NHC-stabilized ruthenium nanoparticles as new catalysts for the hydrogenation of aromatics†
David Gonzalez-Galvez‡a, Patricia Lara‡bc, Orestes Rivada-Wheelaghand, Salvador Conejerod, Bruno Chaudrete, Karine Philippotbc and Piet W. N. M. van Leeuwen*a
aInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007-Tarragona, Spain. E-mail: pvanleeuwen@iciq.es
bLaboratoire de Chimie de Coordination, CNRS, LCC, 205 Route de Narbonne, F-31077 Toulouse, France
cUniversité de Toulouse, UPS, INPT, LCC, F-31077 Toulouse, France
dInstituto de Investigaciones Químicas (IIQ), CSIC and Universidad de Sevilla, Avenida Américo Vespucio 49, 41092 Isla de la Cartuja, Sevilla, Spain
eLPCNO, Laboratoire de Physique et Chimie de Nano-Objets, 135 Avenue de Rangueil, F-31077 Toulouse, France
First published on 16th October 2012
Abstract
The application of ruthenium nanoparticles (RuNPs) stabilized by the N-heterocyclic carbenes (NHC) N,N′-di(tert-butyl)imidazol-2-ylidene (ItBu) and 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene IPr as catalysts in the hydrogenation of several substrates is reported under various reaction conditions (solvent, substrate concentration, substrate/metal ratio, temperature). The RuNHC nanoparticles are active catalysts in the hydrogenation of aromatics and show an interesting ligand effect, RuIPr NPs being generally more active than RuItBu.
Introduction
Metal nanoparticles (MNPs) are widely used nowadays in very different research domains, such as optoelectronics, sensing, medicine, and catalysis.1–6 In particular the investigation of nanoparticles in catalysis is receiving an ever-increasing interest because MNPs present an efficient combination of the advantages of heterogeneous and homogeneous catalysts such as their small size and particular electronic configuration that make them extremely active catalysts. In comparison with traditional molecular complexes, they may catalyze reactions in which molecular systems are less active, such as arene hydrogenation,7 and they are extensively applied by the international scientific community for C–C coupling8,9 and hydrogenation reactions.10–14Metal nanoparticles may be stabilized by the addition of polymers, surfactants or ligands which allows controlling of their size, shape and dispersion and also their surface state. Consequently, an adequate choice of the stabilizer should allow the tuning of the surface properties and, therefore, the catalytic reactivity (activity and/or selectivity). This ligand influence has been recently examined by several research groups.15 Significant efforts have been made in the synthesis of ligand-stabilized nanoparticles to achieve control of their properties. 20 years ago, some of us started using a new strategy for the synthesis of metal NPs that involves the decomposition of an organometallic precursor under mild conditions of pressure and temperature. This methodology provides well-controlled NPs regarding size, dispersion, and shape, the control of which depends on the ligands used for their stabilization.16,17
RuNPs are interesting candidates to be applied in catalytic processes since (i) they can be characterized by different techniques including spectroscopic ones, NMR (due to the absence of a Knight shift) and IR that give information on their surface state and (ii) they are very active catalysts in arene hydrogenation. We have recently described for the first time the synthesis of N-heterocyclic carbenes (NHC)-stabilized RuNPs using the organometallic approach.18 The use of 13C labeled carbenes and also the addition of 13CO to the prepared NPs allowed us to prove the strong binding of the carbenes to the surface of the particles and also to demonstrate the influence of the structure of the carbenes on their location (mainly due to steric effects) and on their reactivity towards styrene hydrogenation.
In the present work, we aim to demonstrate the influence of the nature of NHC ligands attached to the surface of NPs on their catalytic activity in the hydrogenation of different substrates, extending the work recently reported with roof-shaped phosphine stabilized RuNPs.19
Results and discussion
The nanoparticles were prepared following a reported procedure,18 using the organometallic complex [Ru(COD)(COT)] (1,5-cyclooctadiene)(1,3,5-cyclooctatriene)ruthenium(0) as the metal source and the carbenes ItBu20 and IPr21 as stabilizers (Scheme 1). Under all reaction conditions, small and monodisperse nanoparticles are formed, with a mean size of 1.7 (0.2) nm for the particles prepared using 0.5 molar equiv. of ItBu per introduced ruthenium and 1.7 (0.2) and 1.5 (0.2) nm for the particles prepared using 0.2 and 0.5 molar equiv. of IPr, respectively. These nanomaterials, respectively, named colloids RuItBu0.5, RuIPr0.2 and RuIPr0.5 (Scheme 1 and Fig. S1 in ESI†), are extremely air-sensitive and immediately auto-ignite in the presence of oxygen.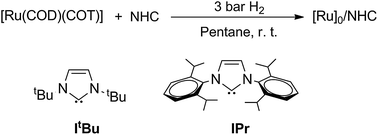 | ||
| Scheme 1 N-heterocyclic carbenes and reaction conditions used for the synthesis of RuNHCs nanoparticles. | ||
Initially, we investigated the hydrogenation of o-methylanisole (Scheme 2; 1o) using the different colloids prepared under various reaction conditions (solvent, concentration, substrate/metal ratio, temperature). We chose first this substrate as a model because its hydrogenation reaction has previously been studied in the presence of RuNPs stabilized with N-donor ligands [aminoalcohol, amino(oxazolines), hydroxy(oxazoline) and bis(oxazolines)],22 P-donor ligands (phosphines,19 and phosphites23,24), and surfactants.25 It has also been performed in the presence of other metal colloids such as Rh24,26,27 and Ir.24
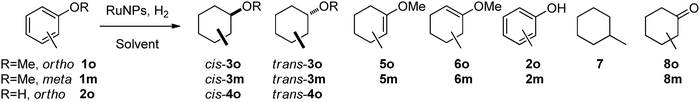 | ||
| Scheme 2 Methylanisoles, 1o and 1m, and o-cresol, 2o, hydrogenation. | ||
As can be seen in Scheme 2, this reaction can lead to a large range of products, from the desired hydrogenation products (3 for total and 5 and 6 for partial hydrogenation) to products derived from hydrogenolysis (2, 7 and 8). However, since chiral products are produced when complete and partial hydrogenation occurs, this reaction represents a good target for studying asymmetric hydrogenation as well.
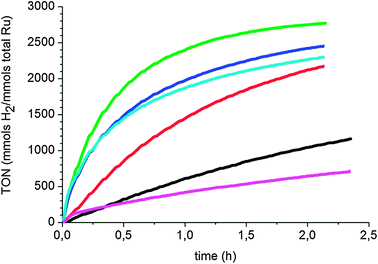
All colloids were tested in the hydrogenation of o-methylanisole, 1o (Fig. 1 and Table 1), and all show a different behavior. As can be seen in the graph, the sample containing RuItBu NPs, as a stabilizing agent, shows a very low rate compared to samples containing RuIPr NPs. The low activity of RuItBu0.5 compared to RuIPr NPs is surprising. Since excess ligand (0.5 equiv.) is necessary to stabilize these particles, it may arise from a high surface coverage when ItBu is used as the ligand. This does not prevent CO from accessing the particle as previously described18 but may hinder the coordination of arenes on the surface. Alternative explanations could be that aryl groups are needed in the ligands to obtain “active” materials, as was found in our study on phosphines;19 or a strong electronic effect, as ItBu is a more basic carbene, it can make the cluster surface electronically richer and avoid rich arenes coordination. RuIPr0.2 was found to exhibit the highest activity. This is in agreement with the observed stabilization of the nanoparticles by the IPr ligand at low concentration and the fact that the faces are readily available for this colloid as deduced from CO coordination reactions, while in RuIPr0.5 both apexes and faces are covered.18
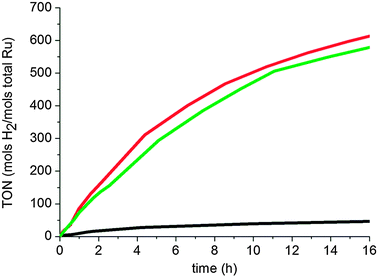 | ||
| Fig. 1 Hydrogenation of o-methylanisole (0.5 M in pentane) under 40 bar of H2 and 298 K in the presence of 0.3% Ru: RuItBu0.5 (black), RuIPr0.2 (red), RuIPr0.5 (green). | ||
| Catalyst | Solvent | t/h | T/K | Conv. (%) | Yield (%) | TONb | TOFc | ||
|---|---|---|---|---|---|---|---|---|---|
| 3o (de)a | 5o + 6o (ratio) | ||||||||
| a Diastereomeric excess between the cis and trans product, cis is always the major one.b Turnover calculated considering mol of H2 consumed per mol of total Ru.c TOF = mol H2 mol Ru−1 h−1. In brackets, initial TOF calculated after 15 min of reaction.d p(H2) = 8 bar. | |||||||||
| 1 | RuItBu0.5 | Pentane | 16 | 298 | 12 | 9 (90) | 2 (57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :43) :43) | 46 | 3 (9) |
| 2 | RuIPr0.2 | Pentane | 16 | 298 | 72 | 69 (88) | 2 (57:43) | 579 | 36 (86) |
| 3 | RuIPr0.5 | Pentane | 16 | 298 | 61 | 57 (88) | 2 (55:45) | 613 | 38 (73) |
| 4 | RuIPr0.2 | THF | 16 | 298 | 25 | 20 (93) | 5 (61:39) | 188 | 12 (38) |
| 5 | RuIPr0.2 | THF | 16 | 353 | 84 | 75 (86) | 7 (57:43) | 714 | 45 (163) |
| 6 | RuIPr0.2 | MeOH | 16 | 298 | 0 | 0 | 0 | 0 | 0 |
| 7 | RuIPr0.2 | — | 2.15 | 298 | 42 | 40 (89) | 2 (56:44) | 1163 | 541 (611–616) |
| 8 | RuIPr0.2 | — | 2.15 | 323 | 79 | 75 (88) | 3 (54:46) | 2176 | 1012 (1696) |
| 9 | RuIPr0.2 | — | 2.15 | 353 | 98 | 95 (85) | 1 (56:44) | 2771 | 1289 (5052) |
| 10 | RuIPr0.2 | — | 2.15 | 373 | 92 | 85 (87) | 6 (56:44) | 2458 | 1143 (4052) |
| 11 | RuIPr0.2 | — | 2.15 | 393 | 89 | 79 (87) | 8 (56:44) | 2303 | 1071 (4084) |
| 12 | RuIPr0.2 | — | 2.15 | 393–353 | 43 | 24 (90) | 6 (57:43) | 717 | 333 (732) |
| 13 | RuIPr0.2d | — | 2.15 | 298 | 13 | 10 (90) | 2 (57:43) | 310 | 135 (140) |
o-Methylanisole hydrogenation was performed in various solvents except those that can be hydrogenated (e.g. acetone, acetonitrile, toluene, etc.) to avoid undesired reactions, and a clear solvent effect can be observed (Fig. 2 and Table 1). The reaction did not work in protic solvents (such as methanol) and worked better in apolar solvents (such as pentane) than in polar non-protic solvents (such as THF). A similar tendency has previously been reported with diphosphite-stabilized RuNPs that showed a better reactivity in pentane than in THF in the hydrogenation of methyl anisole (o and m).23 Especially remarkable is the activity when assaying the reaction under neat conditions; despite the fact that the reactant polarity is similar to that of THF, high rates are obtained. This could be due to a concentration effect and substrate vs. solvent competitive coordination on the surface.
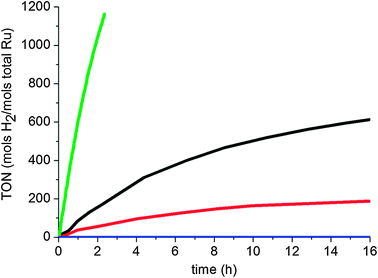 | ||
| Fig. 2 Hydrogenation of o-methylanisole (0.5 M in solvents) under 40 bar of H2 and 298 K in the presence of RuIPr0.2 (0.3% Ru): pentane (black), THF (red), methanol (blue), no-solvent (green, 0.1% Ru). | ||
Pressure and temperature effects were also studied. Hydrogen pressure affects the rate of the reaction, but not the selectivity (Table 1, entries 7 and 13). On the other hand, temperature has a strong effect on rates and selectivity (Fig. 3 and Table 1). The rate increases with temperature, achieving a maximum at approximately 353 K. Nevertheless, the reaction slows at temperatures above 373 K. To check the stability of the colloids with temperature, the RuNPs were heated at 393 K for one hour, cooled to 353 K and then pressurized with dihydrogen. Under these conditions, a much lower rate was observed. These results are consistent with deactivation of the colloids at temperatures higher than 373 K. Interestingly, although the rate is lower at high temperatures, the selectivity towards partial hydrogenation is higher. At 393 K and 20% conversion, over 60% selectivity towards partially hydrogenated products (5o and 6o) was obtained. When performing the reaction at 353 K and using THF as the solvent, also an increased selectivity towards alkenes was observed (Table 1, entries 4 and 5).
 | ||
| Fig. 3 Hydrogenation of o-methylanisole under 40 bar of H2 in the presence of RuIPr0.2 (0.1% Ru): 298 K (black), 323 K (red), 353 K (green), 373 K (dark-blue), 393 K (pale-blue), 353 K-pretreated at 393 K (pink). | ||
o-Cresol, 2o, o-methylanisole 1o and m-methylanisole 1m hydrogenations were studied under several conditions (Table 2). The following conclusions can be extracted from the obtained results: (i) the activity of NHC-stabilized RuNPs is very similar to that reported for phosphine-stabilized ones.19 (ii) These materials hydrogenate phenol derivatives with very good activity. (iii) THF is a better solvent for methylanisole hydrogenation, while cresol is hydrogenated faster in pentane. (iv) m-Methylanisole, 1m, is hydrogenated faster and with lower diastereomeric excess than 1o. (v) During hydrogenation of o-cresol, 46% of chiral ketone 8o is produced at low conversions, a result similar to the previous work by Roucoux and co-workers with RhNPs,28 but far from selectivity of over 99% obtained with Pd—NPs.29–31
| Substrate | Solvent | Ru (%) | Conv. (%) | Yield (%) | TONa | TOFb/h−1 | ||
|---|---|---|---|---|---|---|---|---|
| 3/4 (de) | 3 + 4/8 | |||||||
| a Turnover calculated considering mol of H2 consumed per mol of total Ru.b TOF = mol H2 mol Ru−1 h−1 calculated at the end of the experiment. | ||||||||
| 1 | 1o | Pentane | 0.3 | 100 | 100 (87) | 0 | 1024 | 64 |
| 2 | 1m | Pentane | 0.3 | 100 | 100 (60) | 0 | 1001 | 63 |
| 3 | 2o | Pentane | 0.3 | 100 | 99 (29) | 1 | 1024 | 64 |
| 4 | 1o | Pentane | 0.03 | 1 | 1 (—) | <1 | 79 | 5 |
| 5 | 1m | Pentane | 0.03 | 9 | 8 (65) | 1 | 822 | 51 |
| 6 | 2o | Pentane | 0.03 | 28 | 15 (35) | 13 | 2410 | 151 |
| 7 | 1o | THF | 0.3 | 100 | 100 (90) | 0 | 1024 | 64 |
| 8 | 1m | THF | 0.3 | 100 | 100 (59) | 0 | 1001 | 63 |
| 9 | 2o | THF | 0.3 | 100 | 99 (31) | <1 | 1005 | 63 |
| 10 | 1o | THF | 0.03 | 18 | 15 (92) | 3 | 1746 | 109 |
| 11 | 1m | THF | 0.03 | 22 | 21 (60) | 1 | 2163 | 135 |
| 12 | 2o | THF | 0.03 | 16 | 14 (35) | 2 | 1584 | 99 |
The RuNHC colloids were also investigated in benzene hydrogenation, and the results were similar to those obtained in o-methylanisole hydrogenation (Table 3), i.e. RuIPr0.2 presented the highest activity and the reaction was faster when performed neat. The most active nanomaterial, RuIPr0.2, was also tested in the hydrogenation of benzenes monosubstituted with electron-withdrawing or donating groups (Scheme 3). As previously found,32–35 substrates with electron donating groups are hydrogenated much faster than those with electron withdrawing groups (Table 4). Bromobenzene, 13, and benzonitrile, 15, were not hydrogenated at all, while nitrobenzene, 16, was selectively hydrogenated at the nitro group yielding aniline without altering the aromatic ring (no further hydrogenation is observed). Anisole, 11, shows the highest conversion. The good conversion of dimethylaniline, 10, demonstrates that these RuNPs are stable, active materials in the presence of coordinating groups. However, in the presence of 2% didodecyldisulfide (S/Ru = 14:1) benzene was not hydrogenated by any of these materials, which shows the sensitivity towards certain poisons.
| Catalyst | Solvent | Conv. (%) | TONa | TOFb | |
|---|---|---|---|---|---|
| a Turnover calculated considering mols of H2 consumed per mol of total Ru after 16 h.b TOF = mol H2 mol Ru−1 h−1. In brackets, initial TOF calculated after 15 min of reaction.c 50% v/v mixture. | |||||
| 1 | RuItBu0.5 | — | 66 | 703 | 44 (90) |
| 2 | RuIPr0.2 | — | 100 | 954 | 60 (421) |
| 3 | RuIPr0.5 | — | 93 | 973 | 61 (353) |
| 4 | RuIPr0.2 | THFc | 100 | 1061 | 66 (372) |
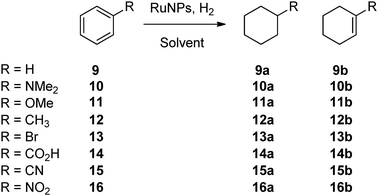 | ||
| Scheme 3 Hydrogenation of benzene derivatives. | ||
Acetophenone (17) was used as the substrate in order to compare the selectivity of the hydrogenation process when other functional groups (carbonyls) are present. As shown in Scheme 4 three possible reaction products can be formed: if the aromatic ring is first hydrogenated, cyclohexylmethylketone, 18, is obtained, while chiral (racemic) phenylethanol, 19, would be formed if the carbonyl group is hydrogenated first. Further hydrogenation of either 18 or 19 would finally produce cyclohexylethanol, 20. In addition, hydrogenolysis products can be obtained, such as ethylbenzene and ethylcyclohexane.
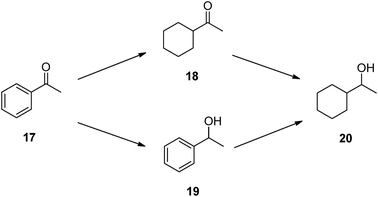 | ||
| Scheme 4 Acetophenone hydrogenation. | ||
The chemoselective reduction of aromatic ketones to benzylalcohols can be carried out effectively by direct hydrogenation and hydrogen transfer reactions, both employing homogeneous catalysts,36 but the selective hydrogenation of the aromatic ring is more complicated. This reaction has been studied in detail,28,32–34,37–51 usually producing a mixture of the three products when heterogeneous catalysts are used and the reaction is not complete.
The effect of several parameters on acetophenone hydrogenation activity and selectivity was studied. The rate of hydrogenation is much higher in THF than that in pentane or MeOH (see Fig. 4). In MeOH, hydrogenation of the aromatic ring stopped after 1 h of reaction time, and ketone hydrogenation after 6 h; very similar results were obtained in EtOH and iPrOH, which shows that the material is not active in alcohols with protons in the α position (2o hydrogenation means that it is stable in front of alcohols), this may be due to decomposition of such alcohols and deactivation of the catalyst by CO poisoning. In pentane, hydrogenation of both the aromatic ring and ketone proceeded slowly perhaps in that case because the product is anchored to the surface and pentane does not remove it properly. On the other hand, in THF, 18 was immediately produced together with some 19, and both disappeared slowly to produce fully hydrogenated 20 (see ESI† for detailed conversions and activities).
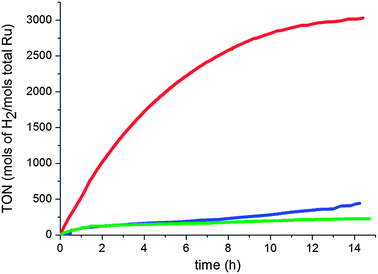 | ||
| Fig. 4 Hydrogenation of acetophenone at 298 K under 40 bar of H2 in the presence of RuIPr0.2 (0.1% Ru) in pentane (blue), THF (red), and MeOH (green). | ||
The effect of substrate/catalyst ratio on the selectivity was also studied (Fig. 5 and ESI†). The activity was higher when lower quantities of Ru were used. However, the conversion was much lower, and, independent of the ratio, the catalyst degraded with reaction time and favored ketone hydrogenation over arene hydrogenation. As we can observe in the selectivity graphs, selectivities of up to 80% were obtained using only 0.03% of total Ru, but with conversions under 50%. On the other hand, at higher Ru loading (0.1–0.3%), over 60% of 18 was obtained at almost full conversion of acetophenone.
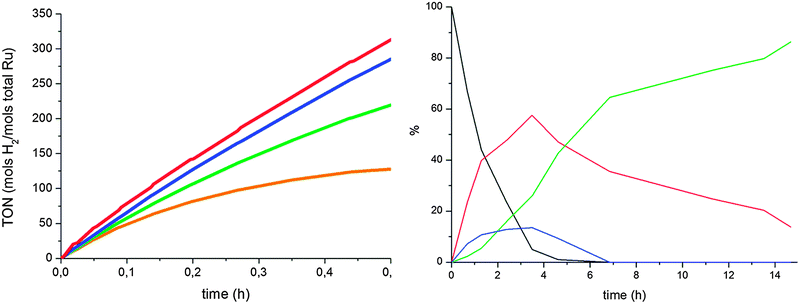 | ||
| Fig. 5 Hydrogenation of acetophenone (0.5 M in THF) at 298 K under 40 bar of H2 in the presence of RuIPr0.2. Left: activity, 1% Ru (brown), 0.3% (green), 0.1% (blue), 0.03% (red). Right: product evolution of 0.3% Ru, 17 (black), 18 (red), 19 (blue), 20 (green). | ||
Pressure did not affect the ketone hydrogenation rate, but it did affect the arene hydrogenation rate, with a low selectivity for 18 obtained at lower pressures (compare Fig. 5 (left) and Fig. 6 (left)). Increasing the temperature had a positive effect on the activity, but not on selectivity as nanomaterial degradation in THF occurs much faster. When the reaction was performed neat, the activity increased (TOF = 4000 h−1 at 353 K), but the selectivity was much lower (45% yield of product 18). This is probably due to a marked change in “solvent” polarity while the reaction proceeds, as alcohols are produced.
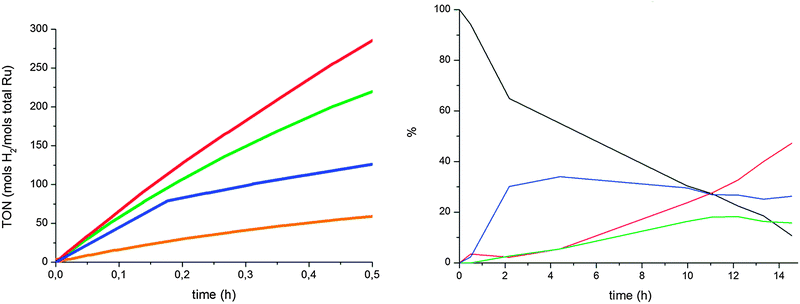 | ||
| Fig. 6 Left: hydrogenation of acetophenone (0.5 M in THF) at 298 K in the presence of RuIPr0.2: 0.1% Ru-40 bar (red), 0.1% Ru-10 bar (blue), 0.3% Ru-40 bar (green), 0.3% Ru-10 bar (brown). Right: product evolution of 0.3% Ru-10 bar, 17 (black), 18 (red), 19 (blue), 20 (green). | ||
Subsequently RuIPr0.2 was tested as a catalyst in the hydrogenation of other aromatic compounds containing ketones in the best solvent so far, THF (Scheme 5). Interestingly, we observed that as the length of the alkyl chain increases the selectivity toward hydrogenation of the arene fragment is enhanced. The selectivity was found over 98% at full conversion for 4-phenyl-2-butanone, 22, and 5-phenyl-2-pentanone, 23. These remarkable results show that nanoparticles may hydrogenate very selectively the arene groups in arylketones as a probable consequence of the thermodynamic preference for arene coordination over ketone coordination on ruthenium surfaces. In the case of acetophenone, it seems likely that the approach through the phenyl ring is also preferred as deduced from the faster production of 18 over 19. The lower selectivity could result from the possibility of concomitant hydrogenation of both the phenyl ring and the CO bond upon coordination via the phenyl ring.
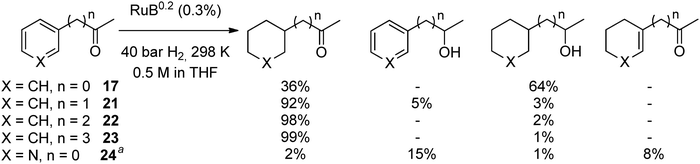 | ||
| Scheme 5 Hydrogenation of compounds containing an arene and a ketone (results at 100% conversion, except 24). a 353K, solvent free and 26% of conversion. | ||
3-Acetylpyridine, 24, was also hydrogenated (Fig. S10, ESI†). The pyridine derivative reacted more slowly than the benzene derivative, but still with a TOF >800 h−1 at 353 K and 40 bar of hydrogen (acetophenone TOF > 4000 h−1). In this case, a complex mixture of compounds was obtained, in which at 26% conversion, the major compounds were the ones derived from ketone hydrogenation (15%) and the one with α,β-saturated ketone (8%), with only 3% of products in which the pyridine ring was fully hydrogenated. This result is in concordance with those found in the literature.52,53
Conclusions
We have shown that RuNPs stabilized by the NHC ligands ItBu and IPr are active nanocatalysts for arene hydrogenation, the latter giving much more active catalysts. RuNPs-ItBu nanoparticles need a higher content of ligand for their stabilization. The presence of this excess ligand coordinated on the surface may lead to a slower catalytic system. The aromatic IPr ligand may be coordinated either on the corner, edges and defects18 at low concentration or additionally on the faces at higher concentration. The excess ligand is removed in both cases and the surface of the particle is accessible. It is likely that the large substituted arene groups interact weakly with the ruthenium surface and allow access of the arene substrates. There is a clear thermodynamic preference for arene coordination over ketone coordination which leads to an interesting selectivity in the case of arylketones. The case of acetophenone is particular perhaps because of its lack of flexibility and the possibility for acetophenone to have both phenyl and carbonyl groups reduced upon coordination to the nanoparticle which would lower the selectivity. The solvent has a major effect on the activity and the most suitable solvent varies with the substrate, probably as a function of the competition between solvent, substrate and product adsorption on the nanoparticle surface. During hydrogenation of ketones (e.g. acetophenone) the medium changes as alcohols are produced, which apparently changes the catalysts thus affecting catalysis.The above results show that ligands used in the preparation of RuNPs and solvents used in catalysis play a major role in determining the properties in hydrogenation reactions, as do ligands and solvents in homogeneous catalysts. A lot has to be learnt yet about how we can control the catalyst properties, but several trends are emerging. First, the presence of aromatic groups in the ligands leads to more stable catalysts. Second, the more electron-donating is the ligand the faster is the hydrogenation of aromatic compounds: NHC > PAlk2Ar ≫ PAr3, but the faster catalysts are also the least stable. The variety of selectivities and rates observed in our studies holds a promise for selective hydrogenation of substituted aromatics to valuable products.
Acknowledgements
The authors thank UPS-TEMSCAN for electron microscopy facilities. The authors also acknowledge financial support from CNRS and the European Union for ERC Advanced Grant (NANOSONWINGS 2009-246763). The Spanish Ministerio de Ciencia e Innovación (projects CTQ2010-17476 and CONSOLIDER-INGENIO 2010 CSD2007-00006, FEDER support) is acknowledged. O. R.-W. thanks the Spanish Ministerio de Ciencia e Innovación for a research grant.References
- Nanoscale Materials in Chemistry, ed. K. J. Klabunde and R. M. Richards, Wiley-Interscience, New York, 2001 Search PubMed.
- Metal Nanoparticles: Synthesis, Characterization and Applications, ed. D. L. Feldheim and C. A. Foss Jr., Marcel Dekker, New York, 2002 Search PubMed.
- G. Schmid, Clusters and Colloids. From Theory to Applications, Wiley VCH, Weinheim, 2004 Search PubMed.
- G. Schmid, Nanoparticles. From Theory to Application, Wiley VCH, Weinheim, 2004 Search PubMed.
- K. J. Klabunde and C. Mohs, Chemistry of Advanced Materials. An Overview, Wiley-VHC, New York, 1998 Search PubMed.
- A. Roucoux, Top. Organomet. Chem., 2005, 16, 261–279 CrossRef CAS.
- Nanoparticles and Catalysis, ed. D. Astruc, Wiley-Interscience, New York, 2008 Search PubMed.
- A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973–4985 RSC.
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757–3778 CrossRef CAS.
- E. Bayram, J. C. Linehan, J. L. Fulton, J. A. S. Roberts, N. K. Szymczak, T. D. Smurthwaite, S. Özkar, M. Balasubramanian and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 18889–18902 CrossRef CAS.
- M. Zahmakıran, Y. Tonbul and S. Özkar, J. Am. Chem. Soc., 2010, 132, 6541–6549 CrossRef.
- A. Gual, C. Godard, S. Castillon and C. Claver, Dalton Trans., 2010, 39, 11499–11512 RSC.
- E. Bayram, M. Zahmakıran, S. Özkar and R. G. Finke, Langmuir, 2010, 26, 12455–12464 CrossRef CAS.
- A. Roucoux and K. Philippot, in The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007, and citations therein Search PubMed.
- J. D. Scholten, B. C. Leal and J. Dupont, ACS Catal., 2012, 2, 184–200 CrossRef CAS and references therein.
- C. Pan, K. Pelzer, K. Philippot, B. Chaudret, F. Dassenoy, P. Lecante and M. J. Casanove, J. Am. Chem. Soc., 2001, 123, 7584–7593 CrossRef CAS.
- K. Philippot and B. Chaudret, C. R. Chim., 2003, 6, 1019–1034 CrossRef CAS.
- P. Lara, O. Rivada-Wheelaghan, S. Conejero, R. Poteau, K. Philippot and B. Chaudret, Angew. Chem., Int. Ed., 2011, 50, 12080–12084 CrossRef CAS.
- D. Gónzalez-Gálvez, P. Nolis, K. Philippot, B. Chaudret and P. W. N. M. van Leeuwen, ACS Catal., 2012, 2, 317–321 CrossRef.
- N. M. Scott, R. Dorta, E. D. Stevens, A. Correa, L. Cavallo and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 3516–3526 CrossRef CAS.
- L. Jafarpour, E. D. Stevens and S. P. Nolan, J. Organomet. Chem., 2000, 606, 49–54 CrossRef CAS.
- S. Jansat, D. Picurelli, K. Pelzer, K. Philippot, M. Gomez, G. Muller, P. Lecante and B. Chaudret, New J. Chem., 2006, 30, 115–122 RSC.
- A. Gual, M. R. Axet, K. Philippot, B. Chaudret, A. Denicourt-Nowicki, A. Roucoux, S. Castillon and C. Claver, Chem. Commun., 2008, 2759–2761 RSC.
- A. Gual, C. Godard, K. Philippot, B. Chaudret, A. Denicourt-Nowicki, A. Roucoux, S. Castillon and C. Claver, ChemSusChem, 2009, 2, 769–779 CrossRef CAS.
- A. Nowicki, V. Le Boulaire and A. Roucoux, Adv. Synth. Catal., 2007, 349, 2326–2330 CrossRef CAS.
- A. Roucoux, J. Schulz and H. Patin, Adv. Synth. Catal., 2003, 345, 222–229 CrossRef CAS.
- J. Schulz, A. Roucoux and H. Patin, Chem.–Eur. J., 2000, 6, 618–624 CrossRef CAS.
- A. Denicourt-Nowicki, B. Leger and A. Roucoux, Phys. Chem. Chem. Phys., 2011, 13, 13510–13517 RSC.
- Y. Z. Xiang, X. N. Li, C. S. Lu, L. Ma, J. F. Yuan and F. Feng, Ind. Eng. Chem. Res., 2011, 50, 3139–3144 CrossRef CAS.
- Y. Wang, J. Yao, H. R. Li, D. S. Su and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 2362–2365 CrossRef CAS.
- X. Yang, L. Du, S. J. Liao, Y. X. Li and H. Y. Song, Catal. Commun., 2012, 17, 29–33 CrossRef CAS.
- K. R. Janusklewick and H. Alper, Organometallics, 1983, 2, 1055–1057 CrossRef.
- G. S. Fonseca, A. P. Umpierre, P. F. Fichtner, S. R. Teixeira and J. Dupont, Chemistry, 2003, 9, 3263–3269 CrossRef CAS.
- Y. Motoyama, M. Takasaki, S. H. Yoon, I. Mochida and H. Nagashima, Org. Lett., 2009, 11, 5042–5045 CrossRef CAS.
- G. Falini, A. Gualandi and D. Savoia, Synthesis, 2009, 2440–2446 CAS.
- P. W. N. M. van Leeuwen, Homogeneous Catalysis—Understanding the Art, Kluwer Academic Publishers, Dordrecht (The Netherlands), 2004 Search PubMed.
- I. Bergault, P. Fouilloux, C. Joly-Vuillemin and H. Delmas, J. Catal., 1998, 175, 328–337 CrossRef CAS.
- F. Gelman, D. Avnir, H. Schumann and J. Blum, J. Mol. Catal. A: Chem., 2001, 171, 191–194 CrossRef CAS.
- M. Casagrande, L. Storaro, A. Talon, M. Lenarda, R. Frattini, E. Rodriguez-Castellon and P. Maireles-Torres, J. Mol. Catal. A: Chem., 2002, 188, 133–139 CrossRef CAS.
- S. Trofimenko, A. L. Rheingold and L. M. L. Sands, Inorg. Chem., 2002, 41, 1889–1896 CrossRef CAS.
- N. Eliau, D. Avnir, M. S. Eisen and J. Blum, J. Sol-Gel Sci. Technol., 2005, 35, 159–167 CrossRef CAS.
- M. Lenarda, M. Casagrande, E. Moretti, L. Storaro, R. Frattini and S. Polizzi, Catal. Lett., 2007, 114, 79–84 CrossRef CAS.
- N. A. Owston, A. J. Parker and J. M. Williams, Org. Lett., 2007, 9, 73–75 CrossRef CAS.
- I. S. Park, M. S. Kwon, K. Y. Kang, J. S. Lee and J. Park, Adv. Synth. Catal., 2007, 349, 2039–2047 CrossRef CAS.
- M. Ito, Y. Endo and T. Ikariya, Organometallics, 2008, 27, 6053–6055 CrossRef CAS.
- F. Jutz, J. M. Andanson and A. Baiker, J. Catal., 2009, 268, 356–366 CrossRef CAS.
- L. Y. Song, X. H. Li, H. N. Wang, H. H. Wu and P. Wu, Catal. Lett., 2009, 133, 63–69 CrossRef CAS.
- J. A. Anderson, A. Athawale, F. E. Imrie, F. M. McKenna, A. McCue, D. Molyneux, K. Power, M. Shand and R. P. K. Wells, J. Catal., 2010, 270, 9–15 CrossRef CAS.
- M. Jahjah, Y. Kihn, E. Teuma and M. Gomez, J. Mol. Catal. A: Chem., 2010, 332, 106–112 CrossRef CAS.
- B. Schimmoeller, F. Hoxha, T. Mallat, F. Krumeich, S. E. Pratsinis and A. Baiker, Appl. Catal., A, 2010, 374, 48–57 CrossRef CAS.
- D. Duraczynska, A. Drelinkiewicz, E. Bielanska, E. M. Serwicka and L. Litynska-Dobrzynska, Catal. Lett., 2011, 141, 83–94 CrossRef CAS.
- M. Freifelder, J. Org. Chem., 1964, 29, 2895–2898 CrossRef CAS.
- E. Wenkert, K. G. Dave, F. Haglid, R. G. Lewis, T. Oshi, R. V. Stevens and M. Terashim, J. Org. Chem., 1968, 33, 747–753 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20561k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |