 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modifications of the metal and support during the deactivation and regeneration of Au/C catalysts for the hydrochlorination of acetylene†
Marco
Conte
*a,
Catherine J.
Davies
a,
David J.
Morgan
a,
Thomas E.
Davies
a,
Albert F.
Carley
a,
Peter
Johnston
b and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: ConteM1@cardiff.ac.uk; Hutch@cardiff.ac.uk
bJohnson Matthey Catalysts, Orchard Road, Royston, Herts SG8 5HE, UK
First published on 7th August 2012
Abstract
The effect of the gold oxidation state and carbon structure on the activity of Au/C catalysts for the hydrochlorination of acetylene was investigated by a combined approach using TPR, XPS and porosimetry determinations. The activity of the catalyst in the synthesis of vinyl chloride monomer was found to be dependent on the presence of Au3+ species in the catalyst. However, by preparing catalysts with different Au3+ content it was possible to determine the existence of a threshold Au3+ amount, beyond which the excess of Au3+ was not active for the reaction. This was explained by the existence of active sites at the Au/C interface, and not just by the presence of Au3+ species on top of Au nanoparticles, as explained by current models for these catalysts. It was also possible to determine the existence of a subset of Au nanoclusters which do not take part in the reaction, as well as changes in the textural properties of the carbon that can affect its long term reusability.
1. Introduction
The hydrochlorination of acetylene using HCl is an important industrial route to the manufacture of vinyl chloride monomer (VCM), which is the building block for polyvinyl chloride. It was previously shown that Au/C catalysts can be the best catalysts to carry out this reaction using the direct acetylene hydrochlorination route.1–3 However, the catalyst was found to deactivate, possibly by reduction of Au3+ species to Au0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4,5 when the reaction is carried out in the 120–180 °C temperature range, and by oligomer formation over its surface when the reaction is carried out in the 60–100 °C temperature range.6 At present, most VCM is obtained by an oxychlorination reaction using ethylene, O2 and Cl2 as reactants.7 Due to the current increase in energy cost, coal-based derived feedstocks, like acetylene, are now becoming economically advantageous. Current industrial catalysts for the direct hydrochlorination reaction make use of mercuric chloride, and the efficiency of this VCM manufacture route is severely affected by the volatility of this metal chlorinated salt. These factors prompted us to explore the nature of the active species present over Au/C catalysts with the aim of identifying which of them are responsible for the catalytic activity. Previous studies showed that it was possible to regenerate the Au catalyst using Cl2 and NO.6 Alternatively, it was possible to demonstrate that an off-line treatment with aqua regia was also an effective regeneration method.8 However in all these cases the attention was on the sole change of the gold oxidation state without considering possible modifications of the support, as well as postulating Au3+ species on top of Au nanoparticles as active sites for the reaction,4 without providing clear insight into the possible correlation between the presence of an excess amount of Au3+ and the catalytic activity.
4,5 when the reaction is carried out in the 120–180 °C temperature range, and by oligomer formation over its surface when the reaction is carried out in the 60–100 °C temperature range.6 At present, most VCM is obtained by an oxychlorination reaction using ethylene, O2 and Cl2 as reactants.7 Due to the current increase in energy cost, coal-based derived feedstocks, like acetylene, are now becoming economically advantageous. Current industrial catalysts for the direct hydrochlorination reaction make use of mercuric chloride, and the efficiency of this VCM manufacture route is severely affected by the volatility of this metal chlorinated salt. These factors prompted us to explore the nature of the active species present over Au/C catalysts with the aim of identifying which of them are responsible for the catalytic activity. Previous studies showed that it was possible to regenerate the Au catalyst using Cl2 and NO.6 Alternatively, it was possible to demonstrate that an off-line treatment with aqua regia was also an effective regeneration method.8 However in all these cases the attention was on the sole change of the gold oxidation state without considering possible modifications of the support, as well as postulating Au3+ species on top of Au nanoparticles as active sites for the reaction,4 without providing clear insight into the possible correlation between the presence of an excess amount of Au3+ and the catalytic activity.
This prompted us to investigate the Au/C catalysts by a series of sequential treatments using H2 as a reducing agent and aqua regia as an oxidizing agent, in order to identify the role of Au3+ and Au0 in the nanoparticles deposited over the carbon support with respect to catalytic activity. The catalysts were therefore investigated by means of high resolution X-ray photoelectron spectroscopy complemented with temperature programmed reduction analysis, the latter being a bulk technique capable of quantifying oxidized gold species and changes in the carbon functional groups that could also affect the final reactivity of the catalyst,9 in the presence of active gold centers at the Au/C interface. In addition, porosimetry determinations were also carried out, as this is proven to be a useful tool for the determination of carbon textural properties.10 In fact, an aspect so far neglected in previous studies is the influence of oxidizing acids in the impregnating mixture that was used to prepare the catalyst. This could affect the formation of oxygenated species over the carbon surface11 as well as the pore structure of the carbon,12 with possible effects on the reusability of this material.
As Au/C catalysts are widely used for reactions other than hydrochlorination e.g. partial oxidation of alcohols13,14 and hydrocarbons,15,16 we foresee that this work may have a broader application to catalyst design for such reactions.
2. Experimental
2.1 Catalyst preparation
All the catalysts were prepared by a wet impregnation method. The gold precursor, HAuCl4·xH2O (Alfa Aesar, 40 mg, assay 49%), was dissolved in aqua regia (3:1 HCl (Fisher, 32%):HNO3 (Fisher, 70%) by volume, 5.4 ml) and the solution added dropwise with stirring to the activated carbon support (Norit ROX 0.8) (1.98 g) in order to obtain a catalyst with a final metal loading of 1 wt%. Stirring was continued at ambient temperature until NOx production subsided, approximately 10 minutes. The product was dried for 16 h at 140 °C and used as a catalyst.
Catalyst oxidation was carried out by stirring the material in a minimum amount of aqua regia to obtain a slurry at room temperature for 10 min (i.e. until NOx production subsided) followed by drying overnight at 140 °C.
Due to the nature of the catalysts preparation procedure used, wet impregnation,17 no filtration of the carbon or catalyst washing was carried out, and the metal loading should be considered as equal to the nominal amount of metal impregnated into the support.18
2.2 Catalytic tests and characterization of the products
Catalysts were tested for acetylene hydrochlorination in a fixed-bed glass microreactor. Acetylene (5 mL min−1, 0.5 bar) and HCl (6 mL min−1, 1 bar) were fed through a mixing vessel and a preheater (70 °C), and further mixed in a N2 flow (10 mL min−1, 1 bar) via calibrated mass flow controllers in a heated glass reactor containing catalyst (200 mg), with a total GHSV of 740 h−1. A reaction temperature of 180 °C was chosen, and blank tests using an empty reactor filled with quartz wool did not reveal any catalytic activity, even at 250 °C with the reactants under these flow conditions, SiC (2 × 2.5 g) was used to extend the bed length, above and below the catalyst itself, separated by quartz wool. The pressure of the reactants, HCl, C2H2 and N2, was chosen both for safety reasons and to test the catalyst under mild conditions. The gas phase products were analyzed on-line by GC using a Varian 450GC equipped with a flame ionisation detector (FID). Chromatographic separation and identification of the products were carried out using a Porapak N packed column (6 ft × 1/8′′ stainless steel).2.3 Characterization of the catalyst
Peak integration was carried out using SpecView Software. The thermogram was subjected to baseline correction and the area integrated using a cumulative counts algorithm (Fig. S1, ESI†).
CO2 from decarboxylation reactions was qualitatively identified by heating the carbon in a He/H2 atmosphere and collecting the effluent gases in a saturated solution of BaCl2·2H2O, the formation of a BaCO3 precipitate was detected.
3. Results and discussion
3.1 Correlation between the catalytic activity and gold oxidation state by TPR determinations
In order to verify the effect of oxidized gold species on Au/C catalysts, a catalytic test was carried out with a fresh catalyst (1 wt% Au) and with this material reduced in a H2 atmosphere prior to the hydrochlorination reaction (Fig. 1). The fresh catalyst displayed a conversion to vinyl chloride monomer of ca. 60% with a selectivity virtually of 100% to VCM, with trace amounts (<0.1%) of 1,2-dichloroethane and chlorinated oligomers only.4,6 In contrast, for the catalyst pre-treated in H2 the activity was limited to only ca. 10% conversion.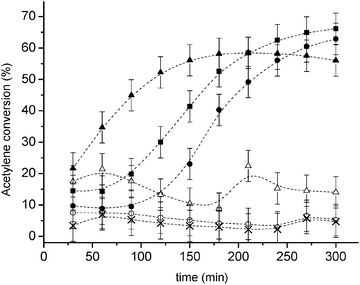 | ||
| Fig. 1 Acetylene conversion over Au/C (1 wt%) catalysts subject to reduction and oxidation cycles, (■) fresh catalyst, (○) reduced catalyst, (●) first catalyst oxidation, (Δ) second catalyst reduction, (▲) second catalyst oxidation, (×) third catalyst reduction. | ||
In order to assess the differences in the Au species for the fresh and the pre-reduced catalyst, temperature programmed reduction (TPR) was used (Fig. 2). It is known that Au3+ can reduce in the range of 120–180 °C, and that the catalyst can form oligomers over its surface in the temperature range of 60–100 °C.6 However, the analyses carried out to date were all focused on pure changes of the oxidation state of the metal. In contrast, by means of TPR we can also probe changes in the carbon support matrix, and therefore acquire a more accurate picture of the catalyst requirements to be active in the case of metal–support interaction. For our catalysts, the fresh Au/C material presents a characteristic reduction band between 230 and 300 °C (with center at 267 °C, Fig. 2 and Fig. S2, ESI†), which is diagnostic of Au3+.22 Comparison of the TCD signal with a standard permits an estimate of the Au3+ amount to be ca. 26% of the total Au loading. In contrast, TPR of the pre-reduced catalyst showed that no Au3+ species remained as this reduction band was absent. Control tests with the carbon support without Au showed that the activity of the pre-reduced Au/C catalyst was identical to that of the support. This residual activity of the carbon can arise from the presence of trace amounts of K+ and Al3+ in the carbon matrix, as these metals can display some activity to the hydrochlorination reaction of acetylene.23 These impurities were identified using SEM-EDX (see ESI†, Fig. S3 and S4) and the following catalyst atomic composition was determined: C 86.44%, O 9.69%, Na 0.11%, Al 0.05%, Si 0.13%, S 0.22%, Cl 2.34%, K 0.04% and Au 1.04%.
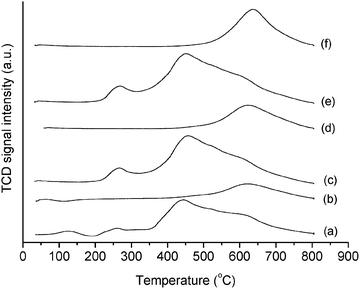 | ||
| Fig. 2 TPR profiles of Au/C catalysts subject to reduction and oxidation cycles, (a) fresh catalyst, (b) reduced catalyst, (c) first catalyst oxidation, (d) second catalyst reduction, (e) second catalyst oxidation, (f) third catalyst reduction. | ||
In view of this, and to evaluate systematically the effect of Au3+ centres on the catalytic activity, the catalyst was treated off-line with aqua regia.8 We have previously demonstrated that a short aqua regia treatment of the catalyst can regenerate the catalytic activity without loss of gold metal (as determined by atomic absorption spectroscopy). This was the case also for the Norit carbon support used in the present study, and the catalytic activity was recovered (Fig. 2). More importantly, if the catalyst was subjected to a reduction treatment the catalytic activity was again lost, but if the catalyst was re-oxidized it was able to recover its original activity without any apparent conversion loss when compared to the fresh catalyst.
Using TPR we were able to monitor the amount of Au3+ present at each step of the reduction–oxidation cycles, and it is clearly possible to observe that at every oxidation Au3+ is restored, and catalytic activity is present, while at each reduction Au3+ is absent, and negligible catalytic activity is detected accordingly (Fig. 2). This is an important result, because it shows a high degree of reversibility of the Au3+–Au0 system, as well as it implies that the catalyst can support a high number of regeneration by sequential oxidation–reduction, and this could be extremely useful in an industrial context.
An important aspect, in the use of TPR tools for the analysis of the catalysts reported in this study, is that they allow the determination of not just the gold oxidation state, and quantification of the bulk Au3+ amount, but also changes in structural groups present in the carbon matrix. In fact, it should be stressed that activated carbon is not a matrix constituted of carbon only as an element, but it has a complex structure characterized by the presence of oxygen in the form of carboxylic, ester, ether and lactone groups12,24 as well as heteroatoms e.g. phosphorus nitrogen or sulphur, present as phosphates, amines and thiols.25
This was evident from careful analysis of the TPR profiles, where the oxidation step in the presence of aqua regia not only regenerated Au3+, but also effected the oxidation of the carbon support (Fig. 2). From the thermograms (Fig. 2), it is inferred that bands in the range from 450 to 800 °C are assigned to decarboxylation reactions of oxygenated carbon functional groups at the carbon surface, which releases CO and CO2 with concomitant reduction of oxygenated groups by H2 in the TPR stream. More specifically, decarboxylation and reduction of oxygenated functional groups in the range of 400–650 °C are attributed to carboxylic acids and carboxylic anhydrides,26 while bands above 650 °C are usually assigned to lactones and phenols.27 Therefore while the pre-treatment of the catalyst in H2 also reduced the surface carboxylic groups on the carbon, the treatment with aqua regia is capable of inducing carboxyl functionality of the carbon surface as well as Au3+ recovery. This effect on the carbon matrix should be considered a consequence of the presence of HNO3 in the regeneration step. In fact, this acid is known to induce modifications on the carbon surface inducing oxygenated functional groups,28 as well as changes in the textural properties of the carbon support24 and this was confirmed by XPS and porosimetry (this will be discussed subsequently in Sections 3.2 and 3.3).
It is worth noting that previous studies on Au/C catalysts for hydrochlorination4,29 showed a monotonic deactivation trend ascribed to the reduction of Au3+ to Au0 and not an activity trend with enhanced activity per time on stream, like the one we are detecting in the current study. On the other hand, for the catalyst reported in the present study, XRPD patterns (Fig. S5, ESI†) permit an estimate of the mean particle size to be around 20 nm while in previous cases catalysts having an average particle size in the range of 4–5 nm were used, and this could affect the observed trend. Additional tests on the differences in drying temperature that could affect the different particle size and the metal/support interface were also carried out. Two catalysts were prepared at a drying temperature of 110 and 180 °C (Fig. S6, ESI†). The latter was chosen because it is the same as the reaction temperature, in order to evaluate if the conversion trend could be affected by the temperature itself. The catalyst dried at 180 °C has a similar trend to those dried at 110 °C, in contrast, the one dried at 140 °C is markedly different with an increase in activity from ca. 10% at the beginning of the reaction to up to 70% after 5 h of time on stream. It is possible that modifications of the Au/C interface, also induced by the particle size, are responsible for the observed phenomenon. XRPD patterns for the set of catalysts dried at 110, 140 and 180 °C (Fig. S5, ESI†) showed a particle size distribution centered at <2 nm, 20 and 3 nm, respectively, suggesting that particles that are too small could not be active for the reaction. On the other hand, previous studies showed that under reaction conditions, the catalyst can be affected by a small, yet detectable, degree of sintering.29
3.2 X-ray photoelectron spectroscopy and particle size distribution
In order to correlate bulk changes of the catalyst structure with the surface of gold nanoparticles, X-ray photoelectron spectroscopy (XPS) was systematically carried out for all these samples (Fig. 4 and Fig. S9 to S15, ESI†). It should be noted that where more than one Au species was evident, curve fitting was employed to determine the ratio of each species. As shown in Table 1, the XPS results confirm the TPR measurements and indicate that the deactivated catalysts contain Au0 only. Similarly, for the active catalysts, the Au3+ content was found to follow the same general trend obtained from TPR. Quantification of the Au3+ content reveals a fluctuation between ca. 12 and 25% of the overall gold content after regeneration (Table 1 and Table S1, ESI†), however regardless of the overall amount all catalysts are capable of reaching similar conversion values of ca. 60%. This result may further indicate that it is not just the presence of Au3+ which is responsible for the observed reactivity, but also the location of such species (the Au3+ amount obtained by XPS are always lower than those obtained by TPR as the former is a surface method, while the latter a bulk technique). The observed increase in surface oxygen content (Table 2) after each regeneration cycle is a consequence of the oxidising effect of the nitric acid in the aqua regia regeneration medium24,30 and could be responsible for stabilising the high Au3+ oxidation state, particularly as there is no decrease in the surface oxygen content of the reduced samples, together with the absence of any Au3+ species.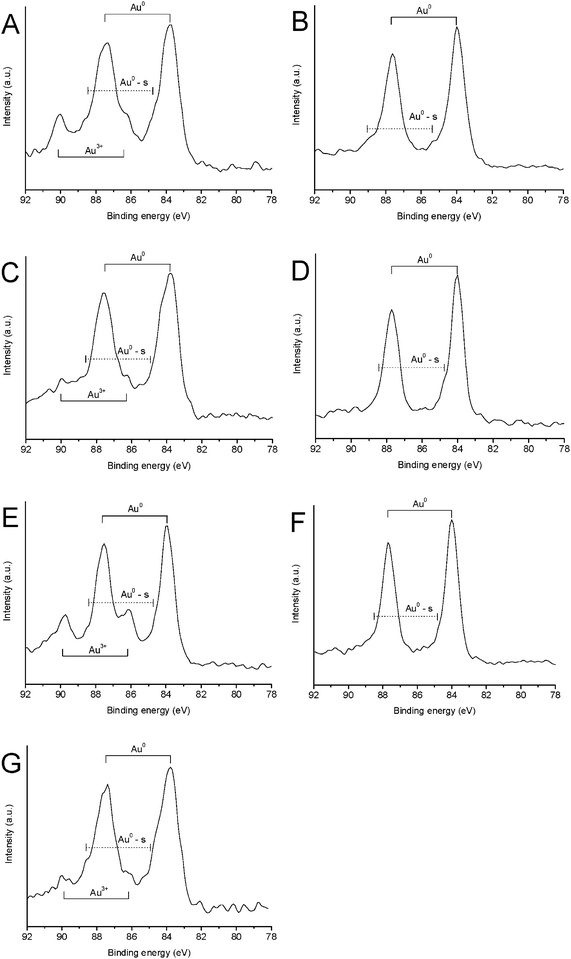 | ||
| Fig. 4 XPS profiles of Au/C catalysts subject to reduction and oxidation cycles, (A) fresh catalyst, (B) reduced catalyst, (C) first catalyst oxidation, (D) second catalyst reduction, (E) second catalyst oxidation, (F) third catalyst reduction and (G) third catalyst oxidation. | ||
| Catalyst treatment | Au species (%) | Binding energies (eV) | ||||
|---|---|---|---|---|---|---|
| Au3+ | Au0 | Au0-s | Au3+ | Au0 | Au0-s | |
| Fresh catalyst | 20.5 | 65.5 | 14.0 | 86.4 | 83.8 | 84.9 |
| 1st reduction | 0 | 87.1 | 12.9 | — | 83.9 | 85.4 |
| 1st oxidation | 13.1 | 75.2 | 11.7 | 86.3 | 83.9 | 85.2 |
| 2nd reduction | 0 | 89.5 | 10.6 | — | 84.0 | 85.1 |
| 2nd oxidation | 24.5 | 67.4 | 8.1 | 86.2 | 83.9 | 85.1 |
| 3rd reduction | 0 | 90.0 | 10.0 | — | 84.0 | 85.3 |
| 3rd oxidation | 12.3 | 70.2 | 17.5 | 86.2 | 83.8 | 84.8 |
| Catalyst treatment | Surface atomic composition (%) | ||||
|---|---|---|---|---|---|
| Au 4f | C 1s | Cl 2p | Na 1s | O 1s | |
| Fresh catalyst | 0.08 | 93.30 | 0.80 | 0.00 | 5.81 |
| 1st reduction | 0.07 | 96.21 | 0.32 | 0.33 | 3.06 |
| 1st oxidation | 0.06 | 93.27 | 1.20 | 0.00 | 5.48 |
| 2nd reduction | 0.04 | 97.42 | 0.13 | 0.18 | 2.23 |
| 2nd oxidation | 0.06 | 94.53 | 1.08 | 0.40 | 3.93 |
| 3rd reduction | 0.04 | 97.25 | 0.13 | 0.05 | 2.54 |
| 3rd oxidation | 0.04 | 92.58 | 1.26 | 0.08 | 6.04 |
However, the most important feature of the XPS spectra is possibly that in the preparation of these catalysts small metallic gold clusters (hereafter labelled Au0-s) are also formed, the binding energy of which is ca. 1 eV higher than the Au(4f) binding energy for the majority of the Au0 species31,32 (Table 2 and Fig. 4). As we as well as other researchers20 have previously observed, Au+ will also reduce to Au0; which is not observed by prolonged X-ray analysis here. However, we consider the Au0-s species to be nothing more than a spectator species, since the reduced catalysts also contain these Au0-s nanoclusters and are inactive, although small gold clusters are active in a range of reactions such as CO oxidation.33
Therefore, considering the XPS and XRD reported in this study, as well as previous work, these data could suggest a particle size range for the particles activity from 529 to 20 nm.
It should also be underlined that the apparent decrease in Au content detected from XPS analysis is an effect induced by pore changes in the carbon structure by nitric acid,16,17 and therefore gold particles are present also inside the carbon pores and not just at the surface of these. In fact when SEM-EDX was used in view of the major penetration depth of this method a composition close to 1% was detected.
3.3 Textural properties of the Au/C catalysts
To corroborate these experimental results, porosimetry determinations were carried out on an untreated carbon support, a fresh catalyst and an catalyst after oxidation treatment with aqua regia.Adsorption of N2 resulted in the formation of a type 1 isotherm typical of microporous material (Fig. S16 and S17, ESI†). The narrow pore size distribution curves we obtained are indicative of uniform pore channels in the small mesopore to micropore region. Application of the BJH method shows that the average pore size is in the range of 2–3 nm but given the high N2 uptake at P/Po < 0.2 the material likely occupies the microporous to mesoporous range (1–2 nm). The untreated carbon has the lowest surface area of 1153 m2 g−1 and the lowest total pore size volume of 0.5318 cm3 g−1 (Table 3). Treatment of the catalyst with acid appears to decrease microporosity indicated by the decrease in the Smic and an increase in Sext, which accounts for the external surface area and mesoporous contribution. The microporosity of the catalyst is further decreased by oxidation. It has been shown previously10,24 that oxidation and acid treatment of activated carbon result in a decrease in the microporous volume due to the formation of surface oxides at the micropore entrance. It was also shown that strong oxidation can result in a total loss of surface area due to destruction of the pore walls and network.12 The blocking of micropores as a result of Au addition should also be considered. These factors have been so far fully neglected in the acetylene hydrochlorination reaction, and they suggest that they could be important for the long term usability of this catalyst.
| Sample | S BET (m2 g−1) | S micro (m2 g−1) | S ext (m2 g−1) | V tot (cm−3 g−1) | V mic (cm−3 g−1) | D (nm) |
|---|---|---|---|---|---|---|
| Carbon (Norit ROX0.8) | 1153 | 938 | 215 | 0.5318 | 0.4859 | 2.2 |
| Au/C fresh | 1181 | 838 | 343 | 0.5391 | 0.4275 | 2.3 |
| Au/C oxidised | 1213 | 827 | 385 | 0.5442 | 0.4251 | 2.4 |
4. Conclusions
It has been confirmed that the activity of Au/C catalysts for the hydrochlorination reaction is invariably associated with the presence of Au3+ species, although our data suggest that the final activity can also be a consequence of the Au3+ location, and not just its concentration, as an excess amount of Au3+ does not contribute to the reaction, with important implications in the catalyst design. In addition, the current work provides insight into the active particle size distribution for this catalyst for this reaction, showing that Au nanoclusters are not active for this reaction. This will have important implications in the catalyst preparation targeted to obtain nanoparticles >1 nm and possibly centered at the size range of 5–20 nm. The catalysts also showed a large extent of reusability, although changes in textural properties of the carbon were detected.Acknowledgements
The authors thank Johnson Matthey Plc and World Gold Council for financial support.References
- G. J. Hutchings, J. Catal., 1985, 96, 292–295 CrossRef CAS.
- B. Nkosi, N. J. Coville and G. J. Hutchings, J. Chem. Soc., Chem. Commun., 1988, 71–72 RSC.
- G. J. Hutchings, Gold Bull., 1996, 29, 123–130 CrossRef CAS.
- M. Conte, A. F. Carley, C. Heirene, D. J. Willock, P. Johnston, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2007, 250, 231–239 CrossRef CAS.
- B. Nkosi, N. J. Coville, G. J. Hutchings, M. D. Adams, J. Friedl and F. E. Wagner, J. Catal., 1991, 128, 366–377 CrossRef CAS.
- B. Nkosi, M. D. Adams, N. J. Coville and G. J. Hutchings, J. Catal., 1991, 128, 378–386 CrossRef CAS.
- I. M. Clegg and R. Hardman, US Patent 5763710, 1998 Search PubMed.
- M. Conte, A. F. Carley and G. J. Hutchings, Catal. Lett., 2008, 124, 165–167 CrossRef CAS.
- P. Arnoldy, E. M. van Oers, O. S. L. Bruinsma, V. H. J. de Beer and J. A. Moulijn, J. Catal., 2985, 93, 231–245 CrossRef.
- N. Zhang, L.-Y. Wang, H. Liu and Q.-K. Cai, Surf. Interface Anal., 2008, 40, 1190–1194 CrossRef CAS.
- H. Tamon and M. Okazaki, Carbon, 1996, 34, 741–746 CrossRef CAS.
- C. Moreno-Castilla, M. A. Ferro-Garcia, J. P. Joly, I. Bautista-Toledo, F. Carrasco-Marin and J. Rivera-Utrilla, Langmuir, 1996, 11, 4386–4392 CrossRef.
- A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657–1712 CrossRef CAS.
- A. Villa, G. M. Veith and L. Prati, Angew. Chem., Int. Ed., 2010, 49, 4499–4502 CrossRef CAS.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS.
- M. D. Hughes, Y.-J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132–1135 CrossRef CAS.
- Supported Metals in Catalysis, ed. J. A. Anderson and M. F. García, Imperial College Press, London, 2005, p. 3 Search PubMed.
- Preparation of Catalysts II, ed. B. Delmon, P. Grange, P. Jacobs and G. Poncelet, Elsevier, Amsterdam, 1979, p. 235 Search PubMed.
- P. Kurr, I. Kasatkin, F. Girgsdies, A. Trunschke, R. Schlögl and T. Ressler, Appl. Catal., A, 2008, 348, 153–164 CrossRef CAS.
- Y. Fong, B. R. Visser, J. R. Gascooke, B. C. C. Cowie, L. Thomsen, G. F. Metha, M. A. Buntine and H. H. Harris, Langmuir, 2011, 27, 8099–8104 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- C. Baatz, N. Decker and U. Prüße, J. Catal., 2008, 258, 165–169 CrossRef CAS.
- K. Shinoda, Chem. Lett., 1975, 219–220 CrossRef CAS.
- M. Gurrath, T. Kuretzky, H. P. Boehm, L. B. Okhlopkova, A. S. Lisitsyn and V. A. Likholobov, Carbon, 2000, 38, 1241–1255 CrossRef CAS.
- J. K. Brennan, T. J. Bandosz, K. T. Thomson and K. E. Gubbins, Colloids Surf., A, 2001, 187–188, 539–568 CrossRef CAS.
- K. Dumbuya, G. Cabailh, R. Lazzari, J. Jupille, L. Ringel, M. Pistor, O. Lytken, H.-P. Steinrück and J. M. Gottfried, Catal. Today, 2012, 181, 20–25 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS.
- S. R. de Miguel, O. A. Scelza, M. C. Román-Martínez, C. Salinas-Martínez de Lecea, D. Cazorla-Amorós and A. Linares-Solano, Appl. Catal., A, 1998, 170, 93–103 CrossRef CAS.
- M. Conte, A. F. Carley, G. Attard, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2008, 257, 190–198 CrossRef CAS.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfão, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- C. N. R. Rao, V. Vijayakrishnan, H. N. Aiyer, G. U. Kulkarni and G. N. Subbanna, J. Phys. Chem., 1993, 97, 11157–11160 CrossRef CAS.
- T. V. Choudhary and D. W. Goodman, Top. Catal., 2002, 21, 25–34 CrossRef CAS.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Catalyst testing, and characterization by XRD, SEM, XPS and porosimetry. See DOI: 10.1039/c2cy20478a |
| This journal is © The Royal Society of Chemistry 2013 |