Comparison of phase transfer agents in the aqueous biphasic hydroformylation of higher alkenes
Received
8th September 2012
, Accepted 5th November 2012
First published on 7th November 2012
Abstract
The Rh-catalyzed aqueous biphasic hydroformylation with the bidentate ligand SulfoXantPhos was investigated for different phase transfer agents (PTA). As such, polymer latices and microemulsions formed by non-ionic surfactants were used. In general, a higher PTA concentration enhances the reaction progress. The feasibility of catalyst recycling by simple phase separation is shown in principle. The Rh losses are low in the surfactant system and promising for a technical approach.
Introduction
The Ruhrchemie–Rhône–Poulenc process from 1984 is the most successful industrial catalytic process performed in an aqueous biphasic reaction medium, which has undoubted economic and environmental benefits. In this process propene is converted in water to butyraldehyde (800·000 tonnes per annum),1 without addition of any phase mediator because of the sufficient water solubility of propene. Due to their extremely low solubility in water, the hydroformylation of higher olefins (≥C6) following the example of the Rhône–Poulenc process is not applicable. Yet, due to the clear economic benefit of a biphasic reaction mixture based on the ease of separation of the product and the catalyst phase and easy catalyst recycling, this is still a very challenging topic. To overcome problems like low space time yields and mass transport inhibition by applying (aqueous) two phase catalysis for higher olefins, different concepts exist in the literature. For instance, fluorous solvent systems, with specific synthesized fluorous phosphine ligands, have been investigated in biphasic hydroformylation of higher alkenes2 even in continuous mode.3 Herein, monodentate ligands perform much better than bidentates e.g. the XantPhos derivatives show less solubility in the fluorous solvent and therefore the organic phase contained high amounts of Rh as well.4
Besides the fluorous solvents, hydroformylation reactions have been carried out in all so-called green solvents, such as ionic liquids5 and supercritical CO2,6 or even in solvent combinations like IL or fluorous and scCO27 and with a solid support.8–11 In all these reaction media a single phase is formed at reaction temperature and phase separation is accomplished by a change of temperature in order to isolate the product and to recycle the catalyst. Microemulsions12,13 and polymers as a phase mediator14 and as a catalyst support15 have already been applied for the hydroformylation of short alkenes and higher alkenes, but with monodentate ligand-based water soluble catalyst complexes only.16,17 Monodentate ligands have a drawback of producing a low linear to branched (l/b) selectivity. There exist a lot of papers well describing the advantageous concept of biphasic catalysis in hydroformylation, but without showing results of recycling experiments and the question of Rh losses into the organic phase remains open sometimes.
In this paper we investigate the aqueous biphasic hydroformylation of 1-octene and 1-dodecene in polymer latices and in microemulsions, respectively. These two different phase transfer methods to increase the interface between water and the organic phase will be discussed for their advantageous as well as disadvantageous aspects in catalysis, phase separation and reuse of the catalyst. The rhodium and phosphor losses were also determined. The hydroformylation experiments were all carried out with Rh(acac)(CO)2 and SulfoXantPhos as a bidentate ligand (Scheme 1).
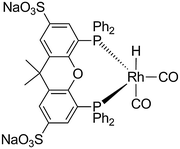 |
| | Scheme 1 The resting state of the water soluble Rh(SulfoXantPhos) catalyst complex. | |
Phase transfer agents
The polymer latex was synthesized by microemulsion polymerization in water with different monomers such as styrene, a cationic styrene salt, a polyethylene glycol styrene and divinylbenzene as a crosslinker. This procedure was already described in an earlier contribution (Fig. 1).14
Thereby, normal micelles are created, which remain in the aqueous phase even at reaction temperature. This means the system is always biphasic with an excess alkene phase (1-octene). This behavior was studied in a glass tube under reaction temperature and stirring up to 600 rpm. The continuous water phase contains polymer micelles with a hydrophobic core, which allows for enclosing the substrate alkene. The reaction takes place in the aqueous phase at the interface of the latex by electrostatic interaction with the catalyst. In this case, the substrate is “transferred” to the water soluble catalyst complex. For the separation of product and reuse of catalyst, the system is cooled to room temperature. Reaction and separation take place in an autoclave in batch mode. In principle the separation can be carried out at any temperature.
Nonionic surfactants that are commercially available were extensively investigated in the hydroformylation of 1-dodecene. The broad phase behavior of surfactant systems to form a microemulsion with excess phases (Fig. 2) was used to (i) ensure a fast reaction progress and (ii) for fast separation at room temperature. At first, the phase behavior was studied extensively in test tubes with a substrate and also a product mixture with different surfactant concentrations by variation of temperature but without gas pressure. After reaching the desired temperature, stirring was stopped to await phase separation. The hydroformylation experiments were performed at temperatures in which a water-in-oil microemulsion (inverse micelles) with excess water phase (2 phase) or a bicontinuous system (3 phase) can be assumed according to the behavior in the test tubes. These systems are characterized as media with low viscosity, low surface tension and strongly increased interfacial area of micelles, especially reverse micelles enclosing the water soluble catalyst in a continuous alkene phase (1-dodecene). This means that under reaction conditions the catalyst is “transferred” to the substrate. The microemulsion systems are thermodynamically stable and their formation is reversible at any gas pressure and stirring speed. After the reaction time, the mixture is cooled to room temperature to give a two-phase system, where normal micelles are stabilized in the continuous water phase with excess alkene phase, for separating the product. It is assumed that the active catalyst complex is quantitatively in the aqueous phase.
Results
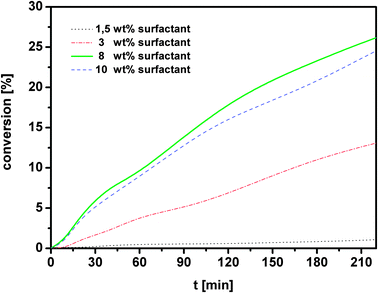
For both systems several screening experiments were performed before the determination of kinetic parameters and limitations in order to find out the optimal reaction conditions for the recycling experiments later. Hydroformylation experiments (a) were carried out mostly at a precursor to ligand ratio (M/L) of 1/2, whereas the hydroformylation experiments (b) were performed mostly at a ratio of 1/4 unless otherwise mentioned (Table 1). Comparing the water soluble ligands TPPTS and SulfoXantPhos in biphasic hydroformylation, it can be summarized that reactions with SulfoXantPhos run better at higher temperatures (≥100 °C), compared to the monodentate TPPTS ligand, where high conversion is obtained under milder conditions (80 °C),17i.e. comparable TOFs for both can be obtained when higher reaction temperatures are chosen with SulfoXantPhos as the ligand. But the major advantage of the bidentate ligand is the much higher selectivity. The organic/water volume ratio is 2 in both hydroformylation systems for the temperature dependent measurements (entries 1 to 4). In general, the higher the reaction temperature, the faster is the reaction progress. For the Hydrof_b two experiments were performed at 80 °C (entries 1 and 2) whereas in the first case a low conversion of 8% is obtained. Here, the reaction mixture is a two phase system at reaction temperature which is characterized by micelles formed in the aqueous phase with a hydrophobic core and an excess oil phase. In the second case another nonionic surfactant was chosen. With Marlipal 24/70 a three phase system is formed at reaction temperature. This three phase system consists of excess water and oil phases and a middle phase which comprises of dodecene, water and a high concentration of the catalyst that allows for a faster reaction progress. In fact, surfactants with a lower degree of ethoxylation n (n = 7 for Marlipal 24/70, see Fig. 2 for comparison) form three phase systems at lower temperatures so that the hydroformylation reactions can be carried out with high rates also at milder reaction temperatures. In the case of Hydrof_a milder reaction conditions (see Table 1, column 3) lead to a higher selectivity, although the reaction cannot be completed to full conversion e.g. at 80 °C. When the reaction is carried out at 110 °C, the selectivity decreases in time, due to the isomerization of 1-octene, more branched products are obtained. When we compare both hydroformylation reactions at the same M/L and temperature (entry 4), the calculated l/b ratios are similar but they seem to decrease at high conversions. It should be considered that lower l/b ratios could be the result of decoordination of one phosphorus ligand, which could be proven by in situ NMR. Surprisingly by comparing entry 5, a M/L ratio of 1/2 leads to a dramatic decrease of the linear aldehyde but parallel to a very fast reaction (especially for Hydrof_b), which shows that the ligand was not strongly coordinated to the metal. The selectivity of 1.4 only is comparable to l/b ratios achieved with monodentate ligands or without ligand. For low M/L ratios, preformation of the catalyst is essential. The hydroformylation with polymer latices results in higher selectivities because the catalyst was preformed first (see Experimental). For hydrof_a it was observed that under same conditions but higher excess of ligand (M/L = 1/4), a TOF value of 65 h−1 was obtained instead of a TOF = 126 h−1 at M/L = 1/2, which is about half the value obtained in the first experiment, showing that the reactions are not mass transport limited but kinetically controlled by the catalyst. On the other hand, the l/b ratio can be increased to 45 by a higher excess of ligand, which can be increased further, when taking into account that in industry an excess of 60 for the monodentate system Rh–TPPTS is used. Another reason for the much higher excess in the industrial process is to keep the metal quantitatively in the aqueous phase, which is an important economical aspect for the recycling that will be discussed later. In a further study, the amount of phase transfer agent was varied (see Fig. 3 and 4). A low amount of the latex polymer is sufficient to facilitate the reaction (Fig. 3), whereas the surfactant containing approach requires higher concentrations. Micelles have to be formed in order to obtain a noticeable reaction acceleration and to allow phase separation after the reaction. Interestingly, in Fig. 4 there is no strong influence of surfactant concentration noticeable for 8 and 10 wt% as soon as enough micelles are available for the transfer of the catalyst to the alkene phase. For the recycling experiments 1.1 wt% of the latex polymer (Fig. 5) was chosen although the reaction progress is not the fastest but the lower amount of the polymer will have advantages in separating the phases. In the microemulsion approach (Fig. 6), 10 wt% of nonionic surfactant and 11 wt% of aqueous phase was chosen, although the reaction is much faster when 36 wt% of aqueous phase is used (wt% referred to the reaction mixture). It is important to mention that for the recycling experiments in microemulsions, the temperature was adjusted again to 80 °C for each run, after the reaction slowed down at 110 °C. With this measure the advantageous three phase state of the reaction mixture was maintained. Therefore, the graphs are twisty. Furthermore, it should be mentioned that the dead time is just the time overnight (not the time for separating the phases) in which no reaction could be performed due to safety reasons. At a first glance both graphs show that after 80 hours four cycles were obtained for the use of the surfactant micelles whereas in the case of the polymer latex the second cycle could be finished at comparable conversions. The main reasons for this are the different concepts of phase transfer. The catalyst transferred to the alkene phase via reverse micelles experiences a much higher local olefin concentration than the catalyst at the latex particles which are only swollen with alkene. In general, the TOFs for both series of experiments decrease from run to run (Table 2). Thereby, it should be noted that all TOF values are calculated at a conversion of 20%. The TOFs in the recycling experiments of the latex polymer are lower than in the single runs, because a lower amount of latex polymer was used here. Comparing these values with literature data, where CTAB and RhCl(CO)(TPPTS)2-BISBIS,19 an ionic liquid with Rh-SulfoXantPhos20 or amphiphilic diphosphines21 were applied, our TOFs are comparable or even higher. Furthermore, the yield of n-nonanal for Hydrof_a was determined only at the end of each run, no sampling during the reaction was possible. Interestingly, the selectivities for Hydrof_b did not change after the first run, whereas in the case of Hydrof_a a significant decrease was obtained. Both observations were made in the temperature dependent experiments and they support the hypothesis that lower selectivities are the result of the parallel isomerization which provides more internal alkenes that react with branched aldehydes when the hydroformylation leads to higher or complete conversions. From this point of view it would be interesting to see if the selectivities in Hydrof_b are similar at complete conversions. Finally, the organic phases of the recycling experiments were analyzed for Rh and P leaching by ICP. The results are summarized in Table 3 with the concentration of rhodium and phosphorus in the organic phase and the percentage of this concentration referred to the initial content of the catalyst in the reaction mixture. It can be seen that the hydroformylations with the polymer latex as a phase transfer agent have much higher losses than the hydroformylation reactions carried out with the nonionic surfactant micelles formed from Marlophen NP 9. The value after the first run of 130 ppb Rh only that leached into the product phase is very low and well in the range of technical feasibility.1 Considering all measured aqueous phases the average can be found at approx. 70 ppb only, which is in the range of the reported ones (less than 1 ppm).19 The higher leaching in the first run could be caused by traces of XantPhos. But the losses of surfactant into the organic phase should also be taken into account. Especially in the first run a non-negligible amount could be detected. Proposals for a solution are under investigation. Furthermore, the conversion should be at least 90%, otherwise the substrate and the aldehyde must be separated by distillation which is detrimental for the long chain aldehydes due to thermal instabilities.
 |
| | Fig. 3 Variation of latex concentration: 23.5 ml 1-octene, 11 ml water, S/M/L = 3500/1/2, 40 bar, 110°C, 600 rpm. | |
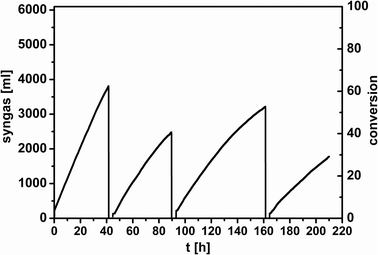 |
| | Fig. 5 Recycling experiment with 23.5 ml 1-octene, 1.1 wt.-% polymer, 11 ml water, M/L/S = 1/2/3500, 55 bar, 110°C. | |
 |
| | Fig. 6 Recycling experiment with 40 ml 1-dodecene, 4 g (10 wt.-%) Marlophen NP 9, 4 ml water, M/L/S = 1/5/3600, 40 bar, 110°C. | |
Table 1 Temperature dependencies and impact of ligand amount
|
T
react
|
Hydrof_a (after 65 h) |
Hydrof_b (after 3 h) |
| M/L |
Conv./Sel. (l/b) |
M/L |
Conv./Sel. (l/b) |
|
4 ml of water only.
Marlipal 24/70 as the surfactant. Hydrof_a: 11 ml water, 1.8 wt% latex, 11 mg Rh(acac)(CO)2 + 67.8 mg SulfoXantPhos, 150 mmol 1-octene (16.8 g, 23.5 ml), S/M/L = 3500/1/2, 40 bar, 600 rpm. Hydrof_b: 20 ml water, 5 g Marlophen NP 9 (9 wt% surfactant), 12.9 mg Rh(acac)(CO)2 + 158 mg SulfoXantPhos, 180 mmol 1-dodecene (30.3 g, 40 ml) S/M/L = 3600/1/4, 40 bar, 1000 rpm. |
| 80 °C |
1/2 |
0.11/100 |
1/4 |
0.08/49 |
| 80 °C |
— |
— |
1/4 |
0.22/32b |
| 100 °C |
1/2 |
0.45/42 |
— |
— |
| 110 °C |
1/4 |
0.72/45 |
1/4 |
0.32/49 |
| 110 °C |
1/2 |
0.95/28 |
1/2 |
0.77/1.4a |
Table 2 Comparison of recycling experiments
|
|
1st run |
2nd run |
3rd run |
4th run |
| Hydrof_a: yield and selectivities estimated after the reaction was stopped. Hydrof_b: yield and selectivities estimated after 8 hours of each cycle. |
| Hydrof_a |
l/b |
50 |
24 |
12 |
12 |
| Yield [%] |
58 |
36 |
43 |
24 |
| TOF [h−1] |
65 |
37 |
38 |
25 |
| Hydrof_b |
l/b |
99 |
49 |
49 |
49 |
| Yield [%] |
42 |
34 |
34 |
29 |
| TOF [h−1] |
266 |
192 |
173 |
152 |
Table 3 Rhodium and phosphor losses in the organic phase
| Hydrof_a (latex) |
Hydrof_b (surfactant) |
| Rh ppm/(%) |
P ppm/(%) |
Rh ppm/(%) |
P ppm/(%) |
| 6.10/(3) |
7.05/(3.1) |
0.13/(0.10) |
1.69/(0.43) |
| 1.74/(1) |
4.60/(2) |
0.04/(0.03) |
1.07/(0.27) |
| 0.75/(0.4) |
5.92/(2.6) |
0.05/(0.04) |
0.87/(0.22) |
| 0.50/(0.3) |
6.38/(2.8) |
0.05/(0.04) |
0.73/(0.19) |
Conclusions
Two different methods of phase transfer were tested in the biphasic hydroformylation of higher alkenes. In method a (Fig. 7), the alkene is enclosed by the polymer micelles and is transferred to the aqueous catalyst phase. In method b (Fig. 8), the water soluble catalyst is enclosed by the micelles formed by nonionic surfactants and is transferred to the alkene. The chosen reaction conditions for both systems are comparable, as well as the calculated selectivities and TOF values for the single batches. The commercially available technical grade surfactants perform well enough for biphasic hydroformylation, which means a fast reaction rate and easy phase separation, since the phase behavior was investigated very well. The fast progress is observed because the water droplets, including the catalyst, are stabilized in the organic phase, so that no mass transfer limitation is assumed (Fig. 8). The hydroformylation with polymer latices is also not mass transfer limited, but much slower, because the alkene has to be enclosed by the latex particles (Fig. 7) and transferred into the aqueous catalyst phase which is assumed to result in a much lower alkene concentration than in the alkene phase with the catalyst inside the reverse micelles. In the recycling experiments we could show that a similar reaction progress was obtained. In the case of using surfactants as phase transfer agents also much lower Rh and P losses were estimated by ICP because of the oil-in-water microemulsion as the continuous phase that is obtained for the separation of the organic product phase. By the use of the polymer latex, higher values for Rh and P were detected in the organic phase. It can be concluded that the polystyrene latices are not stable especially against high temperatures and therefore release catalyst and coagulated material into the organic phase. The property of surfactants, especially their thermodynamic stability and reversible broad phase behavior, makes them an unprecedented material for biphasic catalysis. One has to say that in biphasic hydroformylation with bidentate ligands where higher temperatures are required, polymer latices made of polystyrenes are not suitable as phase transfer agents. However, there is potential for this approach with other types of polymers, showing higher temperature and shear resistance.
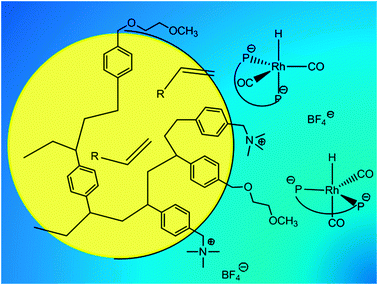 |
| | Fig. 7 Normal micelle enclosing the alkene. | |
 |
| | Fig. 8 Inverse micelle enclosing the water-soluble catalyst. | |
Experimental
The synthesis of polystyrene based latices by microemulsion polymerization has been described in an earlier contribution.14 The biphasic hydroformylation experiments of 1-octene were performed in a stainless steel autoclave, equipped with a gas-impeller stirrer and a dropping funnel. The precursor Rh(acac)(CO)2 (11 mg, 0.043 mmol) and SulfoXantPhos (67.8 mg, 0.086 mmol), giving a catalyst/ligand ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, was stirred in 5.5 ml water and 5.5 ml latex (containing 320 mg solid content) for 1 h. The autoclave was charged with the catalyst solution and preformed at reaction temperature and 40 bar syngas for 1 h at 600 rpm. After that, the substrate 1-octene (150 mmol, 16.8 g, 23.5 ml, 60 wt% organic phase) was added by a dropping funnel (ratio catalyst/substrate of 1/3500) and the conversion was measured by the gas uptake and via GC analysis.
:2, was stirred in 5.5 ml water and 5.5 ml latex (containing 320 mg solid content) for 1 h. The autoclave was charged with the catalyst solution and preformed at reaction temperature and 40 bar syngas for 1 h at 600 rpm. After that, the substrate 1-octene (150 mmol, 16.8 g, 23.5 ml, 60 wt% organic phase) was added by a dropping funnel (ratio catalyst/substrate of 1/3500) and the conversion was measured by the gas uptake and via GC analysis.
For the recycling experiments, the reaction was stopped at a certain time by cooling to room temperature and venting the system. Argon was purged through the reactor while the organic phase was taken out (app. 20 ml) and analyzed via GC and ICP measurements. To start a new cycle, fresh 1-octene was added into the dropping funnel. After a short preformation time under reaction conditions (10 minutes), the substrate was released into the autoclave by opening the valve.
All hydroformylation experiments were carried out in a 100 ml stainless steel autoclave from Premex connected to a 300 ml gas reservoir. The syngas (1:1 mixture) was purchased from Air liquide and dispersed by a gas dispersion stirrer. All basic chemicals were purchased from Roth or Sigma Aldrich and used without further purification. In a general experiment the autoclave was filled with 1-dodecene (180 mmol, 30.3 g, 40 ml) and the nonionic surfactant Marlophen NP 9 (4 g, 10 wt%). Then the reaction mixture was deoxygenated by repeated evacuation and nitrogen purging. Then the catalyst solution (0.05 mmol, 12.9 mg Rh(acac)(CO)2 and 0.25 mmol, 198 mg SulfoXantPhos) in 4 ml water was added to the reaction mixture (catalyst/ligand/substrate = 1/5/3600) under a nitrogen atmosphere over a dosing valve. The autoclave was heated by an oil bath from Huber (CC3) and stirred at 1000 rpm. The initial pressure of syngas was adjusted to 40 bar. At this pressure the systems contain initially 270 mmol of CO and H2 each. During the reaction the pressure decreases according to the conversion of the syngas, at full conversion to 15 bar. Samples were taken at several time intervals and analyzed by gas chromatography. After the reaction was stopped the autoclave was cooled to room temperature, depressurized and flushed with nitrogen. For a next run, new 1-dodecene (40 ml) was filled into the 100 ml stainless steel reactor, after separation of the organic phase from the aqueous catalyst and surfactant containing phase. A new run was started after heating to 110 °C and addition of syngas up to 40 bar.
ICP-OES.
The organic phases were analyzed for rhodium and phosphor losses. Therefore, a certain amount of organic phase has been made accessible for the ICP/OES machine by addition of acids and the use of a microwave (from CEM). In the case of the hydroformylation reactions that were carried out in microemulsions, the whole organic phase was incinerated (for concentrating the Rh and P values) and then the residue was dissolved in acids and pre-treated by a microwave before the measurements.
Acknowledgements
This work has been funded by NANO-HOST (FP 7 programme) (H.N.). We thank Ton Staring for technical support. A.R. and T.H. thank the DFG (this work is part of the Sonderforschungsbereich/Transregio 63), Umicore for the rhodium catalyst and Sasol for the surfactants. The authors thank Astrid Müller-Klauke for the ICP measurements.
Notes and references
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219–225 CrossRef CAS.
-
X. Zhao, D. He, L. Mika and I. Horváth, in Fluorous Chemistry, ed. I. T. Horváth, Springer, Berlin/Heidelberg, 2012, vol. 308, pp. 275–289 Search PubMed.
- E. Perperi, Y. Huang, P. Angeli, G. Manos, C. R. Mathison, D. J. Cole-Hamilton, D. J. Adams and E. G. Hope, Dalton Trans., 2004, 2062 RSC.
- D. J. Adams, D. J. Cole-Hamilton, D. A. J. Harding, E. G. Hope, P. Pogorzelec and A. M. Stuart, Tetrahedron, 2004, 60, 4079–4085 CrossRef CAS.
- S. L. Desset, S. W. Reader and D. J. Cole-Hamilton, Green Chem., 2009, 11, 630 RSC.
- T. J. Koch, S. L. Desset and W. Leitner, Green Chem., 2010, 12, 1719 RSC.
- A. M. Scurto and W. Leitner, Chem. Commun., 2006, 3681 RSC.
- S. Shylesh, D. Hanna, S. Werner and A. T. Bell, ACS Catal., 2012, 2, 487–493 Search PubMed.
- U. Hintermair, Z. Gong, A. Serbanovic, M. J. Muldoon, C. C. Santini and D. J. Cole-Hamilton, Dalton Trans., 2010, 39, 8501 RSC.
- S. Desset, U. Hintermair, Z. Gong, C. Santini and D. Cole-Hamilton, Top. Catal., 2010, 53, 963–968 Search PubMed.
- H. N. T. Ha, D. T. Duc, T. V. Dao, M. T. Le, A. Riisager and R. Fehrmann, Catal. Commun., 2012, 25, 136–141 Search PubMed.
- M. Haumann, H. Yildiz, H. Koch and R. Schomäcker, Appl. Catal., A, 2002, 236, 173–178 Search PubMed.
- H. H. Y. Ünveren and R. Schomäcker, Catal. Lett., 2006, 110, 195–201 Search PubMed.
- K. Kunna, C. Müller, J. Loos and D. Vogt, Angew. Chem., Int. Ed., 2006, 45, 7289–7292 CrossRef CAS.
- D. E. Bergbreiter and S. D. Sung, Adv. Synth. Catal., 2006, 348, 1352–1366 CrossRef CAS.
- F. Van Vyve and A. Renken, Catal. Today, 1999, 48, 237–243 Search PubMed.
- M. Haumann, H. Koch, P. Hugo and R. Schomäcker, Appl. Catal., A, 2002, 225, 239–249 CrossRef CAS.
- R. Schomäcker, M. Schwarze, H. Nowothnick, A. Rost and T. Hamerla, Chem. Ing. Tech., 2011, 83, 1–14 Search PubMed.
- H. Chen, Y. Li, R. Li, P. Cheng and X. Li, J. Mol. Catal. A: Chem., 2003, 198, 1–7 CrossRef CAS.
- J. Dupont, S. M. Silva and R. F. de Souza, Catal. Lett., 2001, 77, 131–133 CrossRef CAS.
- M. S. Goedheijt, B. E. Hanson, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2000, 122, 1650–1657 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.