Palladium catalytic systems with hybrid pyrazole ligands in C–C coupling reactions. Nanoparticles versus molecular complexes†
Daniel Perala, Fernando Gómez-Villarragaa, Xavier Salaa, Josefina Ponsa, J. Carles Bayóna, Josep Rosa, Miguel Guerrerobc, Laure Vendierbc, Pierre Lecanted, Jordi García-Antón*a and Karine Philippot*bc
aDepartament de Química, Unitat de Química Inorgànica, Universitat Autònoma de Barcelona, 08193, Bellaterra, Barcelona, Spain. E-mail: jordi.garciaanton@uab.es; Fax: +34 935812477
bCNRS, LCC (Laboratoire de Chimie de Coordination du CNRS), 205, route de Narbonne, F-31077 Toulouse, France. E-mail: Karine.Philippot@lcc-toulouse.fr; Fax: +33 5 61553003
cUniversité de Toulouse, UPS, INPT, LCC, F-31077 Toulouse, France
dCNRS, CEMES (Centre d'Elaboration de Matériaux et d'Etudes Structurales), 29 rue J. Marvig, F-31055 Toulouse, France
First published on 25th September 2012
Abstract
This paper reports the comparison of the chemoselectivity of two different Pd catalytic systems, namely molecular and colloidal systems, in C–C coupling reactions. For this purpose, new hybrid pyrazole derived ligands containing alkylether, alkylthioether or alkylamino moieties have been synthesized and used to form Pd(II) complexes and to stabilize Pd nanoparticles (Pd NPs). With the aim of studying the coordination mode of the ligands and further to understand their role in catalysis, both types of Pd species were characterized by appropriate techniques. In C–C coupling reactions promoted by different Pd colloidal systems, several reports evidenced that active species are molecular catalysts leached from Pd NPs. The most important feature of this work relies on the differences observed in the output of C–C coupling reactions, depending on the colloidal or molecular nature of the catalyst employed. Thus, molecular systems carry out typical Suzuki–Miyaura cross-coupling, together with the dehalogenation of the substrate in different proportions. In contrast, Pd NPs catalyze either Suzuki–Miyaura or C–C homocoupling reactions depending on the haloderivative used. Interestingly, Pd NPs catalyze the quantitative dehalogenation of 4-iodotoluene. Differences observed in the chemoselectivity of these two catalytic systems support that reactions carried out with Pd NPs stabilized with the hybrid pyrazole ligands employed here take place on the surface of the colloids.
Introduction
The synthesis of nanometre-sized colloidal particles (NPs), especially those of the noble metals, has been intensively investigated in the last few years. This comes not only from their fundamental and technological scientific interest,1–5 but also from their specific properties which are clearly different from those of the bulk metals due to their small sizes and high surface-to-volume ratios,6 which consequently make them interesting candidates for various applications. Among them, catalysis has emerged in the last few years as one of the most relevant applications of metal nanoparticles.7It is well-known that the properties of noble metal NPs are mainly determined by their size, shape and composition.8–10 Therefore, the application of size- and shape-control methodologies for the synthesis of NPs has attracted a great deal of attention.11–15 Among others, the decomposition of organometallic precursors under appropriate conditions represents an efficient way to obtain well-defined metallic nanoparticles.4,16 However, the surface modification of these nanoparticles is crucial to develop applications in different areas such as biotechnology or catalysis.17–21 In this respect, the research carried out during the last decade has evidenced the importance of understanding the surface chemistry of NPs.22–26 In particular, functionalized metal nanoparticles by addition of ligands have been used as catalysts, whose activities and selectivities were shown to be influenced by the nature of the stabilizing ligands.27–29
Palladium plays a key role in many industrial applications, especially those related to hydrogen storage and the reduction of pollutants.30,31 Concerning palladium nanoparticles, they have been mainly used as catalysts in olefin hydrogenation and carbon–carbon coupling reactions that are also catalyzed by organometallic Pd complexes and Pd heterogeneous catalysts.32–39 The interest in nanocatalysis has considerably increased given that nanocatalysis appears to be one of the most promising solutions towards efficient reactions under mild, and in some cases environmentally benign, conditions in the context of green chemistry.40–43
The Suzuki–Miyaura reaction belongs to an indispensable set of palladium-catalyzed C–C coupling reactions and it is nowadays one of the most important methods for the formation of symmetrical and non-symmetrical biaryls,44–50 which play an important role as organic intermediates for different applications, including the preparation of biologically active molecules.51–53 As far as noble metals are concerned, NPs stabilized by polymers, micelles, and ligands have been used as catalysts in Suzuki and other C–C coupling reactions.54–58 It is worth noting that the introduction of ligands as NP stabilizers is of special interest, because it focuses on the precise molecular definition of the catalytic materials.59,60 In spite of the numerous commercial nitrogen-based ligands used in molecular palladium-catalyzed processes, these ligands have not been extensively utilized to stabilize palladium nanoparticles. Moreover, there are very few reports on the surface chemistry of palladium nanoparticles.61–64 In addition, certain controversy about the nature of the catalytically active species in Pd NP-catalyzed C–C coupling reactions has recently arisen. Several authors have demonstrated that the catalytic activity observed is due to molecular complexes leached from the colloidal systems,65–69 while others have claimed that the catalytic reactions occur on the surface of metal NPs.70–73 Furthermore, a recent report strengthens the controversy between molecular or colloidal catalysts differentiation, as it demonstrates the presence of up to 40% of Pd NPs in commercial [Pd2(dba)3], one of the classical Pd precursors for catalytic reactions promoted by Pd complexes.74
Comparative studies on the ligand coordination chemistry between metal nanoparticles and coordination compounds are scarce although they could provide useful information for the catalytic applications of palladium nanoparticles.75,76 In this context, the design of efficient ligands to stabilize Pd NPs, and furthermore the study of their influence on surface properties, appear of high interest to develop novel applications in catalysis for Pd NPs. Recently, Gómez et al. published a report that compares the different behaviour of Pd molecular or colloidal systems in the Suzuki–Miyaura reaction both with the same ligands, dicarboximides or alkylic amines. It is interesting to note that only Suzuki coupling products were observed with both systems.77
Here we report a comparative study of Pd complexes and Pd nanoparticles as catalysts in C–C coupling reactions using haloarenes as substrates. In order to study and compare their effect on the catalytic reactivity, the same new family of hybrid pyrazolic ligands was used to prepare both molecular and colloidal catalytic systems.
Results and discussion
Synthesis of the N-substituted pyrazole ligands (L1–L4)
The following alkylether, alkylthioether or alkylamino N-substituted pyrazole ligands (Fig. 1) have been prepared: 3,5-dimethyl-1-[2-(octyloxy)ethyl]-1H-pyrazole (L1), 3,5-dimethyl-1-[2-(octylthio)ethyl]-1H-pyrazole (L2), 3,5-dimethyl-1-[2-(octylamino)ethyl]-1H-pyrazole (L3) and 3,5-dimethyl-1-[2-(dioctylamino)ethyl]-1H-pyrazole (L4). Compounds L1–L3 are here reported for the first time, while L4 was prepared according to the synthesis described by some of us in a previous report.78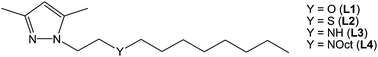 | ||
| Fig. 1 Pyrazole derived ligands. | ||
For the synthesis of L1, 1-bromooctane, 3,5-dimethyl-1-(2-hydroxyethyl)-1H-pyrazole79 and NaH were refluxed in THF for 12 h. Ligands L2 and L3 were prepared by the reaction of 3,5-dimethyl-1-(2-toluene-p-sulfonyloxoethyl)-1H-pyrazole,79 sodium hydroxide and 1-octanethiol in water for L2 or 1-octylamine in a mixture of water–THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for L3. All these ligands have been fully characterized by standard techniques.
:1) for L3. All these ligands have been fully characterized by standard techniques.
Synthesis of Pd(0) nanoparticles (Pd NPs)
Ligands L1–L4 were used as stabilizing agents to prepare nanosized Pd particles (Scheme 1), according to a method previously described.80 The synthesis of the Pd NPs was carried out by reacting, in a Fischer–Porter reactor for 20 h and under 3 bar of dihydrogen, the chosen ligand (L1–L4) with [Pd2(dba)3] (dba = dibenzylideneacetone) as a Pd source at the appropriate molar ratio (ligand/Pd from 0.5 to 2).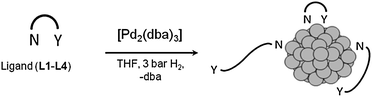 | ||
| Scheme 1 Synthesis of the Pd nanoparticles. | ||
For L1, slightly elongated particles were obtained, organized into roughly spherical and homogeneous in size superstructures. For all the L1/Pd molar ratios used (from 0.5 to 2), the size of the NPs is quite similar, varying from 4.7(1.3) to 4.0(1.1) nm as observed by TEM analysis (Table 1, Fig. 2 and Fig. S1, ESI†). These Pd NPs appear embedded in an organic shell, which may result from a ligand excess. The size of these superstructures varies depending on the ligand/Pd ratio (Table 1). A size of 18(5) nm is observed for a L1/Pd ratio equal to 0.5, which is smaller than those obtained for the L1/Pd ratio of 1 and 2, 32(12) and 41(10) nm, respectively. This difference in the size of the superstructure can be related to the proportion of extra ligand, which is not directly coordinated to the surface of the particles, and thus may form an organic shell around the particles and give rise to the superstructures, as previously observed for other pyrazole-alkyl ether ligands.81 As the proportion of extra ligand increases at higher ligand/metal ratios, this could explain the larger superstructures observed for L1/metal ratios of 1 and 2.
| L | [L]/[Pd] ratio | Nanoparticles, d (nm) | Superstructures, D (nm) |
|---|---|---|---|
| L1 | 0.5 | 4.7(1.3) | 18(5) |
| L1 | 1.0 | 4.0(1.1) | 32(12) |
| L1 | 2.0 | 4.2(0.9) | 41(10) |
| L2 | 0.5 | 3.4(0.9) | — |
| L2 | 1.0 | 2.7(0.7) | — |
| L2 | 2.0 | 2.7(0.5) | — |
| L3 | 0.5 | 3.8(1.1) | — |
| L3 | 1.0 | 3.5(0.9) | — |
| L3 | 2.0 | 3.3(0.9) | — |
| L4 | 0.5 | 3.3 and 5.7 | Layer of NPs |
| L4 | 1.0 | 3.5(1.0) | 66(11) |
| L4 | 2.0 | 3.2(1.0) | — |
![HR-TEM micrographs and the corresponding size-histograms of Pd nanoparticles synthesized as following: (A) [L1]/[Pd] = 1.0; (B) [L2]/[Pd] = 1.0; (C) [L3]/[Pd] = 1.0; (D) [L4]/[Pd] = 1.0.](/image/article/2013/CY/c2cy20517c/c2cy20517c-f2.gif) | ||
| Fig. 2 HR-TEM micrographs and the corresponding size-histograms of Pd nanoparticles synthesized as following: (A) [L1]/[Pd] = 1.0; (B) [L2]/[Pd] = 1.0; (C) [L3]/[Pd] = 1.0; (D) [L4]/[Pd] = 1.0. | ||
In contrast to L1, the L2 ligand allowed us to obtain isolated spherical Pd NPs, presumably due to the presence in this ligand of a sulfur atom with better coordinating properties toward Pd than the oxygen atom of L1. NPs of the same size (ca. 2.7 nm) were obtained for L2/M ratios of 1 and 2. However, slightly larger NPs, 3.4(0.9) nm, were obtained when less ligand (L2/M ratio 0.5) was used in the synthesis (Fig. 2 and Fig. S2, ESI†).
When a L3/Pd ratio of 0.5 was used, elongated worm-like NPs were obtained with a homogeneous mean diameter of 3.8(1.1) nm, but with a non-homogeneous length (around 10 nm). A similar behaviour has been previously observed by some of us in Pd NP systems stabilized by hexadecylamine.80 They also form small aggregates of 10–50 units (Fig. 2 and Fig. S3, ESI†). When the L3/Pd ratio was increased to 1 or 2, isolated spherical NPs were observed, although some of them coalesced into elongated particles with a mean size around 3.5 nm.
Finally, with L4, isolated spherical Pd NPs with an average size of 3.2(1.0) nm were obtained at a L4/Pd ratio of 2, although some of them coalesced to form aggregates with a worm shape, as happened with L3. When a L4/Pd ratio equal to 1 was used, Pd NPs with a size of 3.5(1.0) nm were obtained. They are aggregated into spherical superstructures with a size of ca. 66 nm, although around these superstructures some isolated particles are present. Such superstructures were also obtained at a L4/Pd ratio of 0.5, but in this case, a continuous layer was observed. The NPs embedded in this mosaic display a bimodal size distribution with average sizes of ca. 3.3 nm and 5.7 nm (Fig. 2 and Fig. S4, ESI†).
Elemental microanalyses (C, H, N and Pd) of the NPs (L/Pd ratio equal to 1; N1–N4) have been performed and are in agreement with the following stoichiometries: Pd2057(L1)158(THF)x, Pd561(L2)24(THF)x, Pd1415(L3)149(THF)x, and Pd1415(L4)67(THF)x. Taking into account that the ligands are situated only on the surface of the NPs, ratios (%) of Lper surface Pd atom are 24.6 (L1), 9.5 (L2), 30.3 (L3) and 13.6 (L4). It appears clearly that the quantity of ligand at the surface of the Pd NPs varies depending on the ligand. This can explain the differences observed by TEM as well as the differences in the required amount of ligand to obtain isolated NPs.
Wide-angle X-ray scattering (WAXS) measurements have been performed on nanoparticles N1–N4 (those prepared with L1–L4 in a ligand/Pd molar ratio equal to 1). Radial distribution functions (RDFs) (Fig. S5, ESI†) for N1–N4 are very similar and comparable to the function computed from a 3.5 nm face centred cubic (fcc) model of bulk Pd. The average size of crystalline domains is estimated to be in the 3.5–4 nm range, in agreement with the sizes obtained from TEM (4.0, 2.7, 3.5, and 3.5 nm, for N1–N4 respectively), indicating that the NPs are single crystals.
Scanning electron microscopy-field emission gun (SEM-FEG) experiments showed a good correlation in the mean size of the spherical superstructures (Fig. S6, ESI†) in comparison with TEM.
DLS measurements82,83 were carried out to determine the mean size of the nano-objects in solution. Only the superstructures were observed, with a good correlation with TEM values (Fig. S7, ESI†). For example, for the N4 system, DLS showed a mean size of objects of 79 nm, very close to the size measured by TEM, 66(11) nm for the superstructures.
NMR solution studies could not be performed due to the poor solubility of the systems in common deuterated solvent, except for N3. For this system, NOESY NMR studies show that the ligand is coordinated to the surface of the Pd NPs (Fig. S8, ESI†).
Synthesis of Pd(II) complexes (C1–C4)
Even if there might be differences between the coordination mode of the L1–L4 ligands in molecular complexes and nanoparticles (isolated atoms versus surfaces of atoms), Pd(II) complexes, C1–C4 hereafter, were prepared and characterized. We thus believed that the structure of these complexes, by showing the preferential coordination modes of L1–L4 ligands, would help to rationalize the influence of the ligands on the stabilization and the size control of the Pd NPs.Palladium complexes C1–C4 were prepared by mixing CH2Cl2 solutions of [PdCl2(CH3CN)2] with the corresponding ligands. They were fully characterized by standard techniques, including X-ray analyses of monocrystals obtained from Et2O–CH2Cl2 solutions of C1–C4. The C4 complex, [PdCl2(L4)], was previously reported as a complex where L4 behaves as an N,N-bidentate chelating ligand.78 Concerning complexes C1–C3, L1 forms a [PdCl2(L1)2] complex, while L2 and L3 lead to a [PdCl2(L)] type complex.
In complex C1, L1 behaves as an N-monodentate ligand, coordinating the palladium centre only through the azine nitrogen of the pyrazole ring, with the ether group remaining uncoordinated (Fig. 3 and Fig. S9, ESI†). On the other hand, in C2–C4, the pyrazolic ligands L2–L4 act as chelating N,Y-donors (Y = S (C2), Y = N (C3, C4)), forming a six-membered metallocycle (Fig. 4 and Fig. S10–S11, ESI†). In all cases (C1–C4), the geometry around the palladium centre is square planar with very small distortions. The distances and angles are in agreement with those reported in the literature for similar palladium compounds with a square planar geometry (Table S1, ESI†).84
![ORTEP drawing of [PdCl2(L1)2] (C1) showing all non-hydrogen atoms and the atom numbering scheme; 50% probability amplitude displacement ellipsoids are shown. Only one of the two non-identical molecules present in the X-ray crystal structure is shown. Both of them are shown in the ESI.](/image/article/2013/CY/c2cy20517c/c2cy20517c-f3.gif) | ||
| Fig. 3 ORTEP drawing of [PdCl2(L1)2] (C1) showing all non-hydrogen atoms and the atom numbering scheme; 50% probability amplitude displacement ellipsoids are shown. Only one of the two non-identical molecules present in the X-ray crystal structure is shown. Both of them are shown in the ESI.† | ||
![ORTEP drawing of (A) [PdCl2(L2)] (C2), (B) [PdCl2(L3)] (C3), and (C) [PdCl2(L4)] (C4) showing all non-hydrogen atoms and the atom numbering scheme; 50% probability amplitude displacement ellipsoids are shown. For complexes C2 and C3, only one of the two non-identical molecules present in the X ray crystal structure is shown. For these complexes, both of them are shown in the ESI.](/image/article/2013/CY/c2cy20517c/c2cy20517c-f4.gif) | ||
| Fig. 4 ORTEP drawing of (A) [PdCl2(L2)] (C2), (B) [PdCl2(L3)] (C3), and (C) [PdCl2(L4)] (C4) showing all non-hydrogen atoms and the atom numbering scheme; 50% probability amplitude displacement ellipsoids are shown. For complexes C2 and C3, only one of the two non-identical molecules present in the X ray crystal structure is shown. For these complexes, both of them are shown in the ESI.† | ||
NMR data of C1–C4 confirm that the structure of the complexes observed in the X-ray analysis is preserved in solution (NMR studies of C1–C4, ESI†).
Catalytic experiments
The catalytic performance of the Pd molecular complexes (C1–C4) and the Pd NPs (N1–N4) in C–C coupling reactions was investigated to evaluate the influence of the L1–L4 ligands on both catalytic systems.Catalytic studies with Pd molecular complexes
Palladium complexes of ligands L1–L4 were evaluated as catalysts for the Suzuki–Miyaura reaction (Scheme 2).85–89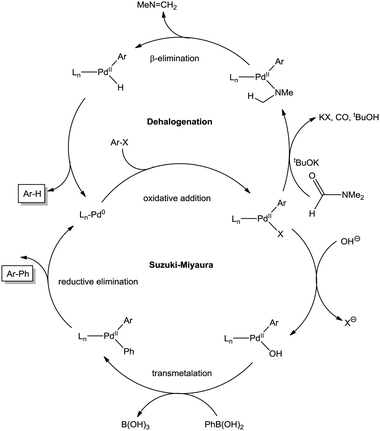 | ||
| Scheme 2 Plausible catalytic cycles for the Suzuki–Miyaura coupling and dehalogenation reactions.44,89,93,94 | ||
Previously described C1–C4 complexes on one hand and, on another hand, complexes formed in situ from [PdCl2(CH3CN)2] and [Pd2(dba)3] precursors in the presence of one or two molar equivalents of ligands L1–L4 were used as catalysts. Moreover, in the case of Pd(0) complexes, DOSY NMR90,91 experiments were performed to corroborate the coordination of the ligand to the metal centre (Fig. S12–S13, ESI†).
4-Halogenotoluene derivatives (4-chlorotoluene, 4-bromotoluene or 4-iodotoluene) and phenylboronic acid were selected as substrates in order to differentiate the cross-coupling product from the homocoupling of two identical aryl moieties. In order to optimize the catalytic reaction conditions, the effect of the solvent, the base, and the substrate on catalyst molar ratio was studied (Table S2, ESI†). This study led to the following conditions: tBuOK/PhB(OH)2/halogenotoluene/Pd = 5000/3125/2500/1 with a [Pd] = 0.1 mM. A mixture of DMF–water (4/1) was used since all reagents and catalysts are soluble in this media. Selected results achieved with molecular catalysts are shown in Table 2, while Table S3 in the ESI† collects the full set of performed experiments.
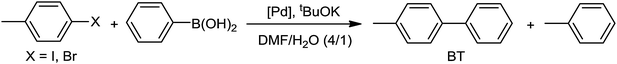
| Catalyst | L ([L]/[Pd])b | X = I | X = Br | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Conv.c (%) | Chem. BTd (%) | Yield BTe (%) | Entry | Conv.c (%) | Chem. BTd (%) | Yield BTe (%) | ||
| a Reaction conditions: 1 × 10−3 mmol of Pd, 2.5 mmol of 4-halogenotoluene, 3.1 mmol of phenylboronic acid, 5.0 mmol tBuOK and 0.5 mmol of naphthalene as internal standard in 8.0 mL of DMF and 2.0 mL of H2O. Temperature 100 °C.b Ligand and equivalents of ligand added.c Conversion after 6 h reaction.d Chemoselectivity in 4-methylbiphenyl (BT).e Yield of 4-methylbiphenyl (BT). | |||||||||
| C1 | 1 | 43 | 61 | 26 | 11 | 19 | 94 | 18 | |
| C2 | 2 | 85 | 57 | 48 | 12 | 26 | 84 | 22 | |
| C3 | 3 | 74 | 58 | 43 | 13 | 52 | 91 | 47 | |
| C4 | 4 | 48 | 70 | 34 | 14 | 25 | 93 | 23 | |
| [PdCl2(CH3CN)2] | — | 5 | 100 | 58 | 58 | 15 | 30 | 82 | 25 |
| [Pd2(dba)3] | — | 6 | 100 | 49 | 49 | 16 | 39 | 95 | 37 |
| [PdCl2(CH3CN)2] | L3 (1/1) | 7 | 64 | 88 | 56 | 17 | 26 | 80 | 21 |
| [PdCl2(CH3CN)2] | L3 (2/1) | 8 | 59 | 74 | 44 | 18 | 32 | 85 | 27 |
| [Pd2(dba)3] | L3 (1/1) | 9 | 64 | 66 | 42 | 19 | 23 | 72 | 17 |
| [Pd2(dba)3] | L3 (2/1) | 10 | 46 | 82 | 38 | 20 | 34 | 70 | 24 |
None of the catalysts used was able to activate 4-chlorotoluene and, as expected, 4-iodotoluene led to better conversions than 4-bromotoluene. In addition to the cross-coupled product (4-methylbiphenyl; BT), both halogenoderivatives yielded a variable amount of toluene arising from the dehalogenation of the substrates. This was the only side product observed in a significant amount. The dehalogenation of aryl halides as a side reaction of the Suzuki–Miyaura coupling has been reported before.92,93 In these cases, the hydrogen arises from a primary or secondary alcohol used as solvent: the alkoxide coordinates the Pd(II) ion, suffering subsequent β-elimination. This produces the palladium hydride species, which reductively eliminates with the already coordinated aryl moiety. In our case, DMF instead of an alcohol was employed as a solvent. Nevertheless, DMF is also capable of acting as a hydrogen source, as it was recently reported94 and illustrated in Scheme 2. Partial decomposition of DMF in basic media leads to carbon monoxide and a Pd(II) coordinated dimethylamide, which via β-elimination generates the palladium hydride intermediate. To corroborate the role of DMF in the catalytic cycle, we have performed an additional experiment using 1,4-dioxane and water (4:1) as solvents (Table S2, ESI†) and no toluene from the dehalogenation pathway was observed.
Interestingly, the dehalogenation was more favoured with 4-iodotoluene than with the bromo derivative. Therefore, even if the preformed C1–C4 complexes presented higher conversions with iodotoluene than with bromotoluene, the chemoselectivities in the cross-coupling products were clearly higher with the latter. Overall outputs of the cross-coupling products reach values close to 50% yield both with bromo- and iodo-derivatives in the case of C3 catalyst (Table 2, entries 3 and 13, respectively). A similar yield is achieved with C2 and iodotoluene (Table 2, entry 2).
When the preformed catalysts C1–C4 were used with 4-iodotoluene, complexes C2 and C3 yielded the best results in terms of turnover numbers (TON in the cross-coupling reaction equal to 1000–1200 in 6 h; Table 2 entries 2 and 3). These TON were slightly worse than those achieved with the [PdCl2(CH3CN)2] and [Pd2(dba)3] precursors without any pyrazolic ligand (TON ca. 1500 in 6 h; Table 2 entries 5 and 6, respectively) which were very fast catalysts, but also showed poor selectivity in the cross-coupled product (ca. 50%). Surprisingly, an equimolar amount of [PdCl2(CH3CN)2] and L3 (a mixture that should lead to C3) rendered a better chemoselectivity (88%) than the preformed complex and an overall output comparable to that of the ligand free catalysts (Table 2, entry 7).
When 4-bromotoluene was used, conversions were low, except in the case of complex C3 (52% conversion and TON ca. 1200 in 6 h; Table 2, entry 13). In situ catalysts prepared from ligand L3 and Pd(II) (Table 2, entry 17) or Pd(0) (Table 2, entry 19) yielded worse results with bromotoluene than the preformed complex C3.
In summary, best conversions both for iodo- and bromotoluene were achieved with molecular catalysts prepared with ligands L2 and L3, likely because these ligands present the best coordinative abilities. Consequently, palladium nanoparticles N2 and N3, stabilized with these two ligands were thoroughly investigated in the Suzuki–Miyaura cross-coupling reaction, using Pd/C as a reference nanocatalyst.
Catalytic studies with Pd nanocatalysts N1–N4
For this study nanoparticles N1–N4, prepared with a pyrazole ligands/Pd metal ratio equal to 1.0, were used. Nanoparticles stabilized by ligands L2 and L3 (N2 and N3, respectively) yielded the best results, and those will be discussed here (Table 3). A complete set of results for N1–N4 is collected in Table S4 (ESI†). As in the case of molecular Pd complexes, 4-chlorotoluene was not reactive with these nanoparticles. When 4-iodotoluene was used as a substrate with N2 and N3 catalysts (0.4 mol% of Pd), complete conversions were achieved in 6 h and the formation of the cross-coupling product, 4-methylbiphenyl (BT), was observed as the main component of the reaction, together with variable amounts of toluene (Table 3, entries 1 and 2, respectively). Pd/C (Table 3, entry 3) yielded similar chemoselectivity to N2 and N3 (ca. 65%), but with a significant lower conversion. With the N3 nanocatalyst, a higher load of the substrate was tested (0.04 mol%), leading to a remarkable improvement in the reaction output (100% conversion in 6 h and 90% chemoselectivity, Table 3, entry 4).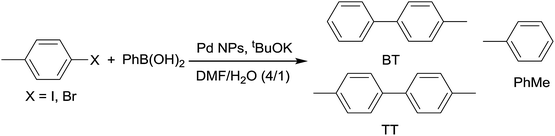
| Cat. | [PhB(OH)2]/[Pd] | X = I | X = Br | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Conv.b (%) | BTc (%) | TTc (%) | PhMec (%) | Yield BTe (%) | Entry | Conv.b (%) | BTc (%) | TTc (%) | PhMec (%) | Yield TTf (%) | ||
| a Reaction conditions: 1 × 10−2 mmol of Pd, 2.5 mmol of 4-halogenotoluene, 5.0 mmol of tBuOK and 0.5 mmol of naphthalene as internal standard in 8.0 mL of DMF and 2.0 mL of H2O. Temperature 100 °C.b Conversion after 6 h reaction.c Product distribution after 6 h reaction.d 4-Halogenotoluene/tBuOK/Pd = 2500/5000/1.e Yield of 4-methylbiphenyl (BT).f Yield of 4,4′-dimethylbiphenyl (TT). | |||||||||||||
| N2 | 315 | 1 | 100 | 69 | — | 31 | 69 | 8 | 12 | — | 14 | 86 | 2 |
| N3 | 315 | 2 | 100 | 61 | — | 39 | 61 | 9 | 37 | — | 53 | 47 | 20 |
| Pd/C | 315 | 3 | 31 | 65 | — | 35 | 20 | 10 | 84 | 17 | 46 | 37 | 39 |
| N3d | 3125 | 4 | 100 | 90 | — | 10 | 90 | 11 | — | — | — | — | — |
| N2 | — | 5 | 100 | — | — | 100 | 0 | 12 | 18 | — | 24 | 76 | 4 |
| N3 | — | 6 | 100 | — | — | 100 | 0 | 13 | 64 | — | 70 | 30 | 45 |
| Pd/C | — | 7 | 100 | — | 1 | 99 | 0 | 14 | 83 | — | 41 | 59 | 34 |
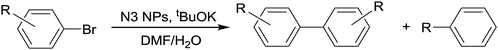
When 4-iodotoluene was replaced by 4-bromotoluene, N2 and N3 nanocatalysts (0.4 mol% of Pd) lead to 4,4′-dimethylbiphenyl (TT), arising from the 4-bromotoluene homocoupling, as the only biarylic product formed. Toluene was the only side product under these conditions, with no traces of the cross-coupled product 4-methylbiphenyl (BT), Table 3, entries 8 and 9, respectively. Pd catalyzed halogenoaryl homocoupling has been investigated in recent years as an alternative to the classical Ullmann reaction that requires stoichiometric amounts of copper and harsh conditions.95 As a consequence, both palladium complexes96,97 and Pd supported in different materials,98 as well as nanoparticles,99 have been used for this reaction. Nevertheless, none of them show enough efficiency for practical applications yet, because of the low turnover numbers achieved and the narrow substrate applicability.
Regardless of the homogenous or heterogeneous nature of the catalyst, the homocoupling of arylhalides requires a stoichiometric amount of a reductive reagent. Electropositive metals (Al, Zn, etc.), as well as organic reductants such as (Me2N)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(NMe2)2, combined with Ni or Pd catalysts, have been used in this reaction.100 Interestingly, in many cases, DMF is used as solvent for these reactions. Since it has been demonstrated that DMF, in the presence of a base, can act as a source of hydrides,94 it is possible that this solvent works as the stoichiometric source of electrons to reduce the Pd(II) to Pd(0) in arylhalide homocoupling, via HX reductive elimination promoted by the base. Furthermore, a reasonable mechanism for this reaction should involve a multimetallic catalyst, such as a metallic nanoparticle surface, in order not to invoke Pd(IV) species arising from double oxidative addition of ArX to a Pd(0). In a metallic surface, two oxidative additions of ArX to two vicinal Pd(0) will lead to two Pd(II) centers. Subsequent halide to hydride conversion, by reaction with DMF, followed by two-center reductive elimination of the HX and biaryl will regenerate the Pd(0) surface.
C(NMe2)2, combined with Ni or Pd catalysts, have been used in this reaction.100 Interestingly, in many cases, DMF is used as solvent for these reactions. Since it has been demonstrated that DMF, in the presence of a base, can act as a source of hydrides,94 it is possible that this solvent works as the stoichiometric source of electrons to reduce the Pd(II) to Pd(0) in arylhalide homocoupling, via HX reductive elimination promoted by the base. Furthermore, a reasonable mechanism for this reaction should involve a multimetallic catalyst, such as a metallic nanoparticle surface, in order not to invoke Pd(IV) species arising from double oxidative addition of ArX to a Pd(0). In a metallic surface, two oxidative additions of ArX to two vicinal Pd(0) will lead to two Pd(II) centers. Subsequent halide to hydride conversion, by reaction with DMF, followed by two-center reductive elimination of the HX and biaryl will regenerate the Pd(0) surface.
In order to confirm that the phenylboronic acid does not play any role in the homocoupling reaction, this was carried out in the absence of the boron reagent, leading in the case of N3 to a remarkable increase in the yield of the homocoupled product (TT; 70% chemoselectivity and 110 TON in 6h, Table 3, entry 13). Interestingly, when 4-iodotoluene was used in the absence of phenylboronic acid, complete conversion of the halogenoderivative to toluene was observed (Table 3, entries 5 and 6). A recent publication by H. Fujihara et al. reports that Suzuki–Miyaura coupling reactions can be catalyzed by Pd NPs at room temperature.101 In this context, we have carried out the homocoupling reaction of 4-bromobenzene catalyzed by N3 at different temperatures. At 25 °C, no homocoupled product was observed, and at 50 °C only 21% of biphenyl was obtained, whereas at 100 °C the reaction yielded 91% of biphenyl (Table 4, entry 2).
| Substrate | Entry | [Substrate]/[Pd] | Conv.b (%) | Sel. in biaryl productc (%) | TON for biaryl productd | Yield for biaryl product (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: 1 × 10−2 mmol of Pd atoms, 1 eq. tBuOK versus substrate and naphthalene as internal standard in 8.0 mL of DMF and 2.0 mL of H2O at 100 °C.b Conversion after 6 h reaction.c Selectivity towards the biaryl product.d Turnover number for the biaryl product.e 100 equivalents of Hg added after 10 minutes of reaction. | ||||||
 | 1 | 250 | 86 | 52 | 112 | 45 |
| 2 | 500 | 92 | 99 | 455 | 91 | |
| 3e | 500 | 23 | 72 | 93 | 17 | |
 | 4 | 250 | 64 | 70 | 112 | 45 |
| 5 | 500 | 29 | 46 | 67 | 13 | |
 | 6 | 250 | 84 | 51 | 107 | 43 |
| 7 | 500 | 49 | 44 | 108 | 22 | |
 | 8 | 250 | 57 | 62 | 88 | 35 |
| 9 | 500 | 46 | 61 | 140 | 28 | |
 | 10 | 250 | 36 | 0 | 0 | 0 |
| 11 | 500 | 21 | 0 | 0 | 0 | |
 | 12 | 250 | 47 | 37 | 43 | 17 |
| 13 | 500 | 20 | 41 | 41 | 8 | |
 | 14 | 250 | 91 | 13 | 30 | 12 |
| 15 | 500 | 90 | 10 | 45 | 9 | |
 | 16 | 250 | 21 | 78 | 41 | 16 |
| 17 | 500 | 15 | 71 | 53 | 11 | |
In the presence of phenylboronic acid and 4-bromotoluene, the Pd/C catalyst (Table 3, entry 10) yielded a mixture of the cross-coupled product (BT; 17%) together with the homocoupling one (TT; 46%) and toluene (PhMe; 37%). In the absence of boronic acid (Table 3, entry 14), a fair conversion was achieved with this catalyst, but toluene was this time the major reaction product (59%). Therefore, N3 nanoparticles show better selectivity towards the homocoupling reaction than the Pd/C catalyst. The homocoupling reaction of 4-bromotoluene catalyzed by the molecular complex C3 in the absence of phenylboronic acid has also been tested but no homocoupled product was observed.
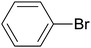
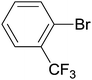
A study of the substrate scope of N3 nanoparticles in the homocoupling reaction was then undertaken. Working at 0.4 and 0.2 mol% Pd, N3 was used with different substituted bromoaryl derivatives (Table 4). The results indicate that conversions and selectivities towards the homocoupled product were not improved when the substrate concentration was doubled, except in the case of bromobenzene, for which the turnover number increased four times, since complete selectivity was achieved at 0.2 mol% Pd (Table 4, entry 2), while at 0.4 mol% it was only 52% (Table 4, entry 1). Except for 4-bromotoluene and 3,5-bis(trifluoromethyl)bromobenzene (Table 4, entries 4/5 and 8/9, respectively) for which the TON decreases and increases ca. 50%, respectively, for the rest of substrates the TON of the homocoupling reaction after 6 h did not significantly change with the increase of substrate concentration, roughly suggesting a zero order dependence on the substrate concentration. Furthermore, the reaction output showed to be very sensitive to the nature and position of the substituents of the bromoarene. As a general trend, substitutions in the aryl ring reduced the TON, since bromobenzene yielded the best result, suggesting that the reaction is very sensitive to steric constraints of the substituents. This can be attributed to an initial interaction between the phenyl ring of the bromoderivative and the surface of the Pd nanoparticle that leads to C–Br bond activation (oxidative addition step). Therefore, any substituent, even at the para position, would have a negative effect. For similar steric hindrance, electron withdrawing groups, such as CF3, led to better results than the electron donating ones, but the sterically hindered ortho-bromotrifluoromethylbenzene did not produce the homocoupled product (Table 4, entries 10 and 11). 2-Bromopyridine showed a fair selectivity in the homocoupled product, but conversions were low (Table 4, entries 16 and 17).
In order to determine the nature of the active catalytic species (molecular complex formed from the nanocatalyst or Pd NPs) in the homocoupling reaction, a mercury poisoning test was carried out (Table 4, entry 3).102 Specifically, 100 equivalents of Hg were added to the homocoupling of bromobenzene with N3 10 min after the addition of the catalyst (0.2 mol% Pd). At this point, the conversion was 23%. After 6 h, the conversion remained at 23%, in contrast to the yield observed for the same reaction performed without mercury addition (92% conversion, Table 4, entry 2). These data are most consistent with Pd NPs being the active species in the catalytic reaction.
The Hg poisoning test was also performed with molecular complexes. Specifically, we carried out the Suzuki–Miyaura reaction between bromobenzene and phenylboronic acid catalyzed by C3. After 10 minutes, an aliquot of the reaction was taken and analyzed by GC (12% of conversion). Then, 100 equivalents of Hg were added and the reaction was stopped after a total time of 6 hours. At this point, the conversion increased to 72%, hinting that C3 acts as a real molecular catalyst.
In order to gain further insight into the mechanism of the homocoupling reaction catalyzed by nanoparticles, the reaction was carried out with equimolar amounts of 4-bromotoluene and bromobenzene in the absence of phenylboronic acid. Results are summarized in Table 5. In all cases, the three expected coupling products were obtained, namely biphenyl (BB), 4-methylbiphenyl (BT) and 4,4′-dimethylbiphenyl (TT), as well as the dehalogenation products (benzene and toluene). Also in this case, N3 nanoparticles yielded the best conversion (Table 5, entry 3). Interestingly, although bromobenzene disappeared from 1.1 to 1.7 times faster than bromotoluene, similar proportions of dehalogenation products (toluene and benzene) were observed. This indicates that the formation of the arene from the metal-aryl intermediate is probably faster for the tolyl fragment than for the phenyl one. Furthermore, the distribution of homocoupling products shows higher proportion of products containing the phenyl than tolyl fragment moieties, suggesting a faster reductive elimination step from the metal surface for the phenyl group in the homocoupling reaction. In summary, both the oxidative addition of the haloderivative and the reductive elimination of the metal-aryl fragment leading to the homocoupling product seem to favour bromobenzene over bromotoluene. However the latter seems to be faster in the dehalogenation process.
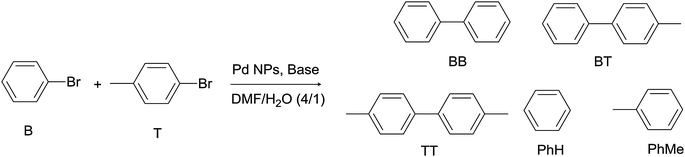
| Catalyst | Entry | Conv. Bb (%) | Conv. Tc (%) | % Homocouplingd (%BB, %BT, %TT) | % Dehalogenatione (%PhH, %PhMe) |
|---|---|---|---|---|---|
| a Reaction conditions: 1 × 10−2 mmol of Pd atoms, 1.3 mmol of 4-bromotoluene, 1.3 mmol of bromobenzene, 5.0 mmol of tBuOK, 0.5 mmol of naphthalene as internal standard, 8.0 mL of DMF, 2.0 mL of H2O, 100 °C, 6 h. BB = biphenyl; BT = 4-methylbiphenyl; TT = 4,4′-dimethylbiphenyl.b Conversion of benzene after 6 h reaction.c Conversion of toluene after 6 h reaction.d Selectivity towards homocoupling products (product distribution).e Selectivity towards dehalogenation products (product distribution). | |||||
| N1 | 1 | 22 | 13 | 36 (44, 42, 14) | 64 (52, 48) |
| N2 | 2 | 18 | 17 | 35 (31, 43, 26) | 65 (48, 52) |
| N3 | 3 | 61 | 38 | 49 (43, 41, 16) | 51 (49, 51) |
| N4 | 4 | 17 | 11 | 13 (46, 38, 16) | 87 (57, 43) |
| Pd/C | 5 | 21 | 15 | 50 (38, 46, 16) | 50 (54, 46) |
In conclusion, three different catalytic reactions have been observed depending on the catalytic system (Scheme 3). The Suzuki–Miyaura reaction takes place with Pd molecular complexes containing L1–L4. The same reaction is also seen with Pd NPs stabilized with these ligands but only when 4-iodotoluene is used as the substrate (Scheme 3A). A secondary reaction, the dehalogenation of the substrate, is always observed (Scheme 3B). Finally, the C–C homocoupling between two molecules of bromoarenes is observed with colloidal catalysts N1–N4 (Scheme 3C).
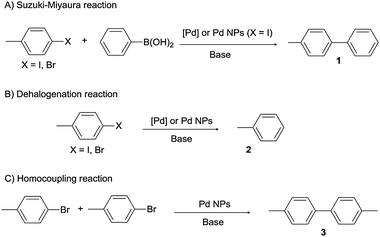 | ||
| Scheme 3 Catalytic reactions. | ||
The different chemoselectivity observed for the reaction with bromoarenes between the molecular and colloidal systems points out the heterogeneous nature of the catalytic species when using pyrazole-stabilized Pd NPs. To confirm this hypothesis, C–C coupling reactions using [Pd2(dba)3] with or without free pyrazolic ligands as catalysts (the most similar species to those that could be obtained from the leaching of molecular complexes from the Pd NPs) were also performed. Under these conditions, these catalysts, as well as the rest of the molecular catalytic systems (C1–C4), did not produce any trace of the homocoupling product (Table 2, entries 6, 9, 10, 16, 19 and 20).
Experimental section
General procedure and reagents
All manipulations were carried out under an argon atmosphere using a standard Schlenk tube or a Fischer–Porter reactor and vacuum line techniques, or in a glove-box. [Pd2(dba)3] was purchased from Strem Chemicals, palladium on activated carbon, 10% Pd (Pd/C), was purchased from Sigma-Aldrich and [PdCl2(CH3CN)2] was prepared as described in the literature.103 Ligand L4 and complex C478 were synthesized following methodologies previously described. Solvents were purchased from SDS and dried through a purification machine (MBraun MB SPS-800) or distilled prior to use: THF, diethyl ether and n-hexane over sodium/benzophenone, and pentane and dichloromethane over calcium hydride.Elemental analyses (C, H, and N) were carried out by the staff of Chemical Analyses Service of the Universitat Autònoma de Barcelona on a Eurovector 3011 instrument. Infrared spectra were run on a Perkin Elmer FT spectrophotometer, series 2000 cm−1 as KBr pellets or polyethylene films in the range of 4000–150 cm−1. 1H NMR, 13C {1H} NMR, HSQC, COSY, DOSY and NOESY spectra were recorded on a Bruker AVANCE360 NMR spectrometer in CDCl3 solutions at room temperature. All chemical shift values (δ) are given in ppm. Electrospray mass spectra were obtained using an Esquire 3000 ion trap mass spectrometer from Bruker Daltonics.
Specimens for TEM/HR-TEM and SEM-FEG analyses were prepared by slow evaporation of a drop of crude colloidal solution deposited under argon onto holey carbon-covered copper grids. TEM/HR-TEM analyses were performed at Servei Microscopia de la UAB using a JEOL JEM 2010 electron microscope working at 200 kV with a resolution point of 2.5 Å. SEM-FEG analyses were performed at the “Service Commun de Microscopie Electronique de l'Université Paul Sabatier” using a MEB JSM6700F microscope. The size distributions were determined via manual analysis of enlarged micrographs by measuring ca. 200 particles on a given grid to obtain a statistical size distribution and a mean diameter.
Data collection for WAXS was performed at the CEMES-CNRS (Toulouse) on small amounts of powder. All samples were sealed in 1 mm diameter Lindemann glass capillaries. The measurements of the X-ray intensity scattered by the samples irradiated with graphite monochromatized MoKα (0.071069 nm) radiation were performed using a dedicated two-axis diffractometer. Measurement time was 15 h for each sample. Scattering data were corrected for polarization and absorption effects, then normalized to one Pd atom and Fourier transformed to obtain the RDFs. To make comparisons with the crystalline structure in real space, a model was generated from bulk Pd parameters. The classic Debye's function was then used to compute intensity values subsequently Fourier transformed under the same conditions as the experimental ones.
Synthesis and characterization of the ligands
L1. Yield: 36% (1.3 g). Anal. calcd for C15H28N2O: C, 71.38; H, 11.18; N, 11.10. Found: C, 71.50; H, 11.56; N, 11.09%. MS m/z (%) = 275.1 (100%) [L1 + Na]+. IR (KBr, cm−1): 2924, 2855 [ν(C–H)al], 1554 [(ν(CC), ν(CN))ar], 1461 [(δ(CC), δ(CN))ar], 1115 [ν(C–O–C)], 773 δ(C–H)oop]. 1H NMR (CDCl3 solution, 360 MHz, 298 K) δ: 5.76 (s, 1H, CH(pz)), 4.12 (t, 2H, 3J = 5.8 Hz, NpzCH2CH2O), 3.72 (t, 2H, 3J = 5.8 Hz, NpzCH2CH2O), 3.34 (t, 2H, 3J = 6.6 Hz, NpzCH2CH2OCH2), 2.24 (s, 3H, CH3(pz)), 2.21 (s, 3H, CH3(pz)), 1.48 (br, 2H, OCH2CH2C5H10CH3), 1.24 (br, 10H, OCH2CH2C5H10CH3), 0.87 (t, 3H, 3J = 7.2 Hz, OCH2CH2C5H10CH3), ppm. 13C{1H} NMR (CDCl3 solution, 91 MHz, 298 K) δ: 147.6 (pz-C), 140.1 (pz-C), 104.9 (CH(pz)), 71.6 (NpzCH2CH2OCH2), 70.0 (NpzCH2CH2OCH2), 48.8 (NpzCH2CH2OCH2), 32.0–26.1 (OCH2CH2C5H10CH3), 14.2 (OCH2CH2C5H10CH3), 13.6, 11.3 (CH3(pz)) ppm.
L2. Yield: 49% (2.1 g). Anal. calcd for C15H28N2S: C, 67.11; H, 10.51; N, 10.43. Found: C, 66.82; H, 10.93; N, 10.34%. MS m/z (%) = 291.1 (100%) [L2 + Na]+. IR (KBr, cm−1): 2924, 2855 [ν(C–H)al], 1555 [(ν(CC), ν(CN))ar], 1462 [(δ(CC), δ(CN))ar], 1030 [ν(C–O–C)], 775 δ(C–H)oop]. 1H NMR (CDCl3 solution, 360 MHz, 298 K) δ: 5.78 (s, 1H, CH(pz)), 4.13 (t, 2H, 3J = 7.2 Hz, NpzCH2CH2S), 2.90 (t, 2H, 3J = 7.2 Hz, NpzCH2CH2S), 2.40 (t, 2H, 3J = 7.5 Hz, NpzCH2CH2SCH2), 2.26 (s, 3H, CH3(pz)), 2.21 (s, 3H, CH3(pz)), 1.53 (q, 2H, 3J = 7.5 Hz, SCH2CH2C5H10CH3), 1.25 (br, 10H, SCH2CH2C5H10CH3), 0.87 (t, 3H, 3J = 7.1 Hz, SCH2CH2C5H10CH3), ppm. 13C{1H} NMR (CDCl3 solution, 91 MHz, 298 K) δ: 147.6 (pz-C), 139.2 (pz-C), 105.0 (CH(pz)), 48.9 (NpzCH2CH2SCH2)), 32.5 (NpzCH2CH2SCH2), 32.3 (NpzCH2CH2SCH2)), 31.9–22.8 (SCH2CH2C5H10CH3), 14.2 (SCH2CH2C5H10CH3), 13.5, 11.3 (CH3(pz)) ppm.
:tetrahydrofuran (1:1). After cooling to room temperature, the solvent was partially removed in vacuo until 25 mL remained. Then, the mixture was extracted with CH2Cl2 (3 × 25 mL). The collected organic layers were dried with anhydrous sodium sulphate and removed in vacuo to give a brown oil that was purified by chromatography (silica gel 60 Å) using ethyl acetate:absolute ethanol (4:1) as the eluent. The product was obtained as pale yellow oil.L3. Yield: 41% (0.7 g). Anal. calcd for C15H29N3: C, 71.66; H, 11.63; N, 16.71. Found: C, 71.44; H, 11.58; N, 16.26%. MS m/z (%) = 252.1 (100%) [L3 + H]+. IR (KBr, cm−1): 3303 [ν(N–H)], 2923, 2853 [ν(C–H)al], 1552 [(ν(CC), ν(CN))ar], 1460 [(δ(CC), δ(CN))ar], 1126 [ν(C–O–C)], 773 δ(C–H)oop]. 1H NMR (CDCl3 solution, 360 MHz, 298 K) δ: 5.77 (s, 1H, CH(pz)), 4.76 (t, 2H, 3J = 6.3 Hz, NpzCH2CH2NH), 3.01 (t, 2H, 3J = 6.3 Hz, NpzCH2CH2NH), 2.60 (t, 2H, 3J = 7.4 Hz, NpzCH2CH2NHCH2), 2.23 (s, 3H, CH3(pz)), 2.20 (s, 3H, CH3(pz)), 1.83 (br, 1H, NH), 1.46 (br, 2H, NHCH2CH2C5H10CH3), 1.25 (br, 10H, NHCH2CH2C5H10CH3), 0.87 (t, 3H, 3J = 7.1 Hz, NHCH2CH2C5H10CH3), ppm. 13C{1H} NMR (CDCl3 solution, 91 MHz, 298 K) δ: 147.7 (pz-C), 139.3 (pz-C), 105.0 (CH(pz)), 49.8 (NpzCH2CH2NHCH2), 49.6 (NpzCH2CH2NHCH2), 48.3 (NpzCH2CH2NHCH2), 32.0–22.4 (NHCH2CH2C5H10CH3), 14.3 (NHCH2CH2C5H10CH3), 13.6, 11.2 (CH3(pz)) ppm.
C1. Yield: 93% (0.12 g). Anal. calcd for C30H56Cl2N4O2Pd: C, 52.82; H, 8.27; N, 8.21. Found: C, 52.58; H, 8.15; N, 8.01%. MS m/z (%) = 647.3 (100%) [C1 − Cl]+. IR (KBr, cm−1): 2917, 2854 [ν(C–H)al], 1556 [(ν(CC), ν(CN))ar], 1469 [(δ(CC), δ(CN))ar], 1124 [ν(C–O–C)], 811 δ(C–H)oop]. 1H NMR (CDCl3 solution, 360 MHz, 298 K) δ: 5.86 (s, 2H, CH(pz)), 4.99 (t, 2.4H, 3J = 5.5 Hz, NpzCH2CH2O syn/anti), 4.92 (t, 1.6H, 3J = 5.5 Hz, NpzCH2CH2O syn/anti), 4.38 (t, 2.4H, 3J = 5.5 Hz, NpzCH2CH2O syn/anti), 4.29 (t, 1.6H, 3J = 5.5 Hz, NpzCH2CH2O syn/anti), 3.41 (t, 4H, 3J = 6.6 Hz, NpzCH2CH2OCH2), 2.87, 2.78, 2.31 (s, 12H, CH3(pz) syn/anti), 1.51 (br, 4H, OCH2CH2C5H10CH3), 1.24 (br, 20H, OCH2CH2C5H10CH3), 0.88 (t, 3H, 3J = 7.1 Hz, OCH2CH2C5H10CH3), ppm. 13C{1H} NMR (CDCl3 solution, 91 MHz, 298 K) δ: 150.2, 149.7, 144.7, 144.5 (pz-C), 107.6, 107.5 (CH(pz)), 71.8 (NpzCH2CH2OCH2), 69.3 (NpzCH2CH2OCH2), 50.2 (NpzCH2CH2OCH2), 32.0–22.8 (OCH2CH2C5H10CH3), 14.3 (OCH2CH2C5H10CH3), 15.0, 12.2 (CH3(pz)) ppm.
C2. Yield: 90% (0.076 g). Anal. calcd for C15H28Cl2N2PdS: C, 40.41; H, 6.33; N, 6.28. Found: C, 39.98; H, 6.20; N, 6.16%. MS m/z (%) = 373.1 (100%) [C2 − HCl − Cl]+. IR (KBr, cm−1): 2924, 2853 [ν(C–H)al], 1554 [(ν(CC), ν(CN))ar], 1466 [(δ(CC), δ(CN))ar], 1038 [ν(C–O–C)], 790 δ(C–H)oop]. 1H NMR (CDCl3 solution, 360 MHz, 223 K) δ: 6.01 (s, 1H, CH(pz)), 5.20, 4.71 (2 br, 1H each, NpzCHHCH2S), 3.48, 3.13 (2 br, 1H each, NpzCH2CHHS), 2.62 (s, 3H, CH3(pz)), 2.49, 2.13 (2 br, 1H each, NpzCH2CH2SCH2), 2.36 (s, 3H, CH3(pz)), 1.85 (br, 2H, SCH2CH2C5H10CH3), 1.21 (br, 10H, SCH2CH2C5H10CH3), 0.85 (br, 3H, SCH2CH2C5H10CH3) ppm. 13C{1H} NMR (CDCl3 solution, 91 MHz, 298 K) δ: 152.9 (pz-C), 143.0 (pz-C), 108.9 (CH(pz)), 49.9 (NpzCH2CH2SCH2)), 40.1 (NpzCH2CH2SCH2), 34.8 (NpzCH2CH2SCH2)), 31.8–22.7 (SCH2CH2C5H10CH3), 15.4 (CH3(pz)), 14.2 (SCH2CH2C5H10CH3), 12.4 (CH3(pz)) ppm.
C3. Yield: 91% (0.074 g). Anal. calcd for C15H29Cl2N3Pd·0.5H2O: C, 41.16; H, 6.91; N, 9.60. Found: C, 40.94; H, 6.73; N, 9.40. MS m/z (%) = 426.1 (100%) [C3 − Cl + CH3OH]+; 356.2 (30%) [C3 – HCl – Cl]+. IR (KBr, cm−1): 3157 [ν(N–H)], 2926, 2855 [ν(C–H)al], 1555 [(ν(CC), ν(CN))ar], 1469 [(δ(CC), δ(CN))ar], 1149 [ν(C–O–C)], 786 δ(C–H)oop]. 1H NMR (CDCl3 solution, 360 MHz, 298 K) δ: 5.92 (s, 1H, CH(pz)), 5.70, 4.32 (2 br, 1H each, NpzCHHCH2NH), 3.51, 2.76 (2 br, 1H each, NpzCH2CHHNH), 2.61 (s, 3H, CH3(pz)), 2.28 (s, 3H, CH3(pz)), 1.96 (br, 2H, NpzCH2CH2NHCH2), 1.59 (br, 2H, NHCH2CH2C5H10CH3), 1.47 (br, 1H, NH), 1.25 (br, 10H, NHCH2CH2C5H10CH3), 0.87 (t, 3H, 3J = 7.1 Hz, NHCH2CH2C5H10CH3) ppm. 13C {1H} NMR (CDCl3 solution, 91 MHz, 298 K) δ: 152.8 (pz-C), 142.0 (pz-C), 108.2 (CH(pz)), 55.3 (NpzCH2CH2NHCH2), 50.1 (NpzCH2CH2NHCH2), 49.3 (NpzCH2CH2NHCH2), 31.9–22.8 (NHCH2CH2C5H10CH3), 15.1 (CH3(pz)), 14.2 (NHCH2CH2C5H10CH3), 11.9 (CH3(pz)) ppm.
150 mg of [Pd2(dba)3] (0.16 mmol) and the chosen quantity of L1 (41 mg for [L1]/[Pd] = 1.0) were dissolved in a Fischer–Porter reactor in THF (150 mL) under argon and vigorous stirring at 196 K. The mixture was pressurized under 3 bar of dihydrogen and left at room temperature. The colour of the solution turned after 1 h from purple to black. The hydrogen pressure and the temperature were maintained for 20 h. After that period of time, the colloidal solution was black and homogeneous. The hydrogen was evacuated and a drop of the crude colloidal solution was deposited under argon on a holey carbon covered copper grid using a paper filter under the grid for TEM and SEM analyses. Then, the colloidal solution was concentrated to ca. 5 mL. Addition of cold pentane (20 mL) allowed the precipitation of the particles as a black solid which was washed with pentane (3 × 20 mL) and dried under reduced pressure. The filtrated pentane from NPs precipitation was slightly yellow, showing dba elimination. This was corroborated by 1H-NMR experiments of the dried pentane solution.
The structures have been solved by direct methods using SIR92,104 and refined by means of least-squares procedures on a F2 with the aid of the program SHELXL97105 included in the software package WinGX version 1.63.106 The atomic scattering factors were taken from International Tables for X-ray Crystallography.107,108 All hydrogen atoms were placed geometrically, and refined by using a riding model.
All non-hydrogen atoms were anisotropically refined, and in the last cycles of refinement a weighting scheme was used, where weights are calculated from the following formula: w = 1/[σ2(Fo2) + (aP)2 + bP] where P = (Fo2+2Fc2)/3.
CCDC reference numbers 836431 (C1), 836430 (C2), 836429, (C3) and 836432 (C4).†
The products obtained from the catalytic reactions were identified using a G1800A Hewlett Packard gas chromatograph with an electron impact ionization detector and a HP-5 column (5% diphenylpolysiloxane and 95% dimethylpolysiloxane). The mass spectra of the catalytic products are in agreement with those published in the literature.109–112
Conclusions
A new family of hybrid pyrazole derived ligands containing alkylether, alkylthioether or alkylamino moieties has been successfully used to prepare Pd(II) coordination compounds and Pd NPs which were fully characterized. These two types of species display highly differentiated catalytic behaviour in C–C coupling reactions. Both in Suzuki–Miyaura and homocoupling reactions, best results were obtained with homogeneous and heterogeneous catalysts containing L2 and L3 ligands. This is probably due to the improved coordinative properties of these ligands compared to L1 and L4.In all cases, the dehalogenation of the substrate was also observed in addition to the C–C coupling reaction. In particular, when 4-iodotoluene was used as the substrate with N2 or N3 nanoparticles, in the absence of phenylboronic acid, the dehalogenation product was obtained in quantitative yield.
Interestingly, Pd complexes catalyze typical Suzuki–Miyaura reactions, whereas Pd NPs lead either to Suzuki–Miyaura or C–C homocoupling reactions depending on the substrate (iodo or bromo derivatives, respectively). These results point out that Pd-hybrid pyrazole molecular or colloidal species can drive the selectivity of the reaction to the cross-coupling, the homocoupling or the dehalogenation product with the appropriate choice of the catalyst.
The fact that the chemoselectivity of the reaction depends on the molecular or colloidal nature of the catalyst strongly suggests that the homocoupling reaction takes place on the surface of our heterogeneous systems. Indeed, the leaching of molecular species from the surface of the NPs to form homogeneous catalysis could produce lower activity than a genuine molecular catalyst at the same total Pd concentration. However the reaction selectivity towards the C–C heterocoupled or homocoupled products must remain identical in both systems, if the colloidal metal would act as a simple molecular catalysts reservoir.
Acknowledgements
This work has been supported by the project CTQ2011-26440 by the Ministerio de Educación y Cultura of Spain and MERQUINSA. F.G.V. and D.P also acknowledge the Universitat Autònoma de Barcelona for their pre-doctoral grants. The authors thank the Microscopy Service of the Universitat Autònoma de Barcelona for technical assistance with TEM and HR-TEM. CNRS is also thanked through the LTPMM-LEA 368 action.Notes and references
- T. K. Sau, A. L. Rogach, F. Jaeckel, T. A. Klar and J. Feldmann, Adv. Mater., 2010, 22, 1805–1825 CrossRef CAS.
- C. N. R. Rao, G. U. Kulkarni, P. J. Thomas and P. P. Edwards, Chem.–Eur. J., 2002, 8, 28–35 CrossRef CAS.
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757–3778 CrossRef CAS.
- K. Philippot and B. Chaudret, C. R. Chim., 2003, 6, 1019–1034 CrossRef CAS.
- J. Cox, Chem. Br., 2003, 21–25 CAS.
- D. L. Feldheim and C. A. Foss Jr., in Metal Nanoparticles: Synthesis, Characterization, and Applications, Marcel Dekker Inc., New York, 2002 Search PubMed.
- D. Astruc, in Nanoparticles and catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- J. E. Millstone, S. Park, K. L. Shuford, L. Qin, G. C. Schatz and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 5312–5313 CrossRef CAS.
- N. C. Bigall, A.-K. Herrmann, M. Vogel, M. Rose, P. Simon, W. Carrillo-Cabrera, D. Dorfs, S. Kaskel, N. Gaponik and A. Eychmueller, Angew. Chem., Int. Ed., 2009, 48, 9731–9734 CrossRef CAS.
- W. Hasan, C. L. Stender, M. H. Lee, C. L. Nehl, J. Lee and W. O. Teri, Nano Lett., 2009, 9, 1555–1558 CrossRef CAS.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS.
- H. Chen, Y. Wang and S. Dong, Inorg. Chem., 2007, 46, 10587–10593 CrossRef CAS.
- C. Wang, H. Daimon, Y. Lee, J. Kim and S. Sun, J. Am. Chem. Soc., 2007, 129, 6974–6975 CrossRef CAS.
- X. Huang and N. Zheng, J. Am. Chem. Soc., 2009, 131, 4602–4603 CrossRef CAS.
- C. Li, R. Sato, M. Kanehara, H. Zeng, Y. Bando and T. Teranishi, Angew. Chem., Int. Ed., 2009, 48, 6883–6887 CrossRef CAS.
- B. Chaudret and K. Philippot, Oil Gas Sci. Technol., 2007, 62, 799–817 CrossRef CAS.
- N. T. K. Thanh and L. A. Green, Nano Today, 2010, 5, 213–230 CrossRef CAS.
- R. Narayanan, Molecules, 2010, 15, 2124–2138 CrossRef CAS.
- N. R. Shiju and N. V. V. Guliants, Appl. Catal., A, 2009, 356, 1–17 CrossRef CAS.
- S. Kidambi, J. Dai, J. Li and M. L. Bruening, J. Am. Chem. Soc., 2004, 126, 2658–2659 CrossRef CAS.
- R. W. J. Scott, A. K. Datye and R. M. Crooks, J. Am. Chem. Soc., 2003, 125, 3708–3709 CrossRef CAS.
- S. K. Basiruddin, A. Saha, N. Pradhan and N. R. Jana, J. Phys. Chem. C, 2010, 114, 11009–11017 CAS.
- D. R. Baer, D. J. Gaspar, P. Nachimuthu, S. D. Techane and D. G. Castner, Anal. Bioanal. Chem., 2010, 396, 983–1002 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2003, 125, 8340–8347 CrossRef CAS.
- A. M. Doyle, S. K. Shaikhutdinov, S. D. Jackson and H.-J. Freund, Angew. Chem., Int. Ed., 2003, 42, 5240–5243 CrossRef CAS.
- D. Astruc, Inorg. Chem., 2007, 46, 1884–1894 CrossRef CAS.
- M. Jahjah, Y. Kihn, E. Teuma and M. Gómez, J. Mol. Catal. A: Chem., 2010, 332, 106–112 CrossRef CAS.
- J. A. Widegren, M. A. Bennett and R. G. Finke, J. Am. Chem. Soc., 2003, 125, 10301–10310 CrossRef CAS.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- A. Kolmakov, X. Chen and M. Moskovits, J. Nanosci. Nanotechnol., 2008, 8, 111–121 CrossRef CAS.
- B. J. Horstein, J. D. Aiken III and R. G. Finke, Inorg. Chem., 2002, 41, 1625–1638 CrossRef.
- F. Diederich and P. J. Stang, in Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 1998 Search PubMed.
- K. Ferré-Filmon, L. Delaude, A. Demonceau and A. F. Noels, Coord. Chem. Rev., 2004, 248, 2323–2336 CrossRef.
- F. Lu, J. Ruiz-Aranzaes and D. Astruc, Angew. Chem., Int. Ed., 2005, 44, 7399–7404 CrossRef.
- D. Astruc, C. Ornelas, A. K. Diallo and J. Ruiz, Molecules, 2010, 15, 4947–4960 CrossRef CAS.
- N. Kim, M. S. Kwon, C. M. Park and J. Park, Tetrahedron Lett., 2004, 45, 7057–7059 CrossRef CAS.
- D. B. Dell'Amico, L. Labella, F. Marchetti and S. Samaritani, Coord. Chem. Rev., 2010, 254, 635–645 CrossRef CAS.
- S. Jansat, J. Durand, I. Favier, F. Malbosc, C. Pradel, E. Teuma and M. Gómez, ChemCatChem, 2009, 1, 244–246 CrossRef CAS.
- L. Rodríguez-Pérez, C. Pradel, P. Serp, M. Gómez and E. Teuma, ChemCatChem, 2011, 3, 749–754 CrossRef.
- J. Liu, F. He, T. M. Gunn, D. Zhao and C. B. Roberts, Langmuir, 2009, 25, 7116–7128 CrossRef CAS.
- M. Studer, H.-U. Blaser and C. Exner, Adv. Synth. Catal., 2003, 345, 45–65 CrossRef CAS.
- U. Heiz and U. Landman, in Nanocatalysis, Springer, Berlin, 2007 Search PubMed.
- B. F. G. Johnson, Top. Catal., 2003, 24, 147–159 CrossRef CAS.
- N. T. S. Phan, M. van der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS.
- V. Polshettiwar, A. Decottignies, C. Len and A. Fihri, ChemSusChem, 2010, 3, 502–522 CrossRef CAS.
- J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS.
- A. N. Cammidge and K. V. L. Crépy, Tetrahedron, 2004, 60, 4377–4386 CrossRef CAS.
- K. Mikami, T. Miyamoto and M. Hatano, Chem. Commun., 2004, 2082–2083 RSC.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS.
- K. Sambasivarao and M. Kalyaneswar, Chem.–Asian J., 2009, 4, 354–362 CrossRef.
- N. Miyaura, Top. Curr. Chem., 2002, 219, 11–59 CrossRef CAS.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS.
- G. Viau, R. Brayner, L. Poul, N. Chakroune, L. E. F. Fiévet-Vincent and F. Fiévet-Vincent, Chem. Mater., 2003, 15, 486–494 CrossRef CAS.
- J. García-Martínez, R. W. J. Scott and R. M. Crooks, J. Am. Chem. Soc., 2003, 125, 11190–11191 CrossRef.
- N. Toshima, Y. Shiraishi, T. Teranishi, M. Miyake, T. Tominaga, H. Watanabe, W. Brijoux, H. Bönnemann and G. Schmid, Appl. Organomet. Chem., 2001, 15, 178–196 CrossRef CAS.
- A. Ohtaka, Y. Tamaki, Y. Igawa, K. Egami, O. Shimomura and R. Nomura, Tetrahedron, 2010, 66, 5642–5646 CrossRef CAS.
- C. Ornelas, A. K. Diallo, J. Ruiz and D. Astruc, Adv. Synth. Catal., 2009, 351, 2147–2154 CrossRef CAS.
- L. Durán-Pachón and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288–299 CrossRef.
- J. Durand, E. Teuma and M. Gómez, Eur. J. Inorg. Chem., 2008, 3577–3586 CrossRef CAS.
- I. Favier, E. Teuma and M. Gómez, C. R. Chim., 2009, 12, 533–545 CrossRef CAS.
- S. U. Son, Y. Jang, K. Y. Yoon, E. Kang and T. Hyeon, Nano Lett., 2004, 4, 1147–1151 CrossRef CAS.
- E. Badetti, A.-M. Caminade, J.-P. Majoral, M. Moreno-Mañas and R. M. Sebastián, Langmuir, 2008, 24, 2090–2101 CrossRef CAS.
- I. Favier, D. Madec, E. Teuma and M. Gómez, Curr. Org. Chem., 2011, 15, 3127–3174 CrossRef CAS.
- M. B. Thathagar, J. E. ten Elshof and G. Rothenberg, Angew. Chem., Int. Ed., 2006, 45, 2886–2890 CrossRef CAS.
- A. V. Gaikwad, A. Holuigue, M. B. Thathagar, J. E. ten Elshof and G. Rothenberg, Chem.–Eur. J., 2007, 13, 6908–6913 CrossRef CAS.
- J. G. de Vries, Dalton Trans., 2006, 421–429 RSC.
- A. K. Diallo, C. Ornelas, L. Salmón, J. Ruiz-Aranzaes and D. Astruc, Angew. Chem., Int. Ed., 2007, 46, 8644–8648 CrossRef CAS.
- M. T. Reetz and E. Westermann, Angew. Chem., Int. Ed., 2000, 39, 165–168 CrossRef CAS.
- T. Maegawa, Y. Kitamura, S. Sako, T. Udzu, A. Sakurai, A. Tanaka, Y. Kobayashi, K. Endo, U. Bora, T. Kurita, A. Kozaki, Y. Monguchi and H. Sajiki, Chem.–Eur. J., 2007, 13, 5937–5943 CrossRef CAS.
- S. Jansat, M. Gómez, K. Philippot, G. Muller, E. Guiu, C. Claver, S. Castillón and B. Chaudret, J. Am. Chem. Soc., 2004, 126, 1592–1593 CrossRef CAS.
- I. Favier, M. Gómez, G. Muller, M. R. Axet, S. Castillón, C. Claver, S. Jansat, B. Chaudret and K. Philippot, Adv. Synth. Catal., 2007, 349, 2459–2469 CrossRef CAS.
- F. Fernández, B. Cordero, J. Durand, G. Muller, F. Malbosc, Y. Kihn, E. Teuma and M. Gómez, Dalton Trans., 2007, 5572–5581 RSC.
- S. S. Zalesskiy and V. P. Ananikov, Organometallics, 2012, 31, 2302–2309 CrossRef CAS.
- J. L. Bolliger and C. M. Frech, Chem.–Eur. J., 2010, 16, 4075–4081 CrossRef CAS.
- E. V. Carino, M. R. Knecht and R. M. Crooks, Langmuir, 2009, 25, 10279–10284 CrossRef CAS.
- D. Sanhes, E. Raluy, S. Rétory, N. Saffon, E. Teuma and M. Gómez, Dalton Trans., 2010, 9719–9726 RSC.
- J. Pons, G. Aragay, J. García-Antón, T. Calvet, M. Font-Bardia and J. Ros, Inorg. Chim. Acta, 2010, 363, 911–917 CrossRef CAS.
- W. G. Haanstra, W. L. Driessen, J. Reedijk, U. Turpeinen and R. Hämäläinen, J. Chem. Soc., Dalton Trans., 1989, 2309–2314 RSC.
- E. Ramirez, S. Jansat, K. Philippot, P. Lecante, M. Gómez, A. Masdeu and B. Chaudret, J. Organomet. Chem., 2004, 689, 4601–4610 CrossRef CAS.
- M. Guerrero, J. García-Antón, M. Tristany, J. Pons, J. Ros, K. Philippot, P. Lecante and B. Chaudret, Langmuir, 2010, 26, 15532–15540 CrossRef CAS.
- B. J. Berne and R. Pecora, in Dynamic Light Scattering, Wiley, New York, 1976 Search PubMed.
- K. Schatztel, J. Mod. Opt., 1991, 38, 1849–1865 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef CAS.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 36, 3437–3440 CrossRef.
- N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866–867 RSC.
- N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513–519 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- C. Amatore, A. Jutand and G. Le Duc, Chem.–Eur. J., 2011, 17, 2492–2503 CAS.
- M. Valentini, H. Rüegger and P. S. Pregosin, Organometallics, 2000, 19, 2551–2555 CrossRef CAS.
- S. U. Son, Y. Jang, K. Y. Yoon, E. Kang and T. Hyeon, Nano Lett., 2004, 4, 1147–1151 CrossRef CAS.
- R.-Y. Lai, C.-L. Chen and S.-T. Liu, J. Chin. Chem. Soc., 2006, 53, 979–985 CAS.
- O. Navarro, N. Marion, Y. Oonishi, R. A. Kelly III and S. P. Nolan, J. Org. Chem., 2006, 71, 685–692 CrossRef CAS.
- J. Muzart, Tetrahedron, 2009, 65, 8313–8323 CrossRef CAS.
- P. E. Fanta, Synthesis, 1974, 9–21 CrossRef CAS.
- W. M. Seganish, M. E. Mowery, S. Riggleman and P. DeShong, Tetrahedron, 2005, 61, 2117–2121 CrossRef CAS.
- S. Nadri, E. Azadi, A. Ataei, M. Joshaghanim and E. Rafiee, J. Organomet. Chem., 2011, 696, 2966–2970 CrossRef CAS.
- M. Zeng, Y. Du, C. Qi, S. Zuo, X. Li, L. Shao and X. M. Zhang, Green Chem., 2011, 13, 350–356 RSC.
- S. Santra, P. Ranjan, S. K. Mandal and P. K. Ghorai, Inorg. Chim. Acta, 2011, 372, 47–52 CrossRef CAS.
- M. Kuroboshi, Y. Waki and H. Tanaka, J. Org. Chem., 2002, 68, 3938–3942 CrossRef , and references therein.
- K. Sawai, R. Tatumi, T. Nakahodo and H. Fujihara, Angew. Chem., Int. Ed., 2008, 47, 6917–6919 CrossRef CAS.
- G. M. Whitesides, M. Hackett, R. L. Brainard, J.-P. P. M. Lavalleye, A. F. Sowinski, A. N. Izumi, S. S. Moore, D. W. Brown and E. M. Staud, Organometallics, 1985, 4, 1819–1830 CrossRef CAS.
- S. Komiya, in Synthesis of Organometallic Compounds: A Practice Guide, Board, New York, 1997 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef CAS.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- T. Hahn, in International Tables for Crystallography, Volume A, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1995 Search PubMed.
- A. J. C. Wilson, in International Tables for Crystallography, Volume C, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1995 Search PubMed.
- X. Xu, D. Cheng and W. Pei, J. Org. Chem., 2006, 71, 6637–6639 CrossRef CAS.
- E. F. Santos-Filho, J. C. Sousa, N. M. M. Bezerra, P. H. Menezes and R. A. Oliveira, Tetrahedron Lett., 2011, 52, 5288–5291 CrossRef CAS.
- Y. Mogli, E. Mascaró, F. Nador, C. Vitale and G. Radivoy, Synth. Commun., 2008, 38, 3861–3874 CrossRef.
- I. J. S. Fairlamb, A. R. Kapdi and A. F. Lee, Org. Lett., 2004, 6, 4435–4438 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for compounds C1–C4, complete catalytic data, HR-TEM micrographs of all Pd NPs, WAX, SEM-FEG and DLS measurements for selected Pd NPs, NOESY NMR spectrum of N3, DOSY experiments for in situ prepared complexes “[Pd(0)(dba)(L1)2]” and “[Pd(0)(dba)(L3)]”, ORTEP drawings of the two non-equivalent molecules of C1–C3, discussion of the NMR studies for C1–C4. CCDC 836431 (C1), 836430 (C2), 836429, (C3) and 836432 (C4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cy20517c |
| This journal is © The Royal Society of Chemistry 2013 |