One-pot photo-reductive N-alkylation of aniline and nitroarene derivatives with primary alcohols over Au–TiO2
David
Stíbal
abc,
Jacinto
Sá
*b and
Jeroen A. van
Bokhoven
ab
aInstitute for Chemical and Bioengineering ETH Zurich, Wolfgang-Pauli Strasse, CH-8093 Zurich, Switzerland
bPaul Scherrer Institute (PSI), CH-5232 Villigen, Switzerland. E-mail: jacinto.sa@psi.ch; Tel: +41 56 310 29 10
cInstitute of Chemical Technology, Technická 5, Prague, 160 00, Czech Republic
First published on 25th September 2012
Abstract
We report the photo-catalytic N-alkylation of aniline by Au–TiO2. We successfully alkylate aniline with several primary alcohols. The combined selectivities of mono- and di-alkylated products were always in excess of 70% and dependent on the alkylating alcohol used. A one-pot reaction from nitrobenzene was found to be possible with several substrates. Preliminary experiments showed that this approach could be adopted for the production of lactams using terminal amino-alcohols.
1. Introduction
Amines and their derivatives are extremely versatile building blocks for a variety of therapeutic applications.1 Amines are also used in the fields of bioorganic, industrial and synthetic organic chemistry.2The most attractive method to prepare amine derivatives is the direct reductive amination of aldehydes and ketones. The carbonyl compound and the amine condense to form an imine that can be hydrogenated to the alkylated product by an appropriate reducing agent in a one-pot reaction.3 However the current methods have some drawbacks, such as lengthy reaction times, high reaction temperatures, reaction must be carried out under inert conditions and reaction often requires harmful reagents and often forms toxic products. Byun et al.4 synthesized N-alkylated amine from anilines and nitroarenes in one-pot using aldehydes and a Pd/C catalyst in aqueous alcoholic solvents with ammonium formate as the in situ source of hydrogen. Despite the high yields and the claim of green process, the reaction produces NH4+ and CO2 as by-products.
Heterogeneous photocatalysis was almost exclusively used in the degradation of unwanted molecules such as pollutants, however recently this technology was adopted for synthetic purposes. Kagiya and co-workers reported the photo-alkylation of linear amines for the first time.5 They reported a 33% yield in the alkylation of ethylamine to diethylamine over Pt–TiO2. They proposed a mechanism based on the condensation of amine and aldehyde leading to the formation of an imine, which was subsequently hydrogenated to the target product by H2 photo-generated in situ. The photo-catalyst was found later to be able to perform N-alkylation of aromatic amines by Matsushita et al.6
Herein, we report an efficient, facile and environmentally benign one-pot reductive N-alkylation of aniline and nitroarenes with linear alcohols, acting both as solvent and reagent, by photo-catalysis with Au–TiO2. To our knowledge, this is the first time that Au–TiO2 is proposed for N-alkylation of aniline and nitroarenes.7 Furthermore the strategy herein proposed can be used for the cyclization of linear amino-alcohols to form industrially relevant lactams.
2. Experimental
Materials
All chemicals have been used as received, without any further purification. TiO2 (P25, Acros Chemicals) was used as a base for the catalyst. HAuCl4 (Sigma-Aldrich) was used as gold source and urea (Sigma-Aldrich) as the precipitating agent. Nitrobenzene 99% (Sigma Aldrich), aniline 99% (ACS Chemicals) and alcohols methanol (anhydrous, 99.8%, Sigma-Aldrich), ethanol (Absolute, VWR Chemicals), propanol (anhydrous, 99.5%, Sigma-Aldrich), butanol (puriss, 99.5%, Sigma-Aldrich) and hexanol (anhydrous 99%, Sigma Aldrich) were used for the reaction. Nitroarenes and aniline derivatives were supplied by Fluka: 2-chloronitrobenzene 99%, 2-chloroaniline 98%, 3-chloronitrobenzene 98%, 4-chloroaniline 99%, 2-bromonitrobenzene 97%, 3-bromonitrobenzene 98%, 4-bromonitrobenzene 98%, 4-bromoaniline 99%, 2-nitrotoluene 99%, 4-nitrotoluene 98% and 2-methoxynitrobenzene 97%. From Acros Chemicals: 3-chloroaniline 99%, 4-chloronitrobenzene 99%, 3-bromoaniline 98%, 3-methylaniline 99%, 4-methylaniline 99%, 3-methoxyaniline 98% and 4-methoxyaniline 97%. From Aldrich: 2,6-dichloroaniline 98%, 2-methylaniline 99%, 3-nitrotoluene 99% and 4-methoxynitrobenzene 97%. From Tokyo Chemical Industry Co: 2,6-dichloronitrobenzene 98% (2,6-diCl-NB) and 2-methoxynitrobenzene 98%. From ABCR Chemicals: 2-bromoaniline 98% and from Merck: 3-methoxynitrobenzene. Mesitylene (99%, Sigma-Aldrich) was used as the GC internal standard.Catalyst preparation
Gold catalyst was prepared by a deposition precipitation method with urea. Briefly, 1 g of TiO2 was added to 100 mL of an aqueous solution of HAuCl4 (4.2 × 10−3 M) and urea (0.42 M). The initial pH was 2. The suspension was equilibrated to 80 °C and vigorously stirred for 16 h (pH increased during that time), then centrifuged, washed and dried. Pretreatment before the reaction consisted of reduction in a hydrogen atmosphere (5% H2/He) for 20 minutes at 60 °C.Characterization
Atomic absorption spectroscopy (AAS) samples were prepared by microwave-assisted digestion. 1 mL of nitric acid, 2 mL of hydrofluoric acid and 3 mL of hydrochloric acid were added to 40 mg of catalyst. The mixture was heated to 160 °C during 20 minutes and kept at that temperature for another 30 minutes while stirring in a microwave oven. AAS measurements were performed on a Varian SpectrAA 200 FS spectrometer with a C2H2–air burner. The Au-content was quantified by calibration with a standard solution (gold solution, 1000 ppm in 1 M hydrochloric acid, Fisher-Scientific). All samples were measured three times.The transmission electron microscopy (TEM) samples were prepared by dispersing the catalyst in ethanol, which was subsequently loaded in a copper grid. TEM measurements were performed on a HD2700CS (Hitachi, aberration-corrected dedicated STEM, cold FEG, 200 kV) and a Tecnai F30 ST (FEI, FEG, 300 kV).
Catalytic tests
The reaction was performed in a photo-catalytic multi-reactor developed in house, which comprised of four quartz three-necked round-bottom reaction flasks horizontally irradiated by four low-cost 16 W UV-A lamps, which have a narrow irradiation peak at around 350 nm. A schematic of the setup is depicted in Fig. 1. The reaction products were analysed by gas chromatography (GC) on an Agilent 6890 GC chromatograph equipped with a Hewlett Packard HP-5 column (30 m, 0.25 mm ID, 0.25 μm film density) and a FID detector. Identification of unknown intermediates and by-products was carried out on an Agilent 7890A GC coupled with an Agilent 5975C mass spectrometer with a Triple-Axis Detector. A DB-5MS column (30 m, 0.25 mm ID) was used. Spectra were compared with the NIST spectral library.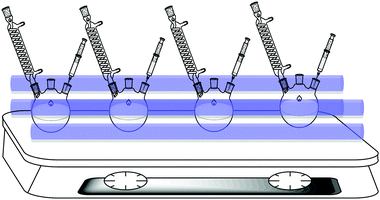 | ||
| Fig. 1 Schematic representation of the photo-reactor. | ||
Aniline N-alkylation
Each quartz round-bottom reactor was charged with 1.1 mmol of aniline, 20 mL of alcohol, 100 mg of catalyst and 30 mg of the internal standard. The mixture was sonicated for 10 minutes and then purged with argon for another 10 minutes with stirring to remove oxygen. A small flow of argon was kept during the whole experiment to prevent the presence of oxygen during the reaction. After these procedures, the UV lamps were switched on, samples (0.5 mL) were taken at pre-defined time intervals, filtered and sealed in GC glass vials for analysis. Each experiment was performed in duplicates.Nitroarenes N-alkylation
Each quartz round-bottom reactor was charged with 100 mg of catalyst, 30 mg of mesitylene, 32 mL of ethanol and 84 mmol of substrate. The mixture was sonicated for 10 minutes and then purged with argon for another 10 minutes with stirring to remove oxygen. A small flow of argon was kept during the whole experiment to prevent the presence of oxygen during the reaction. After these procedures the UV lamps were switched on, samples (0.5 mL) were taken at pre-defined time intervals, filtered and sealed in GC glass vials for analysis. Each experiment was performed in duplicates.5-Aminopentanol cyclization
Each quartz round-bottom reactor was charged with 100 mg of catalyst, 30 mg of mesitylene, 32 mL of ethanol and 84 mmol of 5-aminopentanol. The mixture was sonicated for 10 minutes and then purged with argon for another 10 minutes with stirring to remove oxygen. A small flow of argon was kept during the whole experiment to prevent the presence of oxygen during the reaction. After these procedures, the UV lamps were switched on, samples (0.5 mL) were taken at pre-defined time intervals, filtered and sealed in GC glass vials for analysis. Each experiment was performed in duplicates.3. Results and discussion
The initial gold loading was determined by AAS to be ca. 0.82 wt%. Measurements of gold loading after the reaction showed the same value and therefore metal leaching does not seem to occur during the reaction.Scheme 1 shows the photo-catalytic N-alkylation of aniline with a terminal alcohol over Au–TiO2. The catalytic performance after 4 h on stream is summarized in Table 1. Values close to 100% conversion were measured with all the alcohols after 4 h of reaction, except for 1-hexanol. The selectivity to alkylated products was found to be higher than 70%, which translates to high yield of the desired products. When the reactions were carried out for 24 h, the combined yield of alkylated products was higher than 95%. The yields are comparable to the ones reported by Byun et al.4 however it must be emphasized that they used an aldehyde as the starting reagent and hydrogen was formed from dissociation of ammonium formate, which herein was formed photocatalytically from the alcohol. The reaction was carried out at 50 °C and no irradiation did not yield significant amounts of alkylated products.
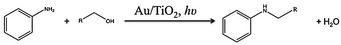 | ||
| Scheme 1 Photo-alkylation of aniline over Au–TiO2. | ||
| Alcohol R–CH2OH | Conversiona (%) | Yield of Ar–NHR′a (%) | Yield of Ar–NR2′a (%) |
|---|---|---|---|
| a Calculated after 4 h on stream. b Reaction rate in mmol (min gcat)−1 calculated after 30 min of reaction; R′ = –CH2–R. | |||
| CH3OH | 100.0 (0.17)b | 5.5 | 60.7 |
| CH3CH2OH | 100.0 (0.15)b | 74.4 | 8.9 |
| CH3(CH2)2OH | 99.7 (0.08)b | 89.1 | 2.4 |
| CH3(CH2)3OH | 99.6 (0.06)b | 87.8 | 0.0 |
| CH3(CH2)5OH | 74.0 (0.04)b | 52.5 | 0.0 |
The proposed reaction mechanism is illustrated in Fig. 2. The process is initiated by the oxidation of the terminal alcohol to its corresponding aldehyde via an alkoxy radical intermediate by the photo-generated holes in TiO2 upon absorption of a photon with enough energy to overcome the semiconductor band gap. Alcohol oxidation can occur via direct interaction with semiconductor surface holes or indirectly via a radical reaction, such as OH˙, formed by the reaction of OH− with the surface holes. The results presented within do not allow discrimination of the individual paths. The photo-generated electrons reduce the resultant protons to molecular hydrogen. The proposed active sites for the formation of hydrogen are the larger gold ensembles (5–15 nm) that act as an electron sink slowing down the rate of charge-pair recombination.8
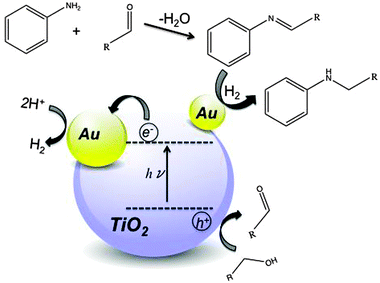 | ||
| Fig. 2 Proposed reaction mechanism for the photo-alkylation of aniline over Au–TiO2. | ||
Small nanoparticles of Au (1–3 nm) are expected to be bad electron reservoirs because they have less metallic character,9 essential for efficient charge transfer. Fig. 3 shows the particle size distribution of the gold catalyst after synthesis and after reduction, which was carried out before the reaction. The reduction sinters some of the Au nano-particles leading to a broadening of the particle size distribution. The catalyst in its state before catalysis is composed roughly by 40% of small gold nano-particles (<4 nm) and the remaining 60% are larger particles with sizes comprised between 5 and 13 nm.
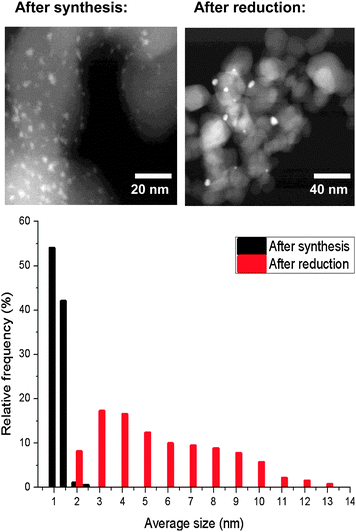 | ||
| Fig. 3 TEM analysis of the catalyst before and after reduction pre-treatment. (top) TEM images; (bottom) histogram of particle size distribution. | ||
After the formation of the aldehyde, an imine is formed from the condensation of the aldehyde and amine. This step is not catalytic and it is observed also with bare TiO2, however with a slightly lower rate. The next step is the hydrogenation of the imine double bond to form the corresponding aniline derivative. This crucial step is carried out on small gold ensembles, which activate the photo-generated molecular hydrogen. In the case of bare TiO2 the process ceases after the imine formation step since the bare support is not able to activate hydrogen. Selvam and Swaminathan10 reported the photo-formation of quinaldines from nitroarenes using Au–TiO2 as the photo-catalyst. In their proposed reaction mechanism, imines are reaction intermediates. However, their photo-catalyst is unable to hydrogenate the imine to the N-alkylated product. The disparity in the result can be easily rationalized if one takes a closer look at the particle size. Their catalyst had no particles smaller than 5 nm, which is fundamental to hydrogenate the photo-formed imines, i.e. split hydrogen. Therefore, the presence of small and large particles in our catalyst confers the essential dual function of gold, namely large gold particles for the photo-formation of hydrogen and gold particles below 5 nm to hydrogenate the imine.
The overall process is governed by the availability of protons, which is governed by the pKa of the alcohol. Methanol is from the alcohols tested the one with the lowest pKa, which increases with the increase of the carbon chain length.10 Therefore, the activity or the reaction rate should decrease with the increase of the carbon chain length, which was the case. Combination of low pKa and low kinetic stability of the methoxy radical promotes alkylation resulting in the appearance of the di-alkylated product. The di-alkylated product is produced at the expense of the mono-alkylated, which can be easily deduced from Fig. 4.
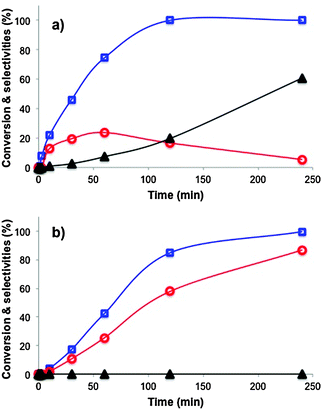 | ||
Fig. 4 Catalytic performance of Au–TiO2 as a function of time in the photocatalytic alkylation of aniline. Alkylating alcohol: (a) methanol and (b) butanol. ( ) aniline conversion, ( ) aniline conversion, ( ) selectivity to N-monoalkylated product and (▲) selectivity to N,N-dialkylated product. ) selectivity to N-monoalkylated product and (▲) selectivity to N,N-dialkylated product. | ||
The aforementioned results confirm that photo-catalytic N-alkylation of aniline with alcohols is possible, however to confer any high degree of flexibility to the methodology, photo-alkylation should be able to start from nitroarenes, which are the typical aniline precursors. The outcomes of the experiments in which nitroarenes were used as starting substrate are summarized in Table 2. The yields are smaller than the ones reported by Byun et al.,4 however they used up to 20 equivalents of ammonium formate to achieve the reported yields. Furthermore, the reaction was only carried out for 4 h in order to compare the effect of the substituent. When some of the reactions were allowed to proceed for 24 h, yields higher than 70% were obtained.
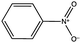
| Substrate | Conv.a (%) | Initial rateb | Yield of Ar–NH2a (%) | Yield of Ar–NR2′a (%) |
|---|---|---|---|---|
| a Conversion and yield after 4 h on stream. b Reaction rate in mmol (min gcat)−1 calculated after 30 min of reaction. c Reaction rate of bare TiO2; R′ = –CH2–R | ||||
|
99.4 | 0.034 (0.090)c | 18.5 | 50.0 |

|
86.8 | 0.060 (0.059)c | 45.2 | 24.1 |

|
97.4 | 0.040 (0.057)c | 12.7 | 5.6 |

|
98.0 | 0.032 (0.071)c | 60.2 | 3.9 |
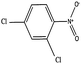
|
100 | 0.090 (0.048)c | 76.5 | 0.0 |

|
99.8 | 0.071 (0.042)c | 52.8 | 24.2 |

|
94.2 | 0.051 (0.028)c | 52.1 | 21.3 |
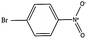
|
99.5 | 0.073 (0.057)c | 9.2 | 56.9 |

|
84.4 | 0.002 (0.055)c | 30.6 | 32.8 |
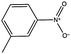
|
100 | 0.009 (0.073)c | 23.7 | 7.6 |

|
100 | 0.043 (0.075)c | 9.5 | 0.6 |
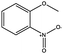
|
100 | 0.039 (0.030)c | 27.7 | 20.3 |

|
54.8 | 0.027 (0.015)c | 5.9 | 10.9 |
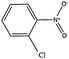
|
99.4 | 0.034 (0.090)c | 18.5 | 50.0 |
Two possible reaction mechanisms can be postulated for the reduction of the nitroarenes to the corresponding aniline. The first is the hydrogenation of the nitro-group over nano-size gold, as previously reported.11 Normally hydrogenation reactions are carried out with 10 bar of hydrogen and at a temperature exceeding 60 °C,11d to ensure meaningful catalytic conversions. Furthermore, the catalytic activity of the bare support is often very low and the overall catalytic performance is not strongly affected by the presence of substituents.12
The other possible mechanism is the photo-reduction of nitroarene. Nitroarene reduction was proposed by Mahdavi et al.,13 who demonstrated that TiO2 under UV light could efficiently reduce nitroarenes to corresponding anilines. Addition of Ag to the catalyst formulation improved the catalytic performance,14 which they attributed to a decrease of charge-recombination, suggesting that nitro reduction occurs primarily on TiO2. Furthermore, the activity was found to be dependent on the nitroarene substituent identity and position with respect to the nitro group,15 which is precisely what the results summarized in Table 2 show. Therefore we assume that conversion of nitro to amine occurs photo-catalytically on TiO2. After the formation of aniline, the reaction proceeds as previously mentioned.
The reported yields are not satisfactory for all nitroarenes, which has to do with the nitroarene reaction rate to aniline. A plethora of species were measured, however it should be pointed out that they were all intermediate species of either aniline formation (slower reaction rates in the nitroarene reduction) or N-alkylation, possibly caused by the competition for the active site, i.e., products related to other reactions were not detected. When some of the reactions were allowed to proceed for 24 h, yields of 70% or better were measured, suggesting that the low yields are related primarily to the short time on stream. However for comparison purposes the reactions were carried out for the same time as that of the alkylation of aniline.
N-heterocycles are important scaffolds for many drugs and natural products.16 As a proof of concept, we tested our catalyst in the photo-cyclization of amino-alcohols for the production of commercially valuable lactams and heterocycle amines, as depicted in Scheme 2 and shown in Table 3.
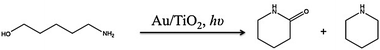 | ||
| Scheme 2 Photo-cyclization of 5-aminopentanol over Au–TiO2. | ||
| Reaction time/h | Conversion (%) | Yield of δ-valerolactam (%) | Yield of piperidine (%) |
|---|---|---|---|
| 4 | 74.0 | 16.3 | 4.8 |
| 17 | 98.5 | 32.5 | 0.3 |
4. Conclusions
The presented results propose that photo-catalysis can be used for the synthesis of secondary and tertiary aniline derivatives in one-pot. The photo-catalysis process is very versatile since the reaction can be carried out either with nitro- or amino-arenes. The reported approach is facile, mild (ambient conditions) and very green since the alkylating and reducing agents are formed in situ from the oxidation of the solvent, reducing drastically the number of side products and/or harmful chemicals. The photo-catalyst is commercially available from the World Gold Council.17 In conclusion, photo-catalysts can be tailored to perform selective and multi-step reactions. This offers great opportunities in the field of organic synthesis.Acknowledgements
The authors thank Dr Frank Krumeich from ETH for the TEM measurements.Notes and references
- J. S. Bradshaw, K. E. Krakowiak and R. H. Izatt, Tetrahedron, 1992, 22, 4475–4515 CrossRef.
- M. J. Bhanushali, N. S. Nandurkar, M. D. Bhor and B. M. Bhanage, Tetrahedron Lett., 2007, 48, 1273–1276 CrossRef CAS.
- (a) A. Heydari, S. Khaksar, M. Esfandyari and M. Tajbakhsh, Tetrahedron, 2007, 63, 3363–3366 CrossRef CAS; (b) A. Heydari, S. Khaksar, J. Akbari, M. Esfandyari, M. Pourayoubi and M. Tajbakhsh, Tetrahedron Lett., 2007, 48, 1135–1138 CrossRef CAS; (c) A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS; (d) H. Kato, I. Shibata, Y. Yasaka, S. Tsumoi, M. Yasuda and A. Baba, Chem. Commun., 2006, 4189–4191 RSC; (e) D. Menche and F. Arikan, Synlett, 2006, 841–844 CrossRef CAS; (f) D. Menche, S. Bohm, J. Li, S. Rudolph and W. Zander, Tetrahedron Lett., 2007, 48, 365–369 CrossRef CAS; (g) M. Suginome, Y. Tanaka and T. Hasui, Synlett, 2006, 1047–1050 CrossRef CAS; (h) D. Gnanamgari, A. Moores, E. Rajaseelan and R. H. Crabtree, Organometallics, 2007, 26, 1226–1230 CrossRef CAS; (i) D. Imao, S. Fujihara, T. Yamamoto, T. Ohta and Y. Ito, Tetrahedron, 2005, 61, 6988–6992 CrossRef CAS.
- E. Byun, B. Hong, K. A. De Castro, M. Lim and H. Rhee, J. Org. Chem., 2007, 72, 9815–9817 CrossRef CAS.
- (a) S. Nishimoto, B. Ohtani, T. Yoshikawa and T. Kagiya, J. Am. Chem. Soc., 1983, 105, 7180–7182 CrossRef CAS; (b) B. Ohtani, H. Osaki, S. Nishimoto and T. Kagiya, J. Am. Chem. Soc., 1986, 108, 308–310 CrossRef CAS; (c) B. Ohtani, H. Osaki, S. Nishimoto and T. Kagiya, Tetrahedron Lett., 1986, 27, 2019–2022 CrossRef CAS.
- (a) Y. Matsushita, N. Ohba, S. Kumada, T. Suzuki and T. Ichimura, Catal. Commun., 2007, 8, 2194–2197 CrossRef CAS; (b) Y. Matsushita, N. Ohba, T. Suzuki and T. Ichimura, Catal. Today, 2008, 132, 153–158 CrossRef CAS.
- (a) A. Primo, A. Corma and H. García, Phys. Chem. Chem. Phys., 2011, 13, 886–910 RSC; (b) G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 3425–3437 RSC.
- P. V. Kamat, J. Phys. Chem. B, 2002, 106, 7729–7744 CrossRef CAS.
- (a) J. A. van Bokhoven, Chimia, 2009, 63, 257–260 CrossRef CAS; (b) A. Tanaka, S. Sakaguchi, K. Hashimoto and H. Kominami, Catal. Sci. Technol., 2012, 2, 907–909 RSC.
- K. Selvam and M. Swaminathan, Catal. Commun., 2011, 12, 389–393 CrossRef CAS.
- For example: (a) A. Corma and P. Serna, Science, 2006, 313, 332–334 CrossRef CAS; (b) A. Corma, P. Concepción and P. Serna, Angew. Chem., Int. Ed., 2007, 46, 7266–7269 CrossRef CAS; (c) A. Corma, P. Serna, P. Concepción and J. J. Calvino, J. Am. Chem. Soc., 2008, 130, 8748–8753 CrossRef CAS; (d) M. Makosch, C. Kartusch, J. Sá, R. B. Duarte, J. A. van Bokhoven, K. Kvashnina, P. Glatzel, D. L. A. Fernandes, M. Nachtegaal, E. Kleymenov, J. Szlachetko, B. Neuhold and K. Hungerbuhler, Phys. Chem. Chem. Phys., 2012, 14, 2164–2170 RSC.
- Unpublished results.
- F. Mahdavi, T. C. Bruton and Y. Li, J. Org. Chem., 1993, 58, 744–746 CrossRef CAS.
- H. Tada, T. Ishida, A. Takao and S. Ito, Langmuir, 2004, 20, 7898–7900 CrossRef CAS.
- J. L. Ferry and W. H. Glaze, Langmuir, 1998, 14, 3551–3555 CrossRef CAS.
- (a) R. B. van Order and H. G. Lindwall, Chem. Rev., 1942, 30, 69–96 CrossRef CAS; (b) G. W. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 1045–1075 RSC; (c) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS; (d) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873–2920 CrossRef CAS; (e) V. V. Kouznetsov, L. Y. V. Méndez and C. M. M. Gómez, Curr. Org. Chem., 2005, 9, 141–161 CrossRef CAS; (f) S. Madapa, Z. Tusi and S. Batra, Curr. Org. Chem., 2008, 12, 1116–1183 CrossRef CAS; (g) J. A. Joule and K. Mills, Heterocyclic Chemistry, Wiley-Blackwell, 5th edn 2010 Search PubMed; (h) J. C. A. Flanagen, L. M. Dornan, M. G. McLaughlin, N. G. McCreanor, M. J. Cook and M. J. Muldoon, Green Chem., 2012, 14, 1281–1283 RSC.
- http://www.gold.org/ .
| This journal is © The Royal Society of Chemistry 2013 |