), MIL-101(Sc) and (to a lesser extent) MIL-88D(Sc) are in the nanopores and not on the surface (as must be the case for MIL-88B(Sc) and socMOF(Sc)). The low activity of MIL-68(Sc) indicates coordinatively unsaturated Sc3+ sites are required for activity, while the poor activity of STA-12(Ni) compared to CPO-27(Ni) could be related to the limited accessibility of its five-fold coordinate Ni2+ cations compared with that of the sites in the more active CPO-27(Ni).
The conversion is found to be strongly solvent-dependent: for MIL-100(Sc) the reaction proceeds readily in the presence of dichloromethane and toluene (Table 6, entries 5 and 6), but not in acetonitrile (39%) or methanol (15%) (entries 7 and 8). This indicates that coordinating solvents compete effectively with the substrate for the Sc3+ sites of MIL-100 when present in large excess. Notably, washing the catalyst with methanol does not reduce catalytic performance, indicating the methanol can be displaced if not in an excess. After reaction, MIL-100(Sc) was filtered off and found to have turned brown. N2 adsorption at 77 K showed a reduction in uptake from 25 mmol g−1 to 20 mmol g−1 indicating that some of the product and/or reactants remained in the pore. Washing of the material with methanol reversed the colour change and the original porosity was restored. NMR analysis of the material recovered in the filtrate after methanol washing confirmed that 8% (0.016 g) of product had been retained in the catalyst. The MIL-100(Sc) catalyst remains crystalline after reaction, and can be recycled successfully (ESI†). MIL-100(Sc) was also found to behave heterogeneously as after catalyst removal from the reaction no further conversion was observed (ESI†).
Other variations of the reaction were attempted (Table 7). MIL-100(Sc) gave high conversions (ca. 90%) when N-methyl indole and indole were used as substrates in reaction with methyl vinyl ketone, and when 2-methylindole was reacted with phenyl vinyl ketone.
Reaction of pyrrole with methyl vinyl ketone over the Lewis acidic MIL-100(Sc), CPO-27(Ni) and HKUST-1 gave mixtures of 2-alkyl pyrrole and 2,5-dialkylpyrrole (Table 8), with the highest conversion and a ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in favour of the mono-substituted pyrrole occurring over MIL-100(Sc).
:1 in favour of the mono-substituted pyrrole occurring over MIL-100(Sc).

Finally for this reaction, the two bulky indoles 5-(4-(tert-butyl)phenyl)-1H-indole and 5-(4-phenoxyphenyl)-1H-indole were prepared and used as substrates in the reaction with methyl vinyl ketone. Successful reaction would result in the conversion of 5-(4-tert-butyl phenyl)-1H-indole (ca. 15 Å × 7 Å) to 4-(5-(4-(tert-butyl)phenyl)-1H-indol-3-yl)butan-2-one (15 Å × 12 Å). Both bulky indoles reacted readily in the presence of homogeneous scandium triflate, as did indole itself, but conversions over MIL-100(Sc) were much lower (and lower still over MIL-88B(Sc), socMOF(Sc) and CPO-27(Ni)) as seen in Tables 9 and 10. Additionally, no colour change was observed for the solids after the experiment, in contrast to the adsorption of products discussed previously, and N2 adsorption isotherms measured before reaction, after reaction and after subsequent methanol washing gave the same uptakes (see ESI†).


The products of these reactions are too large to be formed in the channels of CPO-27(Ni) (10 Å) or, if formed, to diffuse out of the internal pore space of MIL-100(Sc) (the larger cages of which have a diameter of 30 Å but are connected by windows only 9 Å across). The low conversions indicate that the surface activity is very low, even with scandium trimers. By corollary, the high reaction rates of the smaller indoles must result from reaction within the pore space of MIL-100(Sc). It is possible that the conversion of 33% observed over MIL-100(Sc) could have taken place within cages close to the surface.
Here we report the results of catalysis of the reaction of 4′-fluoroacetophenone and benzylamine over MIL-100(Sc) and, for comparison, HKUST-1 (Table 11). It was found that MIL-100(Sc) outperformed other MOF materials, and by increasing the temperature of the reaction conversion could be increased significantly to 85% conversion at 100 °C, considerably higher than if no catalyst is present. This conversion was also seen when MIL-100(Sc) was recovered, washed with methanol, dried and used in another reaction. Simply increasing the concentration and relative amounts of the amine in this equilibrium reaction enables complete conversion to imine to occur (Table 11, entry 11). The pure imine can be isolated by simple filtration and acid washing.

| Entry | Catalyst | Producta (%) |
|---|---|---|
| a Determined by 1H NMR. b Reaction carried out at room temperature. c Reaction carried out at 60 °C. d Recycled MIL-100(Sc) at 100 °C. e 1.5 equivalents of benzylamine. f Using 2 equiv. benzylamine and 1-methylnaphthalene as internal standard. | ||
| 1 | No catalystb | 3 |
| 2 | MIL-100(Sc)b | 49 |
| 3 | MIL-100(Sc)c | 60 |
| 4 | No catalyst | 8 |
| 5 | HKUST-1(Cu) | 20 |
| 6 | MIL-100(Sc) | 85 |
| 7 | MIL-100(Sc)d | 85 |
| 8 | MIL-100(Cr) | 20 |
| 9 | HKUST-1(Cu) | 11 |
| 10 | MIL-100(Sc)e | 92 |
| 11 | MIL-100(Sc)f | >99 |
In order to obtain a preliminary indication of the scope of the reaction, different carbonyl compounds and amines were reacted in the presence of MIL-100(Sc) (Table 12). For the reaction of 4-fluorobenzaldehyde with benzylamine at 20 °C, it was found that MIL-100(Sc) achieved 93% conversion after 1 h and went to complete conversion after 3 h. However, even the uncatalysed reaction achieved 63% conversion after 1 h. For the more challenging ketimine formation, the reaction was carried out with p-anisidine, butylamine, chlorobenzylamine and (S)-1-phenylethanamine. The reaction of p-anisidine at 90 °C gave complete conversion to product when an excess of the amine was used – the pure product could be isolated using acid washing. The aliphatic butyl amine reaction was performed at 70 °C, the boiling point of the amine and gave a conversion of 70%. When using chlorobenzylamine the reaction temperature could be lowered to 70 °C by changing the solvent to hexane and a high conversion was still attained. (S)-1-phenylethanamine required the use of Dean Stark apparatus at high temperatures for high conversion.

| Entry | R1 | R-NH2 | Producta (%) |
|---|---|---|---|
| a Determined by 1H NMR using 1-methylnaphthalene as internal standard. b Stirred at 20 °C, no catalyst. c Stirred at 20 °C. d 2 equivalents of amine. e 2 equivalents of amine used in hexane at 70 °C. f 2 equivalents of amine used with Dean Stark apparatus at 120 °C. | |||
| 1b | H |

|
63 |
| 2c | H | ” | 93 |
| 3 | CH3 |

|
82 |
| 4d | CH3 | ” | >99 |
| 5 | CH3 |

|
70 |
| 6e | CH3 |

|
91 |
| 7d | CH3 | ” | 85 |
| 8f | CH3 |

|
96 |
| 9d | CH3 | ” | 72 |
N bond-forming reactions. It easily outperforms HKUST-1, widely reported as an active Lewis acidic MOF catalyst, and an array of other MOFs with metal sites that can become coordinatively unsaturated upon loss of coordinating solvent, including other scandium and chromium carboxylate MOFs.
The active sites of MIL-100(Sc) are the Sc3+ cations that form part of Sc3O(OH−,(OH2)2)(O2C–)6 trimeric units. Two out of three Sc3+ cations in each trimer are coordinated by water molecules, which are expected to undergo ready exchange with other coordinating solvents and possibly carbonyl O atoms of the substrates. It is notable that in coordinating solvents such as methanol, the activity of MIL-100(Sc) for the conjugate addition of electron deficient olefins to indoles is strongly reduced – one explanation is that the solvent remains bound to the Sc3+ active sites.
None of the other scandium MOFs containing trimers that we have prepared are nearly as active as MIL-100(Sc). For socMOF(Sc) and MIL-88B(Sc) in non-polar solvents this indicates that the activity derives from the internal surface area rather than from trimers at the external surface: low conversions in experiments using bulky substrates confirm (by corollary) that for the reaction of substrates smaller than the window size the majority of the reaction comes from within the pores.
It would not have been possible to predict the greatly superior performance of MIL-100(Sc) compared to that of MIL-101(Sc) or MIL-88D(Sc) from their idealized crystal structures. Instead, the practical advantages of MIL-100(Sc) such as its ease of activation, porosity and stability against recrystallization are the critical factors. MIL-100(Sc) is readily prepared with high surface areas, has a 3-dimensionally connected array of mesoporous cages and is stable in moist air and under the reaction conditions used. By contrast, MIL-101(Sc) is less stable, recrystallizing to MIL-88B(Sc) under certain conditions, and, at least in these experiments, could not be prepared with surface areas close to those observed for its chromium analogue. Its lower catalytic activity is likely to derive from these unsuitable materials properties. The most likely reason for the lower than expected activity of MIL-88D(Sc) is the occurrence of interpenetration, leading to reduction of pore volume and window size, and it may be that modified preparations could give more porous and active catalysts based on this solid. From the current work, however, MIL-100(Sc) is the catalyst of choice.
3. Conclusion
A series of MOFs were investigated for activity in different Lewis acid-catalysed reactions. MIL-100(Sc) was found to be very active in each of these reactions, and outperformed the known and newly-synthesised MOFs tested in this work. In many cases these catalytic reactions achieved full conversion of reactants and are therefore synthetically useful. Not only were high conversions attained but MIL-100(Sc) could be recycled without significant loss in activity or crystallinity. The bulk of the catalysed reactions take place in the pore and not on the external surface of the MOF. These findings point towards MIL-100(Sc) being one of the most useful MOFs for simple Lewis acid catalysis. Further exploration of the activity of MIL-100(Sc) in catalysis in batch and flow mode as-prepared or modified with additional catalytic functionality is in progress.Acknowledgements
We gratefully acknowledge funding from the Engineering and Physical Sciences Research Council (EPSRC) and Johnson-Matthey PLC for a CASE award (LM), from CONACYT, Mexico (BG-S) and Nuffield foundation for vacation scholarship (PW). M. Borges Ordono (Johnson-Matthey) is thanked for providing a sample of MIL-100(Fe). Professor A. M. Z. Slawin (Univ. St. Andrews) is thanked for collection of single crystal diffraction data.References
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- (a) H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS; (b) J. L. C. Rowsell and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 1304–1315 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS.
- A. Corma, H. Garcia and F. X. L. Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- A. D. Burrows, C. G. Frost, M. F. Mahon and C. Richardson, Angew. Chem., Int. Ed., 2008, 47, 8482–8486 CrossRef CAS.
- J. M. Thomas, Design and Applications of Single-Site Heterogeneous Catalysts, Imperial College Press, London, 2012 Search PubMed.
- (a) S. H. Cho, B. Q. Ma, S. T. Nguyen, J. T. Hupp and T. E. Albrecht-Schmitt, Chem. Commun., 2006, 2563–2565 RSC; (b) A. M. Shultz, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2009, 131, 4204 CrossRef CAS; (c) A. M. Shultz, O. K. Farha, D. Adhikari, A. A. Sarjeant, J. T. Hupp and S. T. Nguyen, Inorg. Chem., 2011, 50, 3174–3176 CrossRef CAS; (d) F. Song, C. Wang, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132, 15390–15398 CrossRef CAS; (e) M.-H. Xie, X.-L. Yang, C. Zou and C.-D. Wu, Inorg. Chem., 2011, 50, 5318–5320 CrossRef CAS; (f) A. M. Shultz, A. A. Sarjeant, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2011, 133, 13252–13255 CrossRef CAS.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS.
- (a) C. Prestipino, L. Regli, J. G. Vitillo, F. Bonino, A. Damin, C. Lamberti, A. Zecchina, P. L. Solari, K. O. Kongshaug and S. Bordiga, Chem. Mater., 2006, 18, 1337–1346 CrossRef CAS; (b) S. Bordiga, L. Regli, F. Bonino, E. Groppo, C. Lamberti, B. Xiao, P. S. Wheatley, R. E. Morris and A. Zecchina, Phys. Chem. Chem. Phys., 2007, 9, 2676–2685 RSC.
- (a) L. Alaerts, E. Seguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. De Vos, Chem.–Eur. J., 2006, 12, 7353–7363 CrossRef CAS; (b) N. B. Pathan, A. M. Rahatgaonkar and M. S. Chorghade, Catal. Commun., 2011, 12, 1170–1174 CrossRef CAS.
- K. S. Jeong, Y. B. Go, S. M. Shin, S. J. Lee, J. Kim, O. M. Yaghi and N. Jeong, Chem. Sci., 2011, 2, 877–882 RSC.
- A. Henschel, K. Gedrich, R. Kraehnert and S. Kaskel, Chem. Commun., 2008, 4192–4194 RSC.
- A. Dhakshinamoorthy, M. Alvaro, H. Chevreau, P. Horcajada, T. Devic, C. Serre and H. Garcia, Catal. Sci. Techol., 2012, 2, 324–330 RSC.
- L. Kurfirtova, Y.-K. Seo, Y. K. Hwang, J.-S. Chang and J. Cejka, Catal. Today, 2012, 179, 85–90 CrossRef CAS.
- F. Vermoortele, R. Ameloot, A. Vimont. C. Serre and D. E. de Vos, Chem. Commun., 2011, 47, 1521–1523 RSC.
- M. J. Beier, W. Kleist, M. T. Wharmby, R. Kissner, B. Kimmerle, P. A. Wright, J.-D. Grunwaldt and A. Baiker, Chem.–Eur. J., 2012, 18, 887–898 CrossRef CAS.
- F. G. Cirujano, F. X. Llabres i Xamena and A. Corma, Dalton. Trans., 2012, 41, 4249–4254 RSC.
- (a) S. Kobayashi, Synlett, 1994, 689–701 CrossRef CAS; (b) S. Kobayashi, Eur. J. Org. Chem., 1999, 15–27 CrossRef CAS; (c) A. G. M. Barrett and D. C. Braddock, Chem. Commun., 1997, 351–352 RSC; (d) C. McDonagh and P. O'Leary, Tetrahedron Lett., 2009, 50, 979–982 CrossRef CAS; (e) T. Tsuchimoto, K. Tobita, T. Hiyama and S. Fukuzawa, J. Org. Chem., 1997, 62, 6997–7005 CrossRef CAS; (f) D. A. Evans and J. Wu, J. Am. Chem. Soc., 2005, 127, 8006–8007 CrossRef CAS.
- (a) J. Perles, M. Iglesias, C. Ruiz-Valero and N. Snejko, J. Mater. Chem., 2004, 14, 2683–2689 RSC; (b) F. Gandara, B. Gomez-Lor, M. Iglesias, N. Snejko, E. Gutierrez-Puebla and A. Monge, Chem. Commun., 2009, 2393–2395 RSC.
- J. P. S. Mowat, S. R. Miller, A. M. Z. Slawin, V. R. Seymour, S. E. Ashbrook and P. A. Wright, Micropor. Mesopor. Mater., 2011, 142, 322–333 CrossRef CAS.
- (a) S. Surblé, C. Serre, C. Mellot-Draznieks, F. Millange and G. Férey, Chem. Commun., 2006, 284–286 RSC; (b) C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828–1831 CrossRef CAS.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef.
- (a) P. D. C. Dietzel, R. Blom and H. Fjellvag, Dalton Trans., 2006, 2055–2057 RSC; (b) I. A. Ibarra, X. Lin, S. Yang, A. J. Blake, G. S. Walker, S. A. Barnett, D. R. Allan, N. R. Champness, P. Hubberstey and M. Schröder, Chem.–Eur. J., 2010, 16, 13671–13679 CrossRef CAS.
- Y.-T. Yan-Tao, K.-H. Cui, J. Li, J.-Q. Zhu, X. Wang and Y.-Q. Tian, Chinese J. Inorg. Chem., 2011, 27, 951–956 Search PubMed.
- C. Otero Arean, C. Palomino Cabello and G. Turnes Palomino, Chem. Phys. Lett., 2012, 521, 104–106 CrossRef.
- P. D. C. Dietzel, B. Panella, M. Hirscher, R. Blom and H. Fjellvag, Chem. Commun., 2006, 959–961 RSC.
- S. Chavan, F. Bonino, J. G. Vitillo, E. Groppo, C. Lamberti, P. D. C. Dietzel, A. Zecchina and S. Bordiga, Phys. Chem. Chem. Phys., 2009, 11, 9811–9822 RSC.
- J. A. Groves, S. R. Miller, S. J. Warrender, C. Mellot-Draznieks, P. Lightfoot and P. A. Wright, Chem. Commun., 2006, 3305–3307 RSC.
- S. R. Miller, G. M. Pearce, P. A. Wright, F. Bonino, S. Chavan, S. Bordiga, I. Margiolaki, N. Guillou, G. Férey, S. Bourrelly and P. L. Llewellyn, J. Am. Chem. Soc., 2008, 130, 15967–15981 CrossRef CAS.
- P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. De Weireld, J. S. Chang, D. Y. Hong, Y. K. Hwang, S. H. Jhung and G. Férey, Langmuir, 2008, 24, 7245–7250 CrossRef CAS.
- Y. L. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. C. Kravtsov, R. Luebke and M. Eddaoudi, Angew. Chem., Int. Ed., 2007, 46, 3278–3283 CrossRef CAS.
- K. Barthelet, J. Marrot, G. Férey and D. Riou, Chem. Commun., 2004, 520–521 RSC.
- C. Volkringer, M. Meddouri, T. Loiseau, N. Guillou, J. Marrot, G. Férey, M. Haouas, F. Taulelle, N. Audebrand and M. Latroche, Inorg. Chem., 2008, 47, 11892–11901 CrossRef CAS.
- A. Fateeva, P. Horcajada, T. Devic, C. Serre, J. Marrot, J.-M. Grenèche, M. Morcrette, J.-M. Tarascon, G. Maurin and G. Férey, Eur. J. Inorg. Chem., 2010, 3789–3794 CrossRef CAS.
- Q. Yang, S. Vaesen, M. Vishnuvarthan, F. Ragon, C. Serre, A. Vimont, M. Daturi, G. De Weireld and G. Maurin, J. Mater. Chem., 2012, 22, 10210–10220 RSC.
- M. Dan-Hardi, H. Chevreau, T. Devic, P. Horcejada, G. Maurin, G. Férey, D. Popov, C. Riekel, S. Wuttke, J.-C. Lavalley, A. Vimont, T. Boudewijns, D. de Vos and C. Serre, Chem. Mater., 2012, 24, 2486–2492 CrossRef CAS.
- H. Yang, F. Wang, Y. Kang, T.-H. Li and J. Zhang, Dalton Trans., 2012, 41, 2873–2876 RSC.
- J. M. Newsam, M. M. J. Treacy, W. T. Koetsier and C. B. de Gruyter, Proc. R. Soc. London, Ser. A, 1988, 420, 375–405 CrossRef CAS.
- M. L. Clarke and M. B. France, Tetrahedron, 2008, 64, 9003–9031 CrossRef CAS.
- N. A. Caplan, F. E. Hancock, P. C. B. Page and G. J. Hutchings, Angew. Chem., Int. Ed., 2004, 43, 1685–1688 CrossRef CAS.
- H. Fu, S. Xie, A. Fu and T. Ye, Comput. Theor. Chem., 2012, 982, 51–57 CrossRef CAS; S. Choomwattana, T. Maihom, P. Khongpracha, M. Probst and J. Limtrakul, J. Phys. Chem. C, 2008, 112, 10855–10861 Search PubMed.
- M. L. Clarke, C. E. S. Jones and M. B. France, Beilstein J. Org. Chem., 2007, 3 Search PubMed , art. 24.
- T. Urushima, Y. Yasui, H. Ishikawa and Y. Hayashi, Org. Lett., 2010, 12, 2966–2969 CrossRef CAS; D. A. Evans, S. W. Tregay, C. S. Burgey, N. A. Paras and T. Vojkovsky, J. Am. Chem. Soc., 2000, 122, 7936–7943 CrossRef.
- J. S. Yadav, S. Abraham, B. V. S. Reddy and G. Sabitha, Synthesis, 2001, 2165–2169 CrossRef CAS.
- J. Moskal and P. Milart, Synthesis, 1984, 128–132 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, including diffraction and adsorption data on solids and NMR data on organic compounds is given in the ESI. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cy20577g |
| This journal is © The Royal Society of Chemistry 2013 |