Vanadium(III) phenoxyimine complexes for ethylene or ε-caprolactone polymerization: mononuclear versus binuclear pre-catalysts†
Lucy
Clowes
a,
Mark
Walton
a,
Carl
Redshaw‡
*a,
Yimin
Chao
a,
Alex
Walton
b,
Pertti
Elo
c,
Victor
Sumerin
c and
David L.
Hughes
a
aSchool of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK
bSchool of Physics & Astronomy, University of Leeds, Leeds, LS2 9JT, UK
cBorealis Polymers Oy, P. O. Box 330, Porvoo, FI-06101, Finland
First published on 4th September 2012
Abstract
The mononuclear {[C6H4N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(ArO)]2VCl(THF)} (Ar = 2,4-t-Bu2C6H2 (1), Ar = C6H4 (2)), {O[C6H4NCH(ArO)]2}VCl(THF) (Ar = 2,4-t-Bu2C6H2 (3), Ar = C6H4 (4)) and the binuclear vanadium(III) complexes {[C6H4NCH(ArO)]VCl2(THF)2}2(μ-CH2CH2) (Ar = 2,4-t-Bu2C6H2 (5), Ar = C6H4 (6)), have been synthesized and fully characterized. The compounds [C6H5NCH(ArO)]VCl2(THF)2 (Ar = 2,4-t-Bu2C6H2 (7), Ar = C6H4 (8)), [2,4,6-Me3–C6H2NCH(ArO)]VCl2 (Ar = 2,4-t-Bu2C6H2 (9), Ar = C6H4 (10)) and [2,6-i-Pr2-C6H3NCH(ArO)]VCl2(THF)2 (Ar = 2,4-t-Bu2C6H2 (11), Ar = C6H4 (12)), {μ-CH2CH2[NCH(C6H4O)]2VCl(THF)} (14) and {C6H4[NCH(C6H4O)]2VCl(THF)} (15) were synthesized for comparative polymerization studies. The dizwitterionic compound [2,6-i-Pr2-C6H3N+(H)CH(C6H4O)]2VCl2O (13) was also isolated, and presumably formed via a fortuitous hydrolysis reaction. The complexes 2, 5 and 13 have been structurally characterized; the molecular structure of the parent ligand (L5) in 5 is also reported. All complexes have been screened for ethylene as well as ε-caprolactone polymerization, and results are compared against those for known related mono- and bi-nuclear counterparts to evaluate for possible cooperative effects. The compounds 10 and 12 have been supported on modified SiO2, analysed by XPS and subjected to homo-polymerization (ethylene) and co-polymerization (1-hexene and ethylene) studies.
CH(ArO)]2VCl(THF)} (Ar = 2,4-t-Bu2C6H2 (1), Ar = C6H4 (2)), {O[C6H4NCH(ArO)]2}VCl(THF) (Ar = 2,4-t-Bu2C6H2 (3), Ar = C6H4 (4)) and the binuclear vanadium(III) complexes {[C6H4NCH(ArO)]VCl2(THF)2}2(μ-CH2CH2) (Ar = 2,4-t-Bu2C6H2 (5), Ar = C6H4 (6)), have been synthesized and fully characterized. The compounds [C6H5NCH(ArO)]VCl2(THF)2 (Ar = 2,4-t-Bu2C6H2 (7), Ar = C6H4 (8)), [2,4,6-Me3–C6H2NCH(ArO)]VCl2 (Ar = 2,4-t-Bu2C6H2 (9), Ar = C6H4 (10)) and [2,6-i-Pr2-C6H3NCH(ArO)]VCl2(THF)2 (Ar = 2,4-t-Bu2C6H2 (11), Ar = C6H4 (12)), {μ-CH2CH2[NCH(C6H4O)]2VCl(THF)} (14) and {C6H4[NCH(C6H4O)]2VCl(THF)} (15) were synthesized for comparative polymerization studies. The dizwitterionic compound [2,6-i-Pr2-C6H3N+(H)CH(C6H4O)]2VCl2O (13) was also isolated, and presumably formed via a fortuitous hydrolysis reaction. The complexes 2, 5 and 13 have been structurally characterized; the molecular structure of the parent ligand (L5) in 5 is also reported. All complexes have been screened for ethylene as well as ε-caprolactone polymerization, and results are compared against those for known related mono- and bi-nuclear counterparts to evaluate for possible cooperative effects. The compounds 10 and 12 have been supported on modified SiO2, analysed by XPS and subjected to homo-polymerization (ethylene) and co-polymerization (1-hexene and ethylene) studies.
Introduction
Vanadium catalysts have previously been employed for the production of high molecular weight polyethylene,1 ethylene/propylene co-polymers or ethylene/propylene/diene elastomers.2 However, despite their early commercial importance, it is only in the last few years that highly active α-olefin polymerization catalysts of group V have appeared in the academic literature.3 Of particular note has been the emergence of salicylaldiminato or so-called phenoxyimine-type (FI) ligands as one of the ligand sets of choice for early transition metal catalytic systems. Reports by Fujita et al. described highly active (65.1 kg-PE (mmol V h)−1 at 1 bar ethylene pressure) mononuclear pre-catalysts bearing two phenoxyimines at vanadium (see I, Scheme 1).4 Subsequent studies by Nomura, and by Li et al., led to pre-catalysts of the type II and III, respectively, both of which also proved to be highly active for ethylene polymerization.5 Our laboratory has also recently reported phenoxyimine complexes, where the chelate is either derived from an NO3 tripodal imine ligand set (IV),6 or vanadium complexes possessing N2O2S2-based ligands (V).7 A general feature of this chemistry is the adoption of mononuclear pre-catalysts. However, in a number of other polymerization systems, beneficial cooperative effects are witnessed, leading to either improved catalytic performance or differing polymer properties.8 With this in mind, and given the current interest in vanadium, we have embarked upon a program to develop new multi-metallic vanadium-based pre-catalysts. Furthermore, encouraged by recent work on zinc,9 we herein report the use of the ligand family [C6H4NCH(ArO)]2(μ-CH2CH2) (Ar = 2,4-t-Bu2C6H2L5, C6H4L6), which has previously been shown to act in a tetradentate fashion with either zirconium or zinc centres.9,10 Recently, related group IV and V complexes of tetradentate and bidentate Schiff bases have been screened for their ability to polymerise various α-olefins.11 We also note that a number of bimetallic nickel systems have shown increased activities versus their mononuclear counterparts.12 Herein, polymerization screening results for the bimetallic vanadium pre-catalysts are compared under similar conditions with known and novel related mononuclear related vanadium(III) complexes (Scheme 2).
 | ||
| Scheme 1 Known vanadium pre-catalysts I–V. | ||
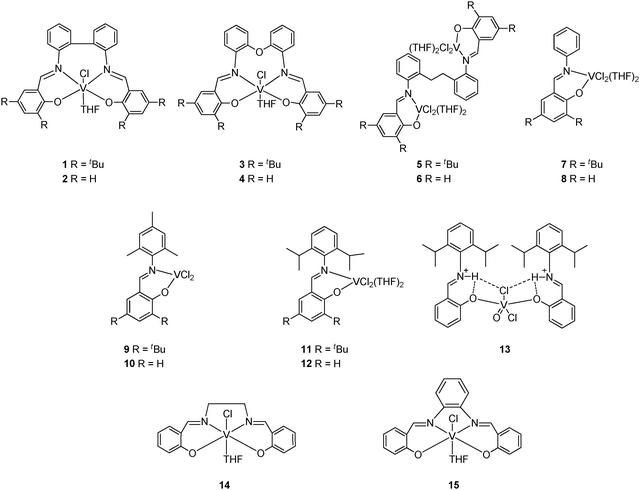 | ||
| Scheme 2 Vanadium pre-catalysts screened in this study. | ||
Results and discussion
The ligands L1–L15 required for each of the corresponding vanadium(III) complexes 1–15 (Scheme 2) were obtained in good yields via standard condensation of the requisite aniline with one or two equivalents of either salicylaldehyde or 3,5-di-tert-butyl-2-hydroxybenzaldehyde. Complexes 1–6, 9–10 were prepared following methods employed by Li and co-workers in their syntheses of the mononuclear phenoxyimine complexes 7, 8, 11 and 12.5b Complex 13 was synthesized using the same method as for compound 12 and was presumably formed via a fortuitous hydrolysis reaction. The known salicylaldehyde bridged phenoxyimine complexes 14 and 15 were synthesised as benchmark catalytic systems.13Mononuclear vanadium(III) phenoxyimines
Complexes 1 and 2 were prepared by treatment of L1 or L214 with excess of VCl3(THF)3 in tetrahydrofuran, in the presence of base (triethylamine), to afford, following work up, the dark red complexes {[C6H4NCH(2,4-t-Bu2C6H2O)]2VCl(THF)} (1) and {[C6H4NCH(C6H4O)]2VCl(THF)} (2) in yields of 47 and 51%, respectively. Crystals of 2 suitable for X-ray crystallography were grown by slow diffusion of the dark red tetrahydrofuran solution into hexane. Compound 2 crystallises with two independent V-complex molecules (together with THF solvent) in the cell. The structure of one of the independent molecules of 2 is depicted in Fig. 1, with selected bond lengths and angles given in the caption; crystallographic data (with those of the other structures reported in this paper) are collated in Table 3.
![View of one of the two very similar molecules in crystals of {[C6H4NCH(C6H4O)]2VCl(THF)} 2 indicating the atom numbering scheme. Hydrogen atoms and solvent molecules have been omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): V(1)–O(1) 1.926(4), V(1)–N(2) 2.107(5), V(1)–N(3) 2.092(5), V(1)–O(4) 1.917(5), V(1)–O(5) 2.127(4), V(1)–Cl(1) 2.353(2); O(1)–V(1)–N(2) 83.8(2), O(1)–V(1)–O(4) 96.41(19), O(1)–V(1)–Cl(1) 169.19(15), N(2)–V(1)–N(3) 89.7(2), N(2)–V(1)–O(4) 198.8(2), N(3)–V(1)–O(4) 90.2(2), N(3)–V(1)–O(5) 176.12(19), O(5)–V(1)–Cl(1) 88.77(13).](/image/article/2013/CY/c2cy20571h/c2cy20571h-f1.gif) | ||
| Fig. 1 View of one of the two very similar molecules in crystals of {[C6H4NCH(C6H4O)]2VCl(THF)} 2 indicating the atom numbering scheme. Hydrogen atoms and solvent molecules have been omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): V(1)–O(1) 1.926(4), V(1)–N(2) 2.107(5), V(1)–N(3) 2.092(5), V(1)–O(4) 1.917(5), V(1)–O(5) 2.127(4), V(1)–Cl(1) 2.353(2); O(1)–V(1)–N(2) 83.8(2), O(1)–V(1)–O(4) 96.41(19), O(1)–V(1)–Cl(1) 169.19(15), N(2)–V(1)–N(3) 89.7(2), N(2)–V(1)–O(4) 198.8(2), N(3)–V(1)–O(4) 90.2(2), N(3)–V(1)–O(5) 176.12(19), O(5)–V(1)–Cl(1) 88.77(13). | ||
The two molecules in 2 are very similar in conformation, each with a six-coordinate vanadium atom with an octahedral coordination pattern. Three of the N2O2 donor atoms of each L22− ligand are arranged in a meridional mode with the remaining O atom bonding normal to the meridional plane. The two remaining sites are filled by a chloride ion (trans to the fourth donor atom) and a THF ligand (trans to an imido N atom). In the folding of the L22− ligand around the metal centre, there is twisting of ca 71.7° about the N(2)–C(21) bond (and equivalent N(7)–C(71) bond in the second molecule) and ca 54.5° about the N(3)–C(31) and its equivalent bond; the rotation between the two phenyl groups, as about C(22)–C(32), is ca 55°. In both molecules, the THF ligand rings have ‘twisted’ conformations.
Compound 12 was synthesized using the reported literature method.5b However, the use of ‘wet’ ligand (L12 that had been dried insufficiently; ligand used for formation of compound 12 had been dried in vacuo in excess of 16 hours) resulted in the formation of a dizwitterionic complex 13 in moderate yield (ca 37%). Single crystals of 13 were grown from a saturated acetonitrile solution on prolonged standing at ambient temperature. The vanadium atom in complex 13 (Fig. 2) is five-coordinate with an approximately square-pyramidal pattern; the vanadyl oxygen, O(5), is at the apex, with the pairs of chloride ligands and phenolate oxygen atoms in opposite corners of the square base. The two phenolate ligands are arranged with approximate mirror symmetry about the central VOCl2 plane. Both NH groups are donors to bifurcated hydrogen bonds; each is linked to the phenolate O atom of its own ligand and to Cl(3). There are also two solvent (acetonitrile) molecules in the asymmetric unit of the cell.
![View of a molecule of the dizwitterionic complex 13, [2,6-i-Pr2-C6H3N+(H)CH(C6H4O)]2VCl2O, indicating the atom numbering scheme. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): V–O(1) 1.987(3), V–O(2) 1.978(3), V–Cl(3) 2.3594(14), V–Cl(4) 2.3265(12), V–O(5) 1.573(3), O(2)–V–O(1) 155.91(14), O(1)–V–Cl(3) 82.08(10), O(1)–V–Cl(4) 90.33(9), O(5)–V–O(1) 101.11(15), O(2)–V–Cl(3) 81.64(9), O(2)–V–Cl(4) 90.26(9), O(5)–V–O(2) 101.43(15), Cl(4)–V–Cl(3) 138.69(5), O(5)–V–Cl(3) 112.20(12), O(5)–V–Cl(4) 109.12(12), C(7)–N(8) 1.312(6), C(27)–N(28) 1.291(6), H(8)⋯O(1) 1.97, N(8)–H(8)⋯O(1) 130.0, H(8)⋯Cl(3) 2.59, N(8)–H(8)⋯Cl(3) 149.1, H(28)⋯O(2) 1.95, N(28)–H(28)⋯O(2) 132.6, H(28)⋯Cl(3) 2.68, N(28)–H(28)⋯Cl(3) 153.2.](/image/article/2013/CY/c2cy20571h/c2cy20571h-f2.gif) | ||
| Fig. 2 View of a molecule of the dizwitterionic complex 13, [2,6-i-Pr2-C6H3N+(H)CH(C6H4O)]2VCl2O, indicating the atom numbering scheme. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): V–O(1) 1.987(3), V–O(2) 1.978(3), V–Cl(3) 2.3594(14), V–Cl(4) 2.3265(12), V–O(5) 1.573(3), O(2)–V–O(1) 155.91(14), O(1)–V–Cl(3) 82.08(10), O(1)–V–Cl(4) 90.33(9), O(5)–V–O(1) 101.11(15), O(2)–V–Cl(3) 81.64(9), O(2)–V–Cl(4) 90.26(9), O(5)–V–O(2) 101.43(15), Cl(4)–V–Cl(3) 138.69(5), O(5)–V–Cl(3) 112.20(12), O(5)–V–Cl(4) 109.12(12), C(7)–N(8) 1.312(6), C(27)–N(28) 1.291(6), H(8)⋯O(1) 1.97, N(8)–H(8)⋯O(1) 130.0, H(8)⋯Cl(3) 2.59, N(8)–H(8)⋯Cl(3) 149.1, H(28)⋯O(2) 1.95, N(28)–H(28)⋯O(2) 132.6, H(28)⋯Cl(3) 2.68, N(28)–H(28)⋯Cl(3) 153.2. | ||
Binuclear vanadium(III) phenoxyimines
The ethylene-bridged ligands, L5 and L6 were prepared in good yields using the literature method.10 It proved possible to obtain X-ray quality crystals of the ligand L5 by crystallization from chloroform/methanol. The molecular structure is shown in Fig. 3, with selected bond lengths and angles in the caption. The molecule lies about a centre of symmetry at the mid-point of the C(27)–C(27′) bond, in an anti, staggered conformation, and thus has an ‘S-shape’ rather than curved in a ‘C-shape’ as it would when enfolding a metal centre. The phenolic hydrogen atom (in each half of the molecule) was refined freely and is directed towards the imino nitrogen forming a hydrogen bond, with H(1o)⋯N(20) 1.63 Å.![View of molecule [C6H4NCH(2,4-t-Bu2C6H2OH)]2(μ-CH2CH2), L5, indicating the atom numbering scheme. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): O(1)–C(1) 1.357(2), O(1)–H(1o) 1.00(3), C(1)–C(2) 1.412(3), C(20)–N(20) 1.283(2), N(20)–C(21) 1.423(2), C(1)–O(1)–H(1o) 105.8(16), O(1)–C(1)–C(6) 119.80(18), C(6)–C(1)–C(2) 120.78(18), N(20)–C(20)–C(2) 122.17(19).](/image/article/2013/CY/c2cy20571h/c2cy20571h-f3.gif) | ||
| Fig. 3 View of molecule [C6H4NCH(2,4-t-Bu2C6H2OH)]2(μ-CH2CH2), L5, indicating the atom numbering scheme. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): O(1)–C(1) 1.357(2), O(1)–H(1o) 1.00(3), C(1)–C(2) 1.412(3), C(20)–N(20) 1.283(2), N(20)–C(21) 1.423(2), C(1)–O(1)–H(1o) 105.8(16), O(1)–C(1)–C(6) 119.80(18), C(6)–C(1)–C(2) 120.78(18), N(20)–C(20)–C(2) 122.17(19). | ||
Complexes 5 and 6 were prepared in the same manner as complex 2. Treatment of L5 or L6 with excess of VCl3(THF)3 in tetrahydrofuran afforded, following work up, the dark red complexes {[C6H4NCH(2,4-t-Bu2C6H2O)]VCl2(THF)2}2(μ-CH2CH2) (5) and {[C6H4NCH(C6H4O)]VCl2(THF)2}2(μ-CH2CH2) (6) in yields of 43 and 44%, respectively.
Crystals of 5 suitable for X-ray crystallography were grown by slow diffusion of n-hexane into a THF solution of 5 at ambient temperature. The molecular structure is depicted in Fig. 4, with selected bond lengths and angles given in the caption. Regardless of whether the ligands L5 or L6 were treated with one or two equivalents of VCl3(THF)3, the di-nuclear species was always obtained and in yields of about 40–45% and 65%, respectively. Interestingly here, the ligand rather than acting in tetradentate fashion as observed previously,10 prefers to act as a bis-bidentate ligand, binding to a vanadium centre at either end of the ligand. The centrosymmetry is retained about the C(27)–C(27′) bond midpoint and there is rotation about the C(21)–N(2) bond to allow the coordination of the V centres. Six-fold coordination, in an octahedral pattern, is achieved by the ligation of two chloride ligands (mutually trans, Cl(3)–V–Cl(4) 176.20(4)°) and two cis-THF molecules (opposite the iminophenolate N and O atoms, O(5)–V–O(6) 85.68(9)°) at each V centre.
![View of molecule {[C6H4NCH(2,4-t-Bu2C6H2O)]VCl2(THF)2}2(μ-CH2CH2) 5 indicating the atom numbering scheme. Hydrogen atoms and the minor disorder component (in one of the THF ligands) have been omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): V–O(1) 1.870(2), V–N(2) 2.087(2), V–Cl(3) 2.3546(9), V–Cl(4) 2.3581(9), V–O(5) 2.107, V–O(6) 2.117(2), O(1)–V–N(2) 89.56(9), O(1)–V–O(5) 92.12(9), O(1)–V–O(6) 177.31(9), N(2)–V–O(5) 177.87(9), N(2)–V–O(6) 92.60(9), Cl(3)–V–Cl(4) 176.20(4).](/image/article/2013/CY/c2cy20571h/c2cy20571h-f4.gif) | ||
| Fig. 4 View of molecule {[C6H4NCH(2,4-t-Bu2C6H2O)]VCl2(THF)2}2(μ-CH2CH2) 5 indicating the atom numbering scheme. Hydrogen atoms and the minor disorder component (in one of the THF ligands) have been omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): V–O(1) 1.870(2), V–N(2) 2.087(2), V–Cl(3) 2.3546(9), V–Cl(4) 2.3581(9), V–O(5) 2.107, V–O(6) 2.117(2), O(1)–V–N(2) 89.56(9), O(1)–V–O(5) 92.12(9), O(1)–V–O(6) 177.31(9), N(2)–V–O(5) 177.87(9), N(2)–V–O(6) 92.60(9), Cl(3)–V–Cl(4) 176.20(4). | ||
Silica immobilisation
Compounds 10 and 12 were immobilised on pre-treated silica by refluxing in toluene to give supported structures S10 and S12; the SiO2 had been heated to 350 °C under dynamic vacuum for 48 h. X-ray photoelectron spectroscopy (XPS) analysis of the silica surface confirmed the percentage coverage of the metal species. Analysis of the resulting photoelectron spectrum of S12 (Fig. 5) showed a V-2p3/2 peak with a single component at 517.0 ± 0.02 eV. The resulting concentration of vanadium was shown to be 0.054% of the bulk sample. The C-1s peak was used to calibrate the spectrum; however the calculated carbon concentration in the sample is much higher than expected, now assigned to the presence of adventitious carbon. Confirmation that the ligand has not been leached from the system was ascertained from the identification of N-1s and Cl-2p peaks at binding energies of 401.0 eV (0.089%) and 199.7 eV (0.162%).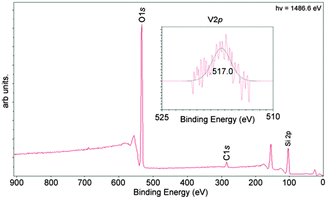 | ||
| Fig. 5 X-ray photoelectron survey spectrum of S12. The vanadium V-2p energy window 525–510 eV is shown inset. | ||
Ethylene polymerization screening
Complexes 1–10 have been evaluated as pre-catalysts for the polymerization of ethylene, employing DMAC (dimethylaluminium chloride) as co-catalyst, and ETA (ethyltrichloroacetate) as a re-activator.15 Polymerizations were carried out in toluene (100 mL) using 4000 molar equiv. of co-catalyst at 25 °C over 30 minutes.In the presence of DMAC and ETA, the new vanadium(III) bridged phenoxyimine complexes 1–6 were highly active for the polymerization of ethylene (Table 1, runs 1–6). However, the activities (<10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mmol−1 h−1 per bar) were disappointing in comparison to other V-based pre-catalyst–DMAC–ETA systems recently reported.11,16 The general trend observed in Table 1 is that the tert-butyl containing pre-catalysts showed higher activities than did their salicylaldehyde counterparts (e.g. runs 1 vs 2); the exception was 3cf.4, whose activities were of the same magnitude. The enhanced performance suggests that the steric hindrance of the tert-butyl group did not hinder the approach of the monomer, or this was possibly simply due to the improved solubility of the tert-butylated pre-catalyst.
000 g mmol−1 h−1 per bar) were disappointing in comparison to other V-based pre-catalyst–DMAC–ETA systems recently reported.11,16 The general trend observed in Table 1 is that the tert-butyl containing pre-catalysts showed higher activities than did their salicylaldehyde counterparts (e.g. runs 1 vs 2); the exception was 3cf.4, whose activities were of the same magnitude. The enhanced performance suggests that the steric hindrance of the tert-butyl group did not hinder the approach of the monomer, or this was possibly simply due to the improved solubility of the tert-butylated pre-catalyst.
| Pre-catalyst/runb | Polymerc | Activityd | M w e | M n f | PDIg | M p h |
|---|---|---|---|---|---|---|
| a Conditions: 1 bar ethylene; toluene (100 mL); ETA (0.1 mL); reaction quenched with dilute HCl, washed with methanol (20 mL) and dried for 12 h in a vacuum oven at 80 °C. b 0.5 μmol. c Grams of polymer. d Grams of polymer mmol−1 h−1 per bar. e Weight average molecular weight. f Number average molecular weight. g Polydispersity index. h °C determined by DSC. All runs carried out at 25 °C, 30 min and Al/V (molar ratio) 4000. | ||||||
| 1 | 2.1 | 8400 | 217000 |
49300 |
4.4 | 143.3 |
| 2 | 0.64 | 2560 | — | — | — | 141.9 |
| 3 | 0.34 | 1360 | 536000 |
136000 |
3.9 | 144.2 |
| 4 | 0.42 | 1680 | 404000 |
288000 |
1.4 | 145.6 |
| 5 | 2.38 | 9520 | 445000 |
181000 |
2.5 | 140.6 |
| 6 | 1.2 | 4800 | 170000 |
30500 |
5.6 | 143.1 |
| 7 | 0.98 | 3920 | 273000 |
47600 |
5.7 | 143.7 |
| 8 | 1.31 | 5240 | 168000 |
39200 |
4.3 | 142.6 |
| 9 | 0.55 | 2210 | — | — | — | — |
| 10 | 0.80 | 3190 | — | — | — | — |
| 11 | 0.48 | 1920 | — | — | — | — |
| 12 | 0.71 | 2850 | — | — | — | — |
| 13 | 0.41 | 1640 | — | — | — | — |
| 14 | 1.14 | 4560 | — | — | — | — |
| 15 | 0.25 | 1000 | — | — | — | — |
Only in the case of 1 (run 1) does the tetra-dentate nature of the ligand appear to be beneficial compared with its bi-dentate counterpart 7 (run 7), though the situation is reversed for the non-tert-butylated species (runs 2 vs 8). These tetra-dentate systems must undergo a rearrangement of the trans disposed chlorines to afford a system which is more catalytically favorable. Bialek et al. have observed enhanced activity in tert-butylated tetra-dentate vanadium(IV) systems compared to their bi-dentate counterparts.11a
In terms of polymer properties, the presence of an oxygen bridge in 3 and 4 led to increased molecular weight (Mw), and in the case of the non-tert-butylated complex 4, better control (Mw/Mn = 1.4, run 4).
When comparing the bi-metallic (5–6) versus the mono-metallic (1–4 and 7–15) vanadium complexes in Table 1, it was evident that in the case of the tetra-dentate ligand sets, activities for the bi-metallic systems were somewhat higher, whilst the molecular weight (Mw) of the polyethylene obtained from 6 was at the lower end of those observed herein.
On comparing complexes 9 and 10 with the bi-metallic complexes 5 and 6, it is evident that for the tert-butyl derivatives (5 and 9), there is evidence of a co-operative effect in 5; observed activities for 11 and 12 suggest that this is not simply a steric effect. In the case of RH (6 and 10), the beneficial cooperative effect in 6 is far less pronounced.
Comparison with the bi-dentate systems 7 and 8 reveals increased activity and molecular weight (Mw) only in the case of the tert-butylated derivative 5 (run 5); the values for 6 closely resemble those of 8. In the case of complexes 14 and 15, the presence of the phenylene backbone was found to be detrimental to activity. Bialek and Liboska have previously observed enhanced activity when using cyclohexylene bridged ligand sets.11c
ε-Caprolactone screening
Complexes 1–15 were also screened, in the presence of benzyl alcohol, for their ability to polymerize ε-caprolactone. Complex 6 was subjected to time, temperature and co-initiator screening to obtain the optimum conditions. The subsequent ε-caprolactone screening of complexes 1–15 were completed using these conditions (Table 2). All complexes, except 6, formed only oligomers with low polydispersity (≤1.4), which suggested that these ‘polymerizations’ occurred without side reactions and, interestingly, only low molecular weight oligomers were obtained under these conditions. Complex 6 deviated, in relation to polydispersity, from the other pre-catalysts screened herein and as such was subjected to lifetime studies which showed an almost linear increase in conversion over time.| Pre-catalyst/run | Conversionb | M n calculated | M n measuredc | PDI |
|---|---|---|---|---|
| a Conditions: monomer/metal = 400; 72 h; 80 °C; 20 mL of toluene; 2.5 mL of ε-caprolactone; 1 equivalent of benzyl alcohol (from a 0.97 M solution in toluene). b Calculated by 1H NMR. c M n measured = 0.58 × Mn GPC.17 | ||||
| 1 | 57 | 26024 |
10751 |
1.4 |
| 2 | 47 | 21458 |
8640 | 1.4 |
| 3 | 24 | 10960 |
7977 | 1.2 |
| 4 | 33 | 15070 |
9249 | 1.2 |
| 5 | 48 | 21900 |
10080 |
1.4 |
| 6 | 70 | 31960 |
11010 |
2.3 |
| 7 | 72 | 32870 |
4973 | 1.2 |
| 8 | 100 | 45660 |
4060 | 1.1 |
| 9 | 87 | 39720 |
13670 |
1.3 |
| 10 | 93 | 42460 |
14260 |
1.1 |
| 11 | 97 | 44290 |
21340 |
1.1 |
| 12 | 19 | 8670 | — | — |
| 13 | 93 | 42460 |
13290 |
1.2 |
| 14 | 94 | 42920 |
4790 | 1.1 |
| 15 | 3 | 1370 | 4560 | 1.4 |
The above results might be described as disappointing; however, they should be put into context by comparison with recent work carried out by Mahha et al.18 who noted that the heteropolyacids HPA-2 produced lower activities under an inert atmosphere; this was attributed to the reduction of the metal centres to V(IV) and/or Mo(V). However, when dioxygen was added, the reduced species were re-oxidized to the active vanadium(V) and Mo(VI). Unlike these heteropolyacid systems, our phenoxyimine complexes show similar conversion rates without the need to re-oxidise the deactivated species. Generally, the salicylaldiminato complexes 2, 4, 6, 8 and 10 slightly out-performed their tert-butylated counterparts; exceptionally, pre-catalyst 11 outperformed complex 12. We note that pre-catalysts 8, 11 and 14 exhibited the best conversion rates and provide excellent control of the molecular weight distribution (PDI = 1.1) of all those reported herein.
Homogenous parallel pressure reactor ethylene screening
Homogenous screening of pre-catalyst 5, in combination with methylaluminoxane (MAO), produced moderate catalysts for the polymerization of ethylene (Table 3). Increasing the amount of catalyst loading was found to be detrimental to the activity. The varying activity in co-catalyst was explored by Nomura et al.,19 who showed that the size of MAO forces the catalyst to form discrete ions, whereas DMAC can form a chloro-bridged species.| Run | Catalystb | Temp.c | Yieldd | Activitye |
|---|---|---|---|---|
| a Conditions: 6.67 bar ethylene, 1 h reaction time, Al/V (molar ratio) 4000. b mmol. c °C. d Grams of polymer. e g mmol−1 h−1 per bar. | ||||
| 1 | 5 (0.10) | 60 | 0.0436 | 65 |
| 2 | 5 (0.15) | 60 | 0.0382 | 38 |
| 3 | 5 (0.20) | 60 | 0.0408 | 31 |
| 4 | 5 (0.25) | 60 | 0.0455 | 27 |
| 5 | 5 (0.10) | 80 | 0.0363 | 54 |
| 6 | 5 (0.20) | 80 | 0.0475 | 36 |
| 7 | 5 (0.25) | 80 | 0.0461 | 28 |
Heterogenous parallel pressure reactor ethylene screening
S10 and S12 were subjected to homo-polymerization (ethylene) and co-polymerization (1-hexene and ethylene), over a one hour period. Triisobutyl aluminium (TIBA) and ethylaluminium dichloride (EADC) were used separately as co-catalysts, with the addition of ethyl trichloroacetate (ETA) where required. The metal content contained in the bulk sample was determined by XPS and this value was used to determine the catalytic activity of the supported catalysts (Tables S1–S3).The results of the polymerization experiments and conditions are shown in the ESI.† Homo-polymerization has shown the two pre-catalysts to be inactive for the polymerization of ethylene using TIBA (Table S1, ESI† runs 1–6); the resulting uptake of ethylene (psi) was due to saturation of heptane rather than polymerisation. Co-polymerization was more successful, where S12 outperformed S10 with peak activities of 1744 and 757 g mmol−1 h−1 bar−1 respectively (Table S3, ESI† runs 25 and 19), using EADC as co-catalyst and the addition of ETA. Use of ETA indeed gave higher activities on a number of runs, but also led to a large divergence of resulting activities (Tables S2 and S3, ESI† runs 7–28).
The problems with repeatability seem to stem from the rapid deactivation of the metal centres; the consumption profiles when using EDAC and ETA show that the active site was dead soon after the polymerization run has commenced (Fig. 6). Co-polymerization using TIBA as co-catalyst was detrimental, lowering the activity by 60–90% (Chart 1), although the active site remained active for a much longer period.
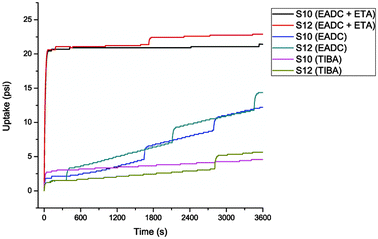 | ||
| Fig. 6 Consumption profiles of S10 and S12. | ||
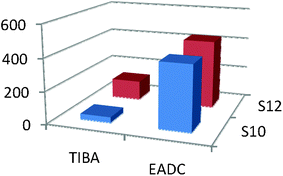 | ||
| Chart 1 TIBA vs EADC as co-catalyst for co-polymerization. | ||
Conclusions
In conclusion, a series of new vanadium(III) complexes bearing bridged phenoxyimines (1–6) have been synthesized and shown to be highly active (<10000 g mmol−1 h−1 per bar) for polyethylene production when used in combination with ETA and DMAC.
For ethylene polymerization, observed activity values for the tert-butylated systems 5 and 7 were suggestive of a cooperative effect operating between the two metal centres in 5; no such effect was observed for the de-tert-butylated systems (runs 6 and 8). Comparatively high activity was also observed when employing the tert-butylated tetra-dentate mono-metallic pre-catalyst 1. The presence of the tert-butyl groups in such systems was thought to play a crucial role by enhancing solubility. Mounting vanadium complexes 10 and 12 upon silica (forming S10 and S12) was detrimental to the polymerization activity.
Screening for ε-caprolactone polymerization revealed that the mononuclear species (complexes 8–14) generally exhibited higher conversions (≥87%) than did the binuclear species (5 and 6) (≤70%), with compounds 7 and 15 exceptions to this rule. Extending the bridge between ligand binding sites in tetra-dentate compounds led to decreased conversion rates (compounds 1–4, 14 and 15). A relationship between the size of the peripheral substituent and percentage conversion was observed, indicating a need for consideration of steric effects when designing pre-catalysts for ε-caprolactone polymerization.
Experimental
All manipulations involving vanadium were carried out under an atmosphere of nitrogen using standard Schlenk and cannula techniques or in a conventional nitrogen-filled glove-box. Solvents were refluxed over an appropriate drying agent, and distilled and degassed prior to use. Elemental analyses were performed by the microanalytical services of the London Metropolitan University. NMR spectra were recorded on a Varian VXR 400 S spectrometer at 400 MHz or a Gemini at 300 MHz (1H); chemical shifts are referenced to the residual protio impurity of the deuterated solvent; IR spectra (nujol mulls, KBr windows) were recorded on Perkin-Elmer 577 and 457 grating spectrophotometers. Solution state magnetic susceptibility was measured by Evans' method on a Varian VXR 400 S spectrometer using a 5% d12-cyclohexane–95% d2-dichloromethane solution. Polymer melting point measurements were carried out using a TA Instruments DSC 2920.Synthesis of ligands
Ligands L1–L15 were synthesised by the standard condensation of the corresponding amine with one or two equivalents as required of either salicylaldehyde or 3,5-di-tert-butyl-2-hydroxybenzaldehyde in ethanol.Synthesis of vanadium complexes
Complexes 1–6 and 9–10, were synthesised following analogous methods to that utilised by Li et al. to synthesise known complexes 7 and 8.5b Complexes 11, 12, 14 and 15 were synthesised by the literature procedures.11 Complex 13 was synthesized using the same method as that for compound 12, however use of ‘wet’ ligand (L12, which had been dried insufficiently) led to a fortuitous hydrolysis reaction.Pre-catalyst 1
To a solution of L1 (0.5 g, 0.8 mmol) in THF (20 ml), VCl3(THF)3 (0.67 g, 1.8 mmol) was added and this solution was stirred for 10 minutes before adding Et3N (0.25 ml, 1.8 mmol). The solution was stirred at room temperature for 4 hours and then concentrated to approximately 15 mL. The mixture was filtered and recrystallised by diffusion of n-hexane (10 mL) to yield complex 1 as a brown powder (0.42 g, 47%). IR/cm−1 1610 (s) (CN). Elemental analysis for VClC46H58N2O3 found (calculated): %C = 71.39 (71.44), H = 7.34 (7.56), N = 3.52 (3.62). MS (EI, m/z): 758 [M-Me]+. Magnetic moment μ = 2.70μB.
Pre-catalyst 2
Procedure as described for complex 1 using L2 (0.5 g, 1.2 mmol), VCl3(THF)3 (0.97 g, 2.6 mmol) and Et3N (0.36 ml, 2.6 mmol). Complex 2 was obtained as maroon crystals (0.56 g, 51%). IR/cm−1 1605(s) (CN). Elemental analysis for VClC26H18N2O2 (bake dried 24 h –THF) found (calculated): %C = 65.40 (65.49), H = 3.87 (3.80), N = 5.78 (5.87). MS (EI, m/z): 476 [M-THF]+. Magnetic moment μ = 2.23μB.
Pre-catalyst 3
Procedure as described for complex 1 using L3 (0.5 g, 0.8 mmol), VCl3(THF)3 (0.34 g, 0.9 mmol) and Et3N (0.13 ml, 0.9 mmol). Complex 3 was obtained as a dark brown powder (0.34 g, 54%). IR/cm−1 1609(s) (CN). Elemental analysis for VClC46H58N2O4 found (calculated): %C = 69.81 (69.99), H = 7.55 (7.41), N = 3.35 (3.55). MS (EI, m/z): 716 [M-THF]+. Magnetic moment μ =1.91μB.
Pre-catalyst 4
Procedure as described for complex 1 using L4 (0.5 g, 1.2 mmol), VCl3(THF)3 (0.97 g, 2.6 mmol) and Et3N (0.36 ml, 2.6 mmol). Complex 4 was obtained as a dark brown powder (0.43 g, 63%). IR/cm−1 1606(s) (CN). Elemental analysis for VClC30H26N2O4 found (calculated): %C = 63.37 (63.78), H = 4.85 (4.64), N = 4.83 (4.96). MS (EI, m/z): 492 [M-THF]+. Magnetic moment μ = 1.79μB.
Pre-catalyst 55b
Procedure as described for complex 1 using L5 (0.25 g, 0.6 mmol), VCl3(THF)3 (0.33 g, 0.9 mmol) and Et3N (0.12 ml, 0.9 mmol). Complex 5 was obtained as dark red crystals (0.57 g, 65%). Mp: 110 °C. IR/cm−1 1609(s) (CN). Elemental analysis for V2Cl4C60H86N2O6 found (calculated): %C = 61.19 (61.33), H = 7.23 (7.38), N = 2.38 (2.38). MS (EI, m/z): 693 [M-V-4Cl-4THF]+. Magnetic moment μ = 1.90μB.
Pre-catalyst 65b
Procedure as described for complex 1 using L6 (0.25 g, 0.6 mmol), VCl3(THF)3 (0.49 g, 1.3 mmol) and Et3N (0.18 ml, 1.3 mmol). Complex 6 was obtained as a red powder (0.34 g, 61%). IR/cm−1 1608(s) (CN). Elemental analysis for V2Cl4C44H54N2O6 found (calculated): %C = 55.65 (55.59), H = 5.60 (5.73), N = 3.07 (2.95). MS (EI, m/z): 468 [M-V-4Cl-4THF]+. Magnetic moment μ = 2.18μB.
Pre-catalyst 9
To a solution of L9 (0.5 g, 1.4 mmol) in THF (20 ml), VCl3(THF)3 (0.57 g, 1.54 mmol) was added and this solution was stirred for 10 minutes before adding Et3N (0.21 ml, 1.54 mmol). The solution was stirred at room temperature for 4 hours and then concentrated to approximately 15 mL. The mixture was filtered and recrystallised by diffusion of n-hexane (10 mL) to yield complex 9 as a brown powder (0.38 g, 57%). IR/cm−1 1612 (s) (CN). Elemental analysis for VCl2C24H32NO found (calculated): %C = 61.15 (61.02), H = 6.70 (6.83), N = 3.08 (2.97). MS (EI, m/z): 452.2 [M-Cl-tBu+H]+. Magnetic moment μ = 1.69μB.
Pre-catalyst 10
To a solution of L10 (0.5 g, 1.4 mmol) in THF (20 ml), VCl3(THF)3 (0.57 g, 1.54 mmol) was added and this solution was stirred for 10 minutes before adding Et3N (0.21 ml, 1.54 mmol). The solution was stirred at room temperature for 4 hours and then concentrated to approximately 15 mL. The mixture was filtered and recrystallised by diffusion of n-hexane (10 mL) to yield complex 10 as a red powder (0.29 g, 36%). IR/cm−1 1631 (s) (CN). Elemental analysis for VCl2C16H16NO (sample dried in vacuo for 12 h, loss of THF) found (calculated): %C = 61.15 (61.02), H = 6.70 (6.83), N = 3.08 (2.97). MS (EI, m/z): 359 [M]+. Magnetic moment μ = 2.05μB.
Pre-catalyst 13
To a solution of L12 (0.5 g, 1.7 mmol), which had been held under dynamic vacuum for 5 hours, in toluene (40 ml), VCl3(THF)3 (0.29 g, 1.0 mmol) was added and this solution was refluxed for 12 hours. The solvents were removed in vacuo and the residue was extracted into hot acetonitrile (35 mL) affording dark orange needles on cooling. (0.49 g, 37%). IR/cm−1 1605 (s) (CN), 927 (VO). Elemental analysis for VCl2C38H46N2O3 (sample dried in vacuo for 12 h, loss of MeCN) found (calculated): %C = 65.00 (65.14), H = 6.70 (6.62), N = 4.12 (4.00). MS (EI, m/z, sample dried in vacuo 12 h, loss of MeCN): 646.2 [M-MeH-HCl]+ 627.3 [M − 2HCl]+. Magnetic moment μ = 0.982μB.
Typical homogenous ethylene polymerization procedure
A flame dried (250 mL) pyrex flask was purged several times with ethylene gas at 1 bar pressure. This pressure was maintained throughout the polymerization run. Dry, degassed toluene (100 mL) and ethyltrichloroacetate (ETA, 0.1 mL, 0.72 mmol) were added and stirred for 10 minutes. The required temperature was achieved via a water bath, and the co-catalyst was added; the pre-catalyst was then injected as a toluene solution. The polymerization time was recorded from injection of pre-catalyst and left for the required time. The polymerization solution was quenched by addition of methanol and acidified water. The solid polyethylene was then filtered and dried.Typical ε-caprolactone ROP procedure
A Schlenk flask (250 mL) previously baked at 160 °C overnight was charged with the required quantity of pre-catalyst under glove box conditions. Dry, degassed toluene (20 mL) was added, the required temperature was achieved, and one equivalent of benzyl alcohol was then added. The polymerization was initiated by addition of the ε-caprolactone and was stirred for the allotted time. Conversion of monomer was determined by 1H NMR, and the polymerization was quenched by addition of methanol.Typical parallel pressure reactor polymerization run
A pre-weighed glass vial with stirring paddles was sealed and purged with ethylene. 5 μmol of co-catalyst from a 100 mM heptane solution was added along with co-monomer (if required). Heptane was then added to reach a volume of 4000 μL in the reaction vessel and heated to 80 °C. The ethylene pressure was set to 92 psi (6.34 bar) and the catalyst (along with ETA) was added as a heptane slurry. The run was left stirring for 60 minutes and quenched with CO2 (35% in N2). The glass vial was dried by vacuum centrifuge and weighed.X-ray photoelectron spectroscopy analysis
A small amount (∼mg) of powdered sample was pressed onto adhesive carbon tape. High resolution XPS core level measurements were performed with a VG Escalab 250 in LENNF, Leeds University, equipped with a conventional hemispherical sector analyser and controlled by a VGX900 data system. XPS experiments were carried out using a high intensity monochromated Al-Kα source (1486.6 eV) operated at 15 kV and 20 mA. V-2p, C-1s, Cl-2p and N-1s spectra were recorded using a pass energy of 20 eV. The energy scale of the spectrometer was calibrated to the Ag-3d5/2 peak at 368.3 eV. The binding energy scale was calibrated to the C-1s at 284.5 eV. High-resolution peak fitting was performed using CasaXPS software (version 2.3.15).Crystallographic analyses
Single-crystal X-ray diffraction intensities for compound 2 were measured at 120 K on a Bruker-Nonius Apex II Kappa-CCD diffractometer (at the EPSRC National Crystallography Service), for L5 and 5 at 140 K on an Oxford Diffraction Xcalibur-3 CCD diffractometer, and for 13 at 100 K on a Rigaku AFC12 Kappa-3-circle, Saturn724 + CCD diffractometer (at the EPSRC National Crystallography Service); all machines were equipped with Mo-Kα radiation and graphite monochromator. Intensity data were measured by thin-slice ω- and φ-scans.The Apex data for 2 were processed with the DENZO/SCALEPACK20 programs and SADABS,21 the Xcalibur data were processed (including correcting for absorption) using the CrysAlis-CCD and -RED programs,22 and the Rigaku data were processed with CrystalClear-SM.23 Structures were determined by the direct methods routines in the SHELXS program24 and refined by full-matrix least-squares methods, on F2's, in SHELXL.24
In compound 2, there is disorder in one of the THF ligands, and one of the THF solvent molecules lies disordered about a centre of symmetry. The non-hydrogen atoms (except the minor component C atom in the disordered ligand, and the half-atoms in the disordered THF molecule) were refined with anisotropic thermal parameters. The phenolic hydrogen atom in L5 was located in a difference map and was refined freely. In 5, there is disorder of a methylene group in one of the THF ligands; the minor occupancy carbon atom was refined isotropically. Except for the phenolic hydrogen atom in L5, all hydrogen atoms were included in idealised positions and their Uiso values were set to ride on the Ueq values of the parent carbon atoms. Scattering factors for neutral atoms in all samples were taken from reference.25 Computer programs used in this analysis have been noted above, and were run through WinGX26 on a Dell Precision 370 PC at the University of East Anglia.
Crystal data and refinement results for the four complexes are collated in Table 4. CCDC 837291–837293 and 895725.†
| Compound | 2 | L5 | 5 | 13 |
|---|---|---|---|---|
| Formula | 2(C30H26ClN2O3V), 1.5THF | C44H56N2O2 | C60H86Cl4N2O6V2 | C38H46Cl2N2O3V, 2 MeCN |
| Formula weight | 1205.99 | 644.9 | 1175.0 | 782.72 |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic | Orthorhombic |
| Space group | P21/a | P21/a | Pbca | P21nb |
| Unit cell dimensions | ||||
| a (Å) | 17.8598(14) | 9.6297(3) | 15.4851(3) | 11.0873(5) |
| b (Å) | 10.7750(8) | 12.1792(4) | 12.3449(2) | 17.9157(8) |
| c (Å) | 31.054(2) | 16.2276(6) | 33.0323(6) | 20.6832(14) |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 101.730(4) | 100.851(4) | 90 | 90 |
| γ (°) | 90 | 90 | 90 | 90 |
| V (Å3) | 5851.2(7) | 1869.18(11) | 6314.5(2) | 4108.4(4) |
| Z | 4 | 2 | 4 | 4 |
| Temperature (K) | 120(2) | 140(1) | 140(1) | 100 |
| D calcd (Mg m−3) | 1.369 | 1.146 | 1.236 | 1.262 |
| Absorption coefficient, μ (mm−1) | 0.470 | 0.069 | 0.512 | 0.414 |
| Crystal size (mm3) | 0.2 × 0.16 × 0.08 | 0.41 × 0.30 × 0.09 | 0.43 × 0.41 × 0.09 | 0.22 × 0.01 × 0.01 |
| 2θmax (°) | 20 | 25 | 25 | 25 |
| Reflections measured | 27495 |
19826 |
104961 |
9744 |
| Unique reflections, Rint | 5438, 0.111 | 3295, 0.106 | 5557, 0.072 | 5871, 0.048 |
| Reflections with F2 > 2σ(F2) | 3866 | 1981 | 4572 | 4516 |
| Transmission factors (max, min.) | 0.746, 0.604 | 1.118, 0.904 | 1.020, 0.982 | 1.000, 0.480 |
| Number of parameters | 737 | 221 | 339 | 471 |
| R 1, wR2 [F2 > 2σ(F2)] | 0.065, 0.116 | 0.050, 0.083 | 0.061, 0.105 | 0.051, 0.090 |
| R 1, wR2 (all data) | 0.101, 0.127 | 0.106, 0.096 | 0.087, 0.113 | 0.078, 0.098 |
| Largest difference peak and hole | ||||
| (e Å−3) | 0.39 and −0.30 | 0.16 and −0.15 | 0.39 and −0.31 | 0.31 and −0.29 |
Acknowledgements
The EPSRC is thanked for financial support. The EPSRC Mass Spectrometry Service (Swansea, UK) and Rapra Smithers Ltd (GPC on polyethylene samples) are thanked for data collection. EPSRC National Crystallography Service (Southampton) is thanked for diffraction data (complexes 2 and 13).Notes and references
- (a) M. H. Lehr, Macromolecules, 1968, 1, 178 CrossRef CAS; (b) W. L. Carrick, J. Am. Chem. Soc., 1958, 80, 6455 CrossRef CAS; (c) W. L. Carrick, R. W. Kluiber, E. F. Bonner, L. H. Wartman, F. M. Rugg and J. J. Smith, J. Am. Chem. Soc., 1960, 82, 3883 CrossRef CAS; (d) M. H. Lehr and C. J. Carman, Macromolecules, 1969, 2, 217 CrossRef CAS; (e) G. Natta, A. Zambelli and I. Pasquon, J. Am. Chem. Soc., 1962, 84, 1488 CrossRef CAS.
- (a) S. C. Davis, Zahalka, H. A. Richter, K. P. Hellens and W. von, in Polymer Material Encyclopedia, CRC Press Inc., Boca Raton, Florida, 1996 Search PubMed; (b) Y. Doi, N. Tokuhiro, M. Nunomura, H. Miyake, S. Suzuki and K. Soga, in Transition Metals and Organometallics as Catalysts for Olefin Polymerization, Springer-Verlag, Berlin, 1988 Search PubMed; (c) D. L. C. G. I. Keim, Macromolecules, 1968, 1, 358 CrossRef; (d) Y. Doi, S. Suzuki and K. Soga, Macromolecules, 1986, 19, 2896 CrossRef CAS; (e) E. Adisson, A. Deffieux, M. Fontanille and K. Bujadoux, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1033 CrossRef CAS.
- For recent vanadium reviews see: (a) K. Nomura, In New Developments in Catalysis Research, ed. L. P. Bevy, NOVA Science Publishers, New York, USA, 2005, p. 199; (b) C. Redshaw, Dalton Trans., 2010, 39, 5595 RSC; (c) K. Nomura and S. Zhang, Chem. Rev., 2011, 111, 2342 CrossRef CAS; (d) C. Redshaw, in Olefin Upgrading Catalysis by Nitrogen-based Metal Complexes I, Catalysis by Metal Complexes, 35, ed. G. Giambastiani and J. Cámpora, Springer Science + Business Media B.V, 2011 Search PubMed; (e) J.-Q. Wu and Y.-S. Li, Coord. Chem. Rev., 2011, 255, 2303 CrossRef CAS.
- (a) Y. Nakayama, H. Bando, Y. Sonobe, Y. Suzuki and T. Fujita, Chem. Lett., 2003, 766 CrossRef CAS; (b) Y. Nakayama, H. Bando, Y. Sonobe and T. Fujita, J. Mol. Catal. A: Chem., 2004, 213, 141 CrossRef CAS.
- (a) Y. Onishi, S. K. atao, M. Fujiki and K. Nomura, Organometallics, 2008, 27, 2590 CrossRef CAS; (b) J. Q. Wu, L. Pan, N. H. Hu and Y. S. Li, Organometallics, 2008, 27, 3840 CrossRef CAS.
- D. Homden, C. Redshaw, J. A. Wright, D. L. Hughes and M. R. J. Elsegood, Inorg. Chem., 2008, 47, 5799 CrossRef CAS.
- D. M. Homden, C. Redshaw and D. L. Hughes, Inorg. Chem., 2007, 46, 10827 CrossRef CAS.
- (a) S. Singh and H. W. Roesky, Dalton Trans., 2007, 1360 RSC; (b) M. P. Weberski Jr., C. Chen, M. Delferro and T. J. Marks, Chem.–Eur. J., 2012, 18, 10715–10732, DOI:10.1002/chem.201200713; (c) M. Delferro and T. J. Marks, Chem. Rev., 2011, 111(3), 2450–2485 CrossRef CAS.
- D. J. Doyle, V. C. Gibson and A. J. P. White, Dalton Trans., 2007, 358 RSC.
- G. J. Clarkson, V. C. Gibson, P. K. Y. Goh, M. L. Hammond, P. D. Knight, P. Scott, T. M. Smit, A. J. P. White and D. J. Williams, Dalton Trans., 2006, 5484 RSC.
- (a) M. Bialek, K. Czaja and E. Syzdlo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 565 CrossRef CAS; (b) M. Bialek and K. Czaja, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6940 CrossRef CAS; (c) M. Bialek and O. Liboska, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 471 CrossRef CAS; (d) J. Q. Wu, L. Pan, N.-H. Hu and Y.-S. Li, Organometallics, 2008, 27, 3840 CrossRef CAS.
- (a) M. Tanabiki, K. Tsuchiya, Y. Motoyama and H. Nagashima, Chem. Commun., 2005, 3409 RSC; (b) D. Zhang and G.-X. Jin, Inorg. Chem. Commun., 2006, 9, 1322 CrossRef CAS; (c) B. K. Bahuleyan, U. Lee, C.-S. Ha and I. Kim, Appl. Catal., A, 2008, 351, 36 CrossRef CAS.
- S. Gambarotta, M. Mazzanti, C. Floriani, A. Chiesi-Villa and C. Guastini, Inorg. Chem., 1986, 25, 2308 CrossRef CAS.
- J. F. Larrow, E. N. Jacobsen, Y. Gao, Y. P. Hong, X. Y. Nie and C. M. Zepp, J. Org. Chem., 1994, 59, 1939 CrossRef CAS.
- (a) D. L. Christman, J. Polym. Sci., Part A1., 1972, 10, 471 CrossRef CAS; (b) H. Hagen, J. Boersma and G. van Koten, Chem. Soc. Rev., 2002, 31, 357 RSC.
- (a) C. Redshaw, L. Warford, S. H. Dale and M. R. J. Elsegood, Chem. Commun., 2004, 1954 RSC; (b) C. Redshaw, M. A. Rowan, L. Warford, D. M. Homden, A. Arbaoui, M. R. J. Elsegood, S. H. Dale, T. Yamato, C. Pérez-Casas, S. Matsui and S. Matsuura, Chem.–Eur. J., 2007, 13, 1090 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114 CrossRef CAS.
- Y. Mahha, A. Atlamsani, J.-C. Blais, M. Tessier, J.-M. Bregeault and L. Salles, J. Mol. Catal. A: Chem., 2005, 234, 63 CrossRef CAS.
- K. Nomura and W. Wang, Macromolecules, 2005, 38, 5905–5913 CrossRef.
- Programs CrysAlisPro, Oxford Diffraction Ltd, Abingdon, UK, 2008 Search PubMed.
- Z. Otwinowski and W. Minor, Processing of X-ray diffraction data collected in oscillation mode, in Methods in Enzymology, Macromolecular Crystallography, Part A, ed. C. W. Carter Jr. and R. M. Sweet, Academic Press, 1997, vol. 276, p. 307–326 Search PubMed.
- G. M. Sheldrick, SADABS – Program for calculation of absorption corrections for area-detector systems, Version 2007/2, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- CrystalClear-SM Expert 2.0 r11: An integrated program for the collection and processing of area detector data, Rigaku Corporation, 2011.
- G. M. Sheldrick, SHELX-97 – Programs for crystal structure determination (SHELXS) and refinement (SHELXL), Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef.
- International Tables for X-ray Crystallography, Kluwer Academic Publishers, Dordrecht, 1992, vol. C, p. 500, 219 and 193 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: PPR polymerisation data from Homo-/Co-polymerization runs (Tables S1–S3). CCDC 837291–837293 and 895725. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cy20571h |
| ‡ Present address: E-mail: C.Redshaw@hull.ac.uk; Tel: +44 (0)1482-465219. |
| This journal is © The Royal Society of Chemistry 2013 |