Catalytic chemical transformations with conformationally dynamic catalytic systems
Received 23rd April 2012, Accepted 31st July 2012
First published on 4th September 2012
Abstract
Proteins, RNA, and other functional biomacromolecules are sophisticated chemical entities that have evolved over billions of years. Their catalytic, constitutional, and communicating functions are intimately related to their three-dimensional molecular architecture and are exquisitely regulated in a spatiotemporal manner by conformational changes through allosteric regulators and a myriad of reversible and unidirectional posttranslational modifications. Their organized functional diversity is the key to the orderly and timely progression of complicated biological events. The main focus in the development of artificial catalysts is to accelerate a specific chemical transformation of interest, and extensive efforts have been devoted to fine-tuning catalytic activity. Here, we review recent developments of multistate catalysts able to perform distinct catalytic functions, with emphasis on the tight link between the structural modification of the conformationally flexible catalysts and the functional change. Although this field is still in its infancy with a simple output, continued advances will allow us to create intriguing systems with organized multitask output in an intricate molecular ensemble.
 Naoya Kumagai | Naoya Kumagai was born in 1978 and raised in Ibaraki, Japan. After he received PhD in Pharmaceutical Sciences at the University of Tokyo in 2005 under the supervision of Prof. Masakatsu Shibasaki, he pursued postdoctoral study in the laboratory of Prof. Stuart L. Schreiber at Harvard University in 2005–2006. He moved to Prof. Shibasaki’s group at the University of Tokyo as an assistant professor in 2006. He is currently a senior researcher at Institute of Microbial Chemistry, Tokyo. He has received the Pharmaceutical Society of Japan Award for Young Scientists in 2010. His research interest is in the development of new methodology in asymmetric catalysis and its application to the bioinspired dynamic processes. |
 Masakatsu Shibasaki | Masakatsu Shibasaki received his PhD from the University of Tokyo in 1974 under the direction of the late Professor Shun-ichi Yamada before doing postdoctoral studies with Professor E. J. Corey at Harvard University. In 1977 he returned to Japan and joined Teikyo University as an associate professor. In 1983 he moved to Sagami Chemical Research Center as a group leader, and in 1986 took up a professorship at Hokkaido University, before returning to the University of Tokyo as a professor in 1991. Currently, He is a director of Institute of Microbial Chemistry, Tokyo. He has received Fluka Prize (Reagent of the Year, 1996), the Elsevier Award for Inventiveness in Organic Chemistry (1998), the Pharmaceutical Society of Japan Award (1999), ACS Award (Arthur C. Cope Senior Scholar Award) (2002), the National Prize of Purple Ribbon (2003), Japan Academy Prize (2005), the Rare Earth Society of Japan Award, (2006), ACS Award for Creative Work in Synthetic Organic Chemistry (2008), Centenary Medal and Lectureship (2008), Prelog Award Medal (2008), Special Award, the Society of Synthetic Organic Chemistry, Japan (2010) and many others. His research interests include asymmetric catalysis and medicinal chemistry of biologically significant compounds. |
1. Introduction
Catalysis is a central concept in general chemistry, appearing in virtually all chemistry disciplines such as organic chemistry, inorganic chemistry, biochemistry, and physical chemistry.1 Because chemistry is the art of transformation of various molecular entities via precise manipulation of chemical bonds, catalysis continues to attract sustained interest from both theoretical and practical viewpoints. Historically, catalysis emerged as a fundamental technology to produce a huge amount of raw materials with detailed theoretical analyses and rational refinement of the overall process. Remarkable achievements toward the mass production of commodity chemicals are exemplified by the Haber–Bosch process for nitrogen fixation from the air and the Ziegler–Natta process for polymerization, and have greatly contributed to thriving human societies throughout the world. As synthetic chemistry matures to allow us to construct fairly complex molecular architectures, asymmetric catalysis, endowed with stereoselective chemical transformations, has emerged to permit stereoselective construction of covalent-bond networks.2 Together with theoretical backgrounds and applications, a myriad of asymmetric catalysts have been produced over the last four decades, and some of them have become indispensable for the industrial-scale production of fine chemicals and therapeutics. In this context, the primary focus in catalyst development is to increase catalytic efficiency for higher conversion and selectivity (e.g., chemo-, regio-, and stereoselectivity) with respect to a specific reaction of interest (Fig. 1). In addition to this reaction-oriented aspect of catalysis, recent research toward the development of function-oriented artificial catalysis has been focused at the interface of the biochemistry and biology disciplines. A vast array of biological processes is exquisitely governed by sophisticated biomacromolecular machineries that harness temporal functional manipulation properties to ensure catalysis and molecular transport with precise timing and spatial control. Functional manipulation of these biomacromolecular machineries plays a vital role in rendering the harmonized biological processes at the molecular level in an extremely complicated molecular ensemble. This is in sharp contrast to the simple and ideal setup of a specific synthetic reaction system in synthetic chemistry, where functional manipulation capabilities are not required and therefore have received much less attention. Obviously, functional diversity of an artificial catalyst under precise control offers unique opportunities and prospective applications, and allows for the timely manifestation of catalytic function in time and spatial dimensions. In this review, we outline the recently disclosed arsenal of artificial catalysts with conformational flexibility as a central means to switch catalytic function and stereoselectivity. Light-gated and allosterically regulated artificial switching systems were recently summarized,3,4 and in this review particular emphasis is placed on conformational changes, leading to a specific catalytic function that can be modulated in the course of the reaction. |
| | Fig. 1 Focus of reaction-oriented catalysis and function-oriented catalysis. | |
2. Allosteric regulation of catalytic function
2-1. Introduction
Allosteric regulation is a well-known, ubiquitously manifested means for timely functional control of biomacromolecules.5 However, artificial low-molecular-weight catalysts with allosteric regulatory capabilities are strikingly rare compared with the large number of artificial catalysts available for the practical synthesis of commodity and fine chemicals. Molecular systems exhibiting allostery have mainly been explored in the field of molecular recognition and sensing,6 and rarely with regard to catalytic chemical transformation.4a,c This section reviews conformationally flexible allosteric catalysts that are multistate-regulated by specific allosteric effectors and whose function is closely linked to the three-dimensional conformation of each state. Recent advances in supramolecular chemistry have found self-assembly systems and semisynthetic systems that exert catalytic turnover in chemical transformation, but these achievements are beyond the scope of this review.4a,6–82-2. Allosteric regulation by metal cations
Krämer and co-workers reported the prototype of an artificial catalyst with allosteric regulation that displays catalytic turnover.9 Their catalyst comprises transition metal cations and polypyridyl ligand 1, which is C2-symmetric and has both a catalytic site and a regulatory site (Fig. 2). Complexation of 1 with transition metals Ni2+, Cu2+, and Pd2+ as “structural” metals (Ms) in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 produces the 1/1 complex [1/Ms]+, which was characterized by NMR, UV/Vis, EPR, MALDI-MS, and X-ray crystallographic analysis to confirm installation of the metal at the central tetradentate position, an allosteric regulation site. Further coordination of Cu2+ cations as “functional” metals (Mf) to two bipyridyl arms of 1, which are catalytic sites, was monitored by spectrophotometric titration, and the formation of trinuclear complexes [1/Cu/Cu2]5+, [1/Ni/Cu2]5+, and [1/Pd/Cu2]5+ was observed without cation exchange at the tetradentate coordination site. The thus-formed trinuclear complexes were evaluated as catalysts for the hydrolysis of 2-(hydroxypropyl)-p-nitrophenyl phosphate (2: HPNP) under pH 7.0, a model substrate for RNA cleavage (Scheme 1).10 Hydrolysis of 2 was slow with Cu2+ alone or mononuclear complexes [1/Cu]+, [1/Ni]+, and [1/Pd]+ under similar conditions. The trinuclear complexes exhibited a significantly higher hydrolysis rate and a Lineweaver–Burk plot showed that [1/Cu/Cu2]5+ was the most active, with a rate constant 77 times higher than that of the slowest catalyst [1/Pd/Cu2]5+.9b X-ray analysis of the mononuclear complex indicated that the central tetradentate plane was not flat but distorted, suggesting that the installation of a metal with a different ionic radius has a significant impact on the spatial arrangement and distance of the two catalytically active Cu–bipyridine sites. An electronic effect of the central metal cation toward the active site is unlikely and a slight conformational change by the central cation as an allosteric effector results in a remarkable difference in catalytic activity. This strategy was further extended to the redox allosteric system, where a Co cation was used as a “structural” metal and its oxidation state (II/III) was utilized to alter the conformation of the catalyst.11 The Co3+ complex [1/Co(NO2)2/Cu2]4+ exhibited a five times higher reaction rate than the Co2+ counterpart [1/Co/Cu2]5+ in the catalytic cleavage of 2, probably because the shorter Co3+–N bond brought the two “functional” Cu2+ ions closer together, thereby increasing the reactivity.
:1 produces the 1/1 complex [1/Ms]+, which was characterized by NMR, UV/Vis, EPR, MALDI-MS, and X-ray crystallographic analysis to confirm installation of the metal at the central tetradentate position, an allosteric regulation site. Further coordination of Cu2+ cations as “functional” metals (Mf) to two bipyridyl arms of 1, which are catalytic sites, was monitored by spectrophotometric titration, and the formation of trinuclear complexes [1/Cu/Cu2]5+, [1/Ni/Cu2]5+, and [1/Pd/Cu2]5+ was observed without cation exchange at the tetradentate coordination site. The thus-formed trinuclear complexes were evaluated as catalysts for the hydrolysis of 2-(hydroxypropyl)-p-nitrophenyl phosphate (2: HPNP) under pH 7.0, a model substrate for RNA cleavage (Scheme 1).10 Hydrolysis of 2 was slow with Cu2+ alone or mononuclear complexes [1/Cu]+, [1/Ni]+, and [1/Pd]+ under similar conditions. The trinuclear complexes exhibited a significantly higher hydrolysis rate and a Lineweaver–Burk plot showed that [1/Cu/Cu2]5+ was the most active, with a rate constant 77 times higher than that of the slowest catalyst [1/Pd/Cu2]5+.9b X-ray analysis of the mononuclear complex indicated that the central tetradentate plane was not flat but distorted, suggesting that the installation of a metal with a different ionic radius has a significant impact on the spatial arrangement and distance of the two catalytically active Cu–bipyridine sites. An electronic effect of the central metal cation toward the active site is unlikely and a slight conformational change by the central cation as an allosteric effector results in a remarkable difference in catalytic activity. This strategy was further extended to the redox allosteric system, where a Co cation was used as a “structural” metal and its oxidation state (II/III) was utilized to alter the conformation of the catalyst.11 The Co3+ complex [1/Co(NO2)2/Cu2]4+ exhibited a five times higher reaction rate than the Co2+ counterpart [1/Co/Cu2]5+ in the catalytic cleavage of 2, probably because the shorter Co3+–N bond brought the two “functional” Cu2+ ions closer together, thereby increasing the reactivity.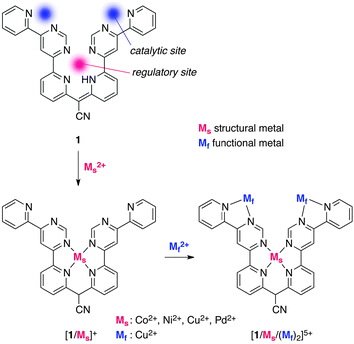 |
| | Fig. 2 The structure of polypyridyl allosteric catalyst 1. | |
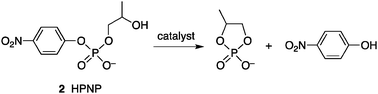 |
| | Scheme 1 Hydrolytic cleavage reaction of 2-(hydroxypropyl)-p-nitrophenyl phosphate (2: HPNP). | |
Kubo et al. reported that a conformationally flexible dibenzo-diaza-30-crown-10 scaffold 3 (Fig. 3) bearing two catalytically active thiourea moieties exhibits significant rate enhancement in the cleavage reaction of HPNP (2) (Scheme 1).10,12 Based on an energy-minimized model, the addition of alkali metal cations induces a conformational change to form two thiourea functionalities in proximal positions, which significantly accelerates the cleavage reaction. When combined with a K+ cation, a 400-fold rate enhancement was observed, whereas combination with Na+ or Cs+ produced a lower acceleration. A downfield shift in the 31P NMR spectrum of the 3/K+/diphenyl phosphate complex suggested that the rate enhancement was due to the increased electrophilicity at the phosphorus atom of 2 through a hydrogen-bond interaction with thiourea functionalities.
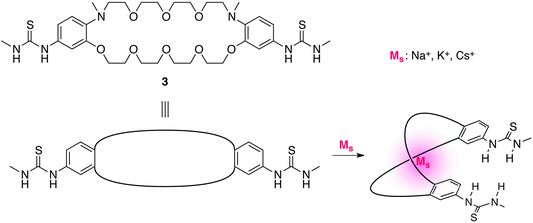 |
| | Fig. 3 The structure of crown-ether-based allosteric catalyst 3. | |
Scrimin and colleagues reported a cooperative catalytic system derived from ligand 4 comprising a tris(2-aminoethyl)amine (tren) core and peripheral azacrown units tethered with a simple amide or peptide linker (Fig. 4).13 The former serves as a regulatory site upon complexation with a structural metal (Ms) and the latter incorporates a functional metal (Mf) to produce catalytic activity toward HPNP (2) cleavage. Preferential coordination of tren over the azacrown moieties to Cu2+ was verified by spectrophotometric titration, indicating that sequential addition of the metal cation induces a conformational change of 4 with Ms, followed by the installation of Mf to peripheral azacrown units to produce catalytic activity. The kinetic profile of the cleavage of HPNP (2) under pH 7.0 at 40 °C by the Zn complex of 4 indicated 30-fold acceleration compared with the unassembled Zn complexes.10
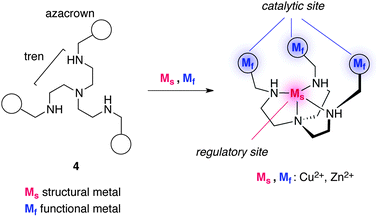 |
| | Fig. 4 The structure of allosteric catalyst 4 with tris(2-aminoethyl)amine (tren) core and peripheral azacrown units. | |
Shinkai et al. showed a different polyamine platform, 5, exhibiting increased catalytic activity by allosteric effect in the cleavage of HPNP (2) upon complexation with Zn2+ or Cu2+ (Fig. 5).10,145 has a central 2,2′-bipyridine (bpy) core armed with 2,2′-dipicolylamine (dpa) groups that act as a regulatory site and catalytic site, respectively. Spectrophotometric titration revealed that the first two equivalents of metal cations (Cu2+ or Zn2+) were incorporated to tridentate dpa moieties. This observation is consistent with the similar and low catalytic efficiency of [5/(Mf)2] and [6/(Mf)2], in which the bpy core was replaced by a nonfunctional biphenyl core. Further addition of metal cations to [5/(Mf)2] led to a significant increase (19.4-fold in Cu2+ complex) in the reaction rate because of the formation of [5/(Mf)2/Ms] and [(5)2/(Mf)4/Ms], in which the dpa units containing Mf are fixed in close proximity to work in a cooperative manner. In sharp contrast, platform 6 lacks a regulatory site and rate enhancement was hardly detected with [6/(Mf)2] upon addition of over two equivalents of metal cations.
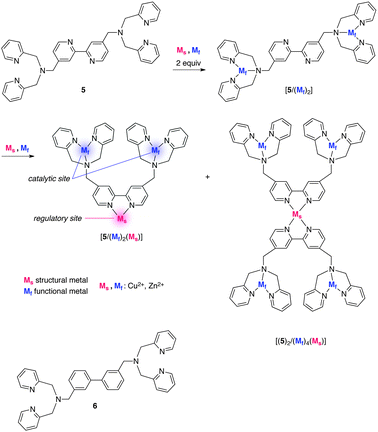 |
| | Fig. 5 The structure of allosteric catalyst 5 with a bpy core and dpa units. | |
The obvious drawback of enhancing catalytic activity by allosteric effect is their unidirectional nature and irreversibility. In the allosteric regulation manifested in biomacromolecular systems, reversible structural perturbation is linked with functional diversification. Irreversibility is avoided by changing the allosteric effectors from accumulating metal cations to specific small molecules or photoirradiation, allowing for reversible structural and functional transitions as well as temporal control of the overall processes.
2-3. Allosteric regulation by specific molecules
2-3-1. Layered systems. Mirkin and co-workers devised double-layered metal complexes linked by two Rh cations (or Cu2+) and 7 (Fig. 6).15 The layer-shaped metal complexes are metal–salen (Cr3+, Zn2+), Zn–porphyrin, or Zn–bis(2,6-diimino)pyridine complexes, which function as the catalytic site. On the other hand, two Rh cations located at the edge of the layer-shaped catalytic metal complexes serve as an Ms with the ability to induce reversible structural modifications. In the closed state, thioether and diphenylphosphine groups coordinate to Rh+ in a bidentate fashion to form bis-Rh-double-layered complex 8, in which two layered catalytic sites are located too close to each other to allow for their catalytic function. In contrast to the absence/presence of an Ms for structural transition in the previous section, the coordination mode of Rh+ was exploited to modulate the overall structure of the catalyst, which is manipulated by external allosteric effectors (CO) and specific anions (Cl− or AcO−). Selective coordination of these allosteric effectors to Rh+ drives the structural change from the closed state 8 to the opened state 9, where two Rh cations are tethered only through diphenylphosphine groups in a monodentate fashion. Removal of CO in vacuo or by nitrogen purge reverses the process to give closed catalyst 8. A series of allosteric catalysts endowed with this mechanism is readily synthesized via a weak-link approach (WLA)16 for supramolecular architecture17 to streamline the synthetic process and allow for convergent access.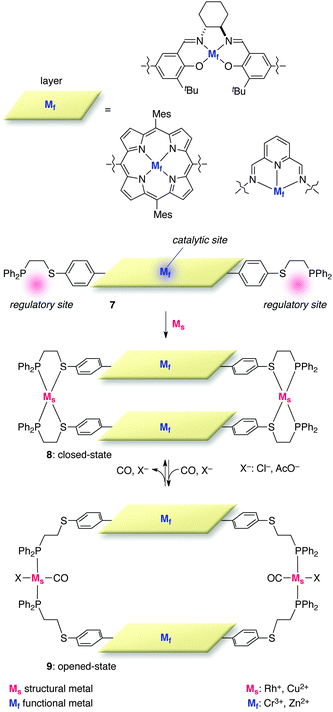 |
| | Fig. 6 The general structure of the double-layer allosteric catalysts. | |
The reversible structural transition between the closed/opened state was initially verified with a layered allosteric catalyst 10 equipped with two chiral Cr3+Cl–salen catalytic sites and Rh+ regulatory sites (Mf = [CrCl]2+, Ms = Rh+) (Fig. 7).15a Diagnostic Rh-bound CO bands in the IR spectrum and a square planar trans-phosphine complex of Rh+ in the 31P NMR spectrum confirmed the opened structure of 11. The catalytic function of chiral [CrCl]2+–salen in this allosteric system was evaluated in the catalytic ring-opening reaction of cyclohexene oxide (12) reported by Jacobsen et al. (Scheme 2).18 Upon addition of bis(triphenylphosphine)iminium chloride (PPNCl) followed by CO purge,19 the closed catalyst 10 was transformed quantitatively into opened catalyst 11 and the reaction rate of 12 with TMSN3 was doubled.20
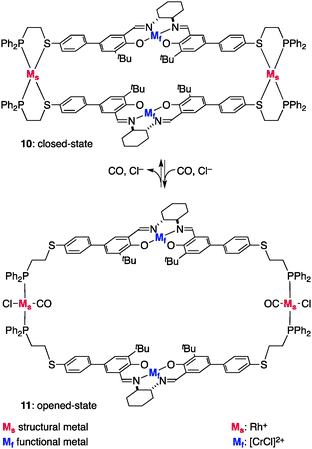 |
| | Fig. 7 The structure of the double-layer allosteric catalysts 10 and 11. | |
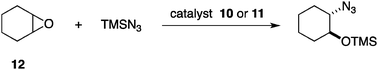 |
| | Scheme 2 Epoxide-opening reaction of cyclohexene oxide (12) with TMSN3. | |
This closed/opened system, in which the opened state exhibits enhanced catalytic activity for a specific reaction, has a wide range of applications.15,21 With an allosteric catalyst comprising Zn–bis(2,6-diimino)pyridine catalytic sites and the identical Rh-based regulatory mechanism, significant rate enhancement by the structural transition from the closed state to the opened state was observed in the hydrolytic cleavage of HPNP (2) (Scheme 1).10,15e Bubbling N2 to expel CO on Rh+ turned the catalytic activity off through a transition to the closed state, verifying the reversibility of the allosteric regulation. Another intriguing application of the layered allosteric system is the demonstration of a signal-amplification system operated by allosterically regulated catalysis.6,15b,d A notable achievement in the development of more sophisticated systems was the synthesis of a triple-layer allosteric catalyst for regulated catalysis.21c
2-3-2. Self-locking systems. Schmittel et al. used a self-locking system22 for reversible capture/release of an organocatalyst.23 The designed catalyst 13 comprises a Zn–porphyrin unit, a HETPHEN complexation motif,24 and an internal inhibitory unit 4-(2-pyridyl)pyrimidine (py-pym) (Fig. 8). 13 inherently prefers its locked conformation because of a favorable internal coordination of the py-pym end to Zn–porphyrin to give a triangular complex, 13-locked. When 2,9-bis(9-anthranyl)-1,10-phenanthroline (14) and [Cu(CH3CN)4]PF6 were added to 13-locked, preferential formation of a tetrahedral Cu complex between py-pym and 14 flipped the py-pym inhibitory unit away to produce the unlocked state (13-unlocked), thereby allowing the Zn–porphyrin unit to accept the coordination of another monodentate ligand. By choosing an organocatalyst as the external guest ligand, the self-locking system allows for reversible capture/release of catalytically active species and functions as a switchable catalyst. In the presence of piperidine (15), the unlocked system comprising 13-unlocked, [Cu(CH3CN)4]PF6, 14, and 15 afforded a ternary complex of [13-unlocked/15/14]+ to capture 15, as shown by 1H NMR. Addition of 2-ferrocenyl-9-mesityl-1,10-phenanthroline (16) trapped the Cu+ to form a highly stable homoleptic complex [Cu(16)2]+, and hence the inhibitory py-pym unit flipped to coordinate to the Zn–porphyrin to constitute the locked state (13-locked). The Zn–porphyrin locked state exhibited a significantly smaller binding constant to piperidine (15) and the released 15 promoted the Knoevenagel reaction of p-nitrobenzaldehyde (17) and diethyl malonate (18) (Scheme 3).25 Arresting the catalysis in the unlocked state and reviving the catalysis in the locked state has been repeatedly demonstrated.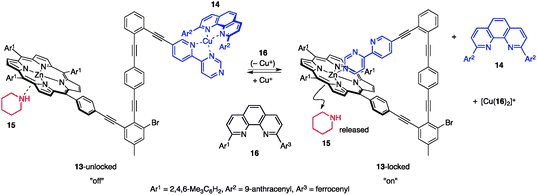 |
| | Fig. 8 The structure of self-locking allosteric catalyst 13. | |
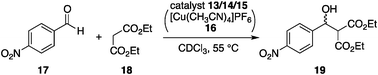 |
| | Scheme 3 Knoevenagel reaction of p-nitrobenzaldehyde (17) and diethyl malonate (18). | |
3. Photoswitchable catalytic systems
3-1. Introduction
Photoirradiation with specific wavelengths is particularly advantageous for conformational control of the three-dimensional structure of a catalyst.26 In contrast to chemical triggers (organic molecules, cations, and anions), photoirradiation is noncumulative in the whole catalytic system and can be orthogonal to the reaction of interest by appropriate selection of the irradiation wavelength and specific chromophores. This favorable property is well exploited for modifying biomacromolecules with a photochromic unit to regulate their functions via photoirradiation.27,28 In contrast to the related photocaged catalysis,29 in which an inactive precatalyst is irreversibly activated by photoirradiation to produce an active catalyst capable of promoting a certain chemical reaction, photoswitchable catalysts are dual-state catalysts, and the catalytically active (ON) and inactive (OFF) states can interconvert by photoirradiation with a specific wavelength. Although early examples of photoswitchable catalysts were constructed based on the context of the intermolecular interaction of a catalytic component and a photochromic component to regulate the catalytic function,30 the specific focus of this section is on photoswitchable catalysts in which the catalytic site and a photochromic moiety are covalently linked. Photoirradiation induces conformational changes in the catalyst to elicit its inherent catalytic function and to induce a reversible structural transition.3-2. Acceleration by photoinduced deshielding
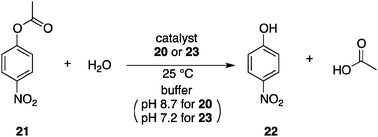
Ueno et al. disclosed a cyclodextrin-based approach toward a photoswitchable catalyst.31 The designed photoswitchable catalyst 20 contains a photochromic azobenzene unit32 that is covalently linked at the lower rim of β-cyclodextrin (β-CD) (Fig. 9). β-CD serves as a host for the hydrophobic aryl group of p-nitrophenyl acetate (21) and the bound 21 undergoes facilitated hydrolysis by the peripheral hydroxyl groups at the upper rim of β-CD.33 This property has been exploited to construct a photoswitchable catalytic system with intermolecular inhibition of binding of 21 through competitive binding of the photochromic azobenzene inhibitor.30a Photoswitchable catalyst 20 provides a two-photoisomeric state because of the covalently bound photochromic azobenzene unit. In the state of 20-E, a flat E-azobenzene entity is located right below the lower rim of β-CD, making the internal cavity shallow and thereby decreasing the binding affinity of 21 to the hydrophobic pocket. In contrast, in the state of 20-Z with a bent Z-azobenzene unit after photoirradiation at 320–390 nm, a deeper cavity is created that offers stronger binding affinity toward 21. Michaelis–Menten analysis of the hydrolytic cleavage of 21 demonstrated the approximate binding affinity and reaction rate (Scheme 4), and revealed the absence of a synergistic effect. Although 20-Z has a more than 10-fold higher binding affinity to 21 than 20-E, 20-E promotes hydrolysis 2-fold faster than 20-Z, presumably because the bound 21 in 20-E is located at a favorable position for nucleophilic attack by the peripheral hydroxyl group. The effect of binding affinity dominates and a maximum 5.5-fold faster hydrolysis is observed upon irradiation, although the content of 20-Z was only 38% in the photostationary state.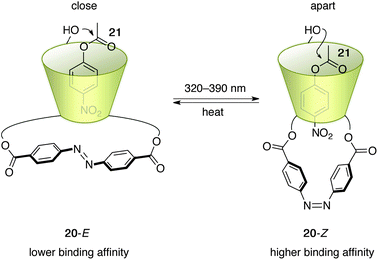 |
| | Fig. 9 The structure of photoswitchable catalyst 20. | |
 |
| | Scheme 4 Hydrolysis of p-nitrophenyl acetate (21). | |
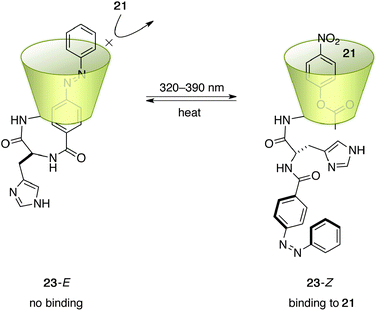
Based on the above β-CD system, a more advanced photoswitchable catalyst 23 was reported by Ueno et al. (Fig. 10).34 By connecting a conformationally flexible azobenzene unit with a histidine linker, the extended E-azobenzene moiety of 23-E occupies the hydrophobic cavity of β-CD to block the catalytic function of the hydrolysis of 21 (Scheme 4). After 12 min of irradiation at 320–390 nm, photostationary equilibrium (2.5 × 10−5 M) was achieved and the 23-Z content reached 79.7%. No evidence of complexation between 23-E and 21 was detected in a detailed kinetic analysis of the hydrolytic cleavage of 21. In contrast, Michaelis–Menten analysis of the reaction with 23-Z indicated that hydrolysis is facilitated through binding of 21 in the inner hydrophobic cavity.
 |
| | Fig. 10 The structure of photoswitchable catalyst 23. | |
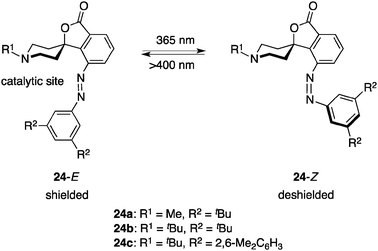
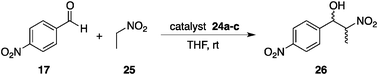
Hecht et al. produced photoswitchable catalysts to regulate catalytic efficiency using a more direct approach, reversible shielding/deshielding of a catalytically active site upon photoirradiation. The well-designed catalyst 24 comprises an aliphatic piperidine base as the catalytic site and an azobenzene shield, which are assembled with isobenzofuranone in a spirolactone ring system to construct a rigid molecular framework (Fig. 11).35,36 Photoirradiation at 365 nm yields a 24-E:24-Z = 10:90 mixture in a photostationary state and 24-Z reverts to 24-E thermally or by photoirradiation at > 400 nm. The three-dimensional structures of both E and Z isomers of 24a–c have been fully characterized by single-crystal X-ray crystallography, revealing the efficient shielding/deshielding architecture. The photoswitchable catalyst functions as a general base catalyst in a nitroaldol (Henry) reaction of p-nitrobenzaldehyde (17) and nitroethane (25) (Scheme 5).37 When more sterically demanding R1 and R2 substituents are employed, the R1 group more predominantly occupies the equatorial position to prevent N-inversion38 and the azobenzene terminus equipped with the R2 group more efficiently shields the piperidine catalytic site, thereby displaying better switching performance. The ratio of the rate constants, k24c-Z/k24c-E, reached 35.5.39 The photoswitchable piperidine base was successfully immobilized on a silica gel surface.40
 |
| | Fig. 11 The structure of photoswitchable catalysts 24. | |
 |
| | Scheme 5 Nitroaldol (Henry) reaction of p-nitrobenzaldehyde (17) and nitroethane (25). | |
3-3. Acceleration via photoinduced molecular recognition
Rebek et al. reported a photoswitchable catalytic system in which molecular recognition plays a key role.41 The catalyst 27 consists of a central azobenzene unit as a photochromic element and imide functionalities derived from Kemp’s triacid, which are tethered by carbazole spacers (Fig. 12). These imides exhibit high binding affinity toward the adenine nucleobase (Ka = 104–105 M−1 in chloroform).42 The linked adenine receptors based on the imides promote the bimolecular amide formation of amine 28 and p-nitrophenyl ester 29 bearing the adenine unit by increasing the effective concentration (Scheme 6).43 Catalyst 27 is a sophisticated version of these template-based catalysts endowed with a conformational change capability by photoirradiation. Irradiation with 366 nm wavelength light on the E-form of 27 induces remarkable conformational changes caused by photoisomerization of the azobenzene unit to give the Z-form of 27. Although a photostationary equilibrium with irradiation at 366 nm affords 27-E:27-Z = 1:1, the rate of the amide coupling reaction of 28 and 29 is enhanced more than 10-fold through the formation of a termolecular complex of 27-Z/28/29 in which 28 and 29 are located in close proximity. The addition of competitive binders for the imides cancels the rate enhancement, supporting the above mechanistic model.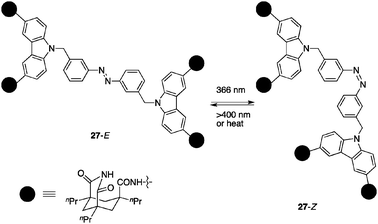 |
| | Fig. 12 The structure of photoswitchable catalyst 27. | |
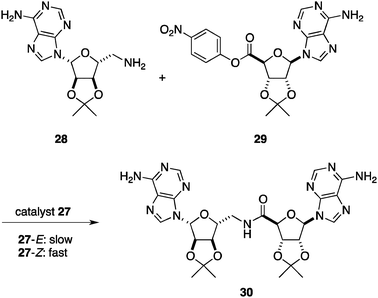 |
| | Scheme 6 Amide bond formation of adenine-containing amine 28 and p-nitrophenyl ester 29. | |
Cacciapaglia and co-workers reported a similar photoswitchable catalysis utilizing metal coordination.44 Based on their earlier findings of size-selective hydrolysis of esters and anilides by dinuclear alkaline-earth metal complexes of bis-crown ether45 and azobenzene-based photoswitching of binding affinity, they devised a bis-crown ether-type photoswitchable catalyst 31 with a central azobenzene spacer hosting two barium cations (Fig. 13). In the ethanolysis of activated anilide 32 bearing para carboxylate (Scheme 7), the Z-form of 31 provides a suitable platform for two-point binding to the substrate 32 through two Ba cations incorporated by 18-crown-6 units, whereas coordination of 32 in a similar fashion is not topologically attainable by the E-form of 31. Photoirradiation of 31-E at 370 nm leads to 31-E:31-Z = 5:95 in the photostationary state and photoirradiation at 480 nm reverts this Z-major mixture to 31-E:31-Z = 81:19. A higher reaction rate was observed with 31-Z because of two-point binding, but 31-E with an extended conformation continues to exhibit catalytic activity, probably because of the high conformational flexibility of 18-crown-6 units, and the observed ratio of the rate constants is around 3 for each state of the photoswitchable catalyst for ethanolysis of 32.
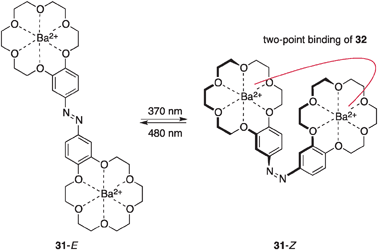 |
| | Fig. 13 The structure of bis-crown ether-based photoswitchable catalyst 31. | |
 |
| | Scheme 7 Ethanolysis of activated amide 32. | |
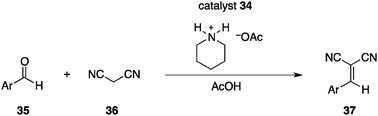
Rebek et al. reported a docking approach for photoswitching catalysis.46 A commonly adopted strategy for supramolecular catalysis is that catalysts host a substrate to accelerate a specific reaction, but there are some reports in which catalysts are guest molecules and their catalytic functions are elicited, or are substantially enhanced, upon incorporation.47 The designed cavitand 34 armed with an isopropyl-substituted azobenzene unit incorporated piperidinium acetate in its 34-E state with an association constant Ka of 4.3 × 103 M−1 (Fig. 14).48 On the other hand, photoisomerized cavitand 34-Z, obtained by photoirradiation at 365 nm, exhibited a self-filling structure in which the isopropyl-substituted azobenzene unit was located inside the cavity, as confirmed by X-ray crystallographic analysis, with unusually high stability (half-life at room temperature is 75 days). The cavitand 34 and piperidinium acetate system functioned as a photoswitchable catalyst in a Knoevenagel reaction of aldehyde 35 and malononitrile (36) (Scheme 8). NOESY analysis indicates that the amino function of the incorporated piperidinium acetate in 34-E heads for the open end of the cavitand. The peripheral amide groups of the cavitand are assumed to facilitate the reaction together with the piperidinium salt and a higher reaction rate was observed for 34-E, despite the relatively small difference in the reaction rate observed between 34-E and 34-Z. Substrates are not guest molecules and a range of aldehydes can be used in this photoswitchable catalytic system.
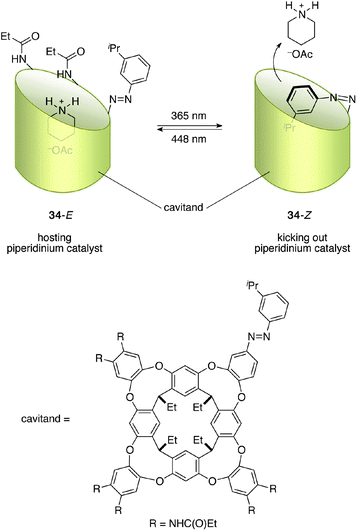 |
| | Fig. 14 The structure of cavitand-based photoswitchable catalyst 34. | |
 |
| | Scheme 8 Knoevenagel reaction of aromatic aldehyde 35 and malononitrile (36). | |
3-4. Acceleration by photoinduced deaggregation of metal nanoparticles
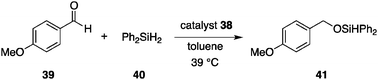
Grzybowski et al. demonstrated that reversible aggregation/dispersion of Au nanoparticles regulated by photoirradiation offers an efficient means of switchable catalysis.49 The temporal manipulation of the surface area of Au nanoparticles (AuNPs) is exploited. The AuNPs (core diameter = 5.5 ± 0.6 nm) are decorated with a mixed self-assembled monolayer (mSAM) comprising primary amines with a long linear alkyl chain and linear alkylthiols installed with an azobenzene terminus (Fig. 15).50 When the azobenzene geometry is E (38-E), the decorated AuNPs are well dispersed in dry toluene and the solvent-exposed surface area promotes the hydrosilylation of p-anisaldehyde (39) and diphenylsilane (40) with a bimolecular rate constant of k38-E = 9 × 10−4 mM−1 min−1 (Scheme 9).51 Irradiation at 365 nm isomerizes the E-azobenzene terminus of the thiol ligand to Z to develop electric dipoles, which allows AuNPs to attract each other to form aggregates in a nonpolar solvent (toluene) within ≈1 min. Much lower catalytic activity is observed with the aggregated AuNPs with a rate constant of k38-Z = 1 × 10−5 mM−1 min−1, displaying a remarkably large rate constant ratio, k38-E/k38-Z = 90. The aggregates are dispersed upon irradiation with visible light. To drive the aggregation/dispersion cycle efficiently, the composition of the mixed self-assembled monolayer mSAM should be controlled. The surface coverage (χ) of the thiol ligand should be controlled in the range 0.23 ≤ χ ≤ 0.33 to ensure light-driven aggregation/dispersion and prevent poisoning of the catalytic activity,52 while alkylamine ligands are loosely bound to AuNP, serving as a background ligand (0.05 ≤ χ ≤ 0.5).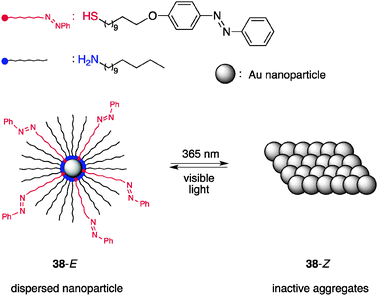 |
| | Fig. 15 The structure of nanoparticle-based photoswitchable catalyst 38. | |
 |
| | Scheme 9 Hydrosilylation of p-anisaldehyde (39) and diphenylsilane (40). | |
4. Switchable catalysis for stereoselectivity
4-1. Introduction
In previous sections, the main focus was on the manipulation of the reaction rate with multistate catalysts. As shown by the broad expansion of asymmetric catalysts in both a scientific and practical sense,2 stereoselectivity as well as promotion of a specific reaction are central issues in catalysis. In this section, we briefly review a recent development in switchable multistate catalysts that promote chemical transformation with distinct stereoselectivities stemming from the different temporal molecular architectures of the catalyst.4-2. Manipulation of stereoselectivity by modulating the catalyst conformation
Branda et al. reported a photochromic multistate catalyst capable of displaying different stereoselectivities.53 The designed photochromic ligand 42 contains two chiral oxazoline groups on the photochromic dithienylethene framework that function as chiral ligands for Cu+ complexes with photoswitchable properties (Fig. 16).54 In the opened state, 42-open coordinated to CuOTf 1/2benzene in a bidentate fashion to construct a rigid asymmetric environment around Cu+, and the thus-obtained Cu complex promoted the cyclopropanation of styrene (43) and ethyl diazoacetate (44) (Scheme 10).55 The reaction with 42-open/Cu+ afforded the desired cyclopropane 45 as a trans–cis mixture with moderate enantioselectivity, indicating that the reaction proceeded in the asymmetric environment of the Cu+/bisoxazoline complex. In contrast, the closed-state catalyst 42-closed/Cu+, in which photocyclized 42-closed was prepared after purification and 97% of 42 was in the closed form, produced 45 with substantially lower enantioselectivity, probably because of the insufficiently asymmetric environment produced by monodentate coordination of the oxazoline unit to Cu+. The major drawback of this catalytic system is the low efficiency for photocyclization in the presence of Cu+; only 23% of 42-closed is formed at the photostationary state and stereoselectivity is only slightly disrupted.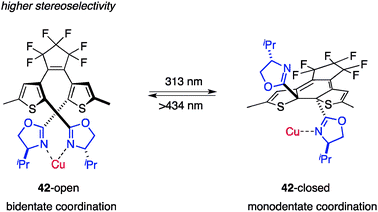 |
| | Fig. 16 The structure of dithienylethene-based photoswitchable catalyst 42. | |
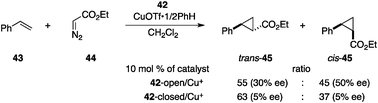 |
| | Scheme 10 Catalytic asymmetric cyclopropanation of styrene (43) and ethyl diazoacetate (44). | |
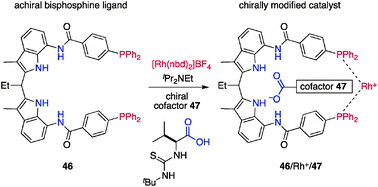
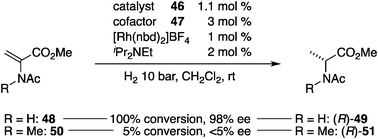
Reek et al. reported a phosphine–Rh+ catalytic system in which the chiral information is transmitted through a noncovalent bond, and coined the term cofactor-controlled catalysis.56,57 The concept allows for in situ conformational regulation of achiral and conformationally flexible phosphine ligand 46 by concomitant use of a chiral cofactor (Fig. 17).58 The central framework of 46 is a symmetric diamidodiindolylmethane59 that serves as an anion receptor,60 and diphenylphosphino groups installed on its termini capture Rh+ in a bidentate fashion. X-ray crystallographic analysis of a single crystal obtained from 46 and [Rh(nbd)]BF4 confirmed that two phosphine groups coordinated to Rh+ in a cis fashion and that the BF4− anion was located at the internal anion binding site. Because of the higher binding affinity of the carboxylate anion to diamidodiindolylmethanes, quantitative replacement of BF4− with a chiral carboxylate anion derived from the carboxylic acid cofactor 47 and iPr2NEt took place, as confirmed by diastereotopic P atoms and amide protons in NMR studies. The preferential association of the chiral carboxylate anion manifested in situ chirogenesis through noncovalent interactions.61 Screening of the chiral cofactors in the asymmetric hydrogenation62 of methyl 2-acetamidoacrylate (48) identified the L-valine-derived cofactor 47 armed with a thiourea functionality as the optimal cofactor, affording the corresponding methyl N-acetyl-D-alanine (49) in remarkably high enantioselectivity (98% ee) (Scheme 11). Asymmetric hydrogenation of the N-methylated derivative 50 resulted in remarkably decreased enantioselectivity to give a virtually racemic product 51, suggesting that the thiourea functionality in chiral cofactor 47 played a crucial role in the enantiodiscrimination step of the reaction, probably through a hydrogen-bonding interaction with the acetamide moiety of 48.63
 |
| | Fig. 17 The structure of achiral ligand 46 and chiral cofactor 47. | |
 |
| | Scheme 11 Catalytic asymmetric hydrogenation of 48 and 50 by cofactor-based catalyst 46/Rh+/47. | |
Leeuwen et al. constructed a Ti–Rh bimetallic complex for enantioselective hydrogenation in which a remote chiral induction through metal coordination is operative.64 Complexation of achiral Schiff bases bearing a diphenylphosphine group and chiral diols with Ti(OiPr)4 generated Δ and Λ isomers having C1 or C2 symmetry irrespective of the order of addition. The use of TADDOLs as chiral diols showed a strong preference to form a single isomer and the combination of Schiff base 52/TADDOL 53a/Ti(OiPr)4/[Rh(nbd)2]BF4 produced a heterobimetallic complex 54 in 92% yield (Fig. 18). The complex was characterized by 1H and 31P NMR, and the bridgehead protons of the nbd ligand appeared diastereotopic, indicating that chirality of 53a was transduced over more than 14 Å through 13 bonds. The heterobimetallic catalyst served as an enantioselective Rh catalyst in asymmetric hydrogenation of (Z)-methyl 2-acetamido-3-phenylacrylate (48).62 TADDOL 53b was found to be the best chiral diol to afford the product 49 in 92% ee with full conversion (Scheme 12). Control experiments using a catalyst lacking one of the catalyst components produced no enantioselectivity, supporting the contention that remote chiral induction is operative to construct an asymmetric environment around the Rh center.
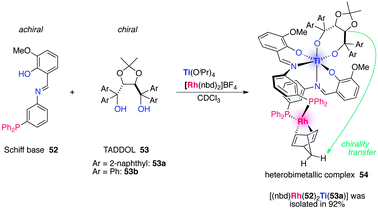 |
| | Fig. 18 Remote chiral induction in a Ti–Rh heterobimetallic complex 54. | |
![Catalytic asymmetric hydrogenation of 48 by heterobimetallic catalyst prepared from 52/53b/Ti(OiPr)4/[Rh(nbd)2]BF4.](/image/article/2013/CY/c2cy20257c/c2cy20257c-s12.gif) |
| | Scheme 12 Catalytic asymmetric hydrogenation of 48 by heterobimetallic catalyst prepared from 52/53b/Ti(OiPr)4/[Rh(nbd)2]BF4. | |
A limitation of the previous examples is the lack of timely control of the stereoselectivity; the fate of the stereochemical outcome could not be altered during the reaction. Although the last example exploits noncovalent interactions for chirogenesis and holds great potential for timely control of the stereoselectivity, catalytic systems with distinct stereoselection processes remain to be developed.65 Our group reported a catalytic system comprising an amide-based ligand 55 and rare earth metal alkoxide that exhibits different stereoselectivities depending on the rare earth metal, and more importantly, the stereoselectivity of the catalyst can be altered during the reaction (Fig. 19).66–68 A catalyst prepared from 55/Sc(OiPr)3 in a ratio of 2:1 was originally reported as an efficient catalyst for an anti-selective asymmetric Mannich-type reaction69 of α-cyanoketone 56 and N-Boc-aldimine 57 to give the Mannich product anti-58 bearing two consecutive stereocenters with high stereoselectivity (Scheme 13a).67c The analogous catalyst 55/Er(OiPr)3 in a ratio of 2:1 displays remarkable diastereoselectivity reversal to give syn-58 with high enantioselectivity under otherwise identical reaction conditions (Scheme 13b). Both the anti-selective 55/Sc catalyst and the syn-selective 55/Er catalyst are assumed to have an undefined structure and to be in dynamic equilibrium with respect to ligand exchange and coordination/dissociation. Slight differences in the ionic radius and coordination mode between Sc3+ and Er3+ are presumably leveraged by the conformational flexibility of the amide-based ligand 55, thereby showing different stereoselectivities. Although the dynamic nature and paramagnetic properties of the Er3+ hampered NMR analysis, CD spectra of each catalyst produced distinct patterns, suggesting that chiroptically different catalysts were formed depending on the rare earth metal used. Particularly noteworthy is that the 55/Er catalyst can be transformed into a 55/Sc catalyst by the addition of Sc(OiPr)3, as shown by the clear change in the CD spectral pattern.66 This is probably due to the dynamic nature of the 55–rare earth metal complex, and manipulation of the stereoselectivity of the catalyst is possible during the reaction (Scheme 13c); (1) initial reaction of 56 and 59 was performed with syn-selective 55/Er catalyst to produce syn-60; (2) after consumption of 59, Sc(OiPr)3 was added to modify the catalyst and another aldimine 57 was added; (3) the catalyst switched gears to use the 55/Sc catalyst and engaged in anti-selective reaction to produce anti-58, and both syn-60 and anti-58 were obtained with high stereoselectivity. Unfortunately, the change in the stereoselectivity was only unidirectional and a reversible switching system remains to be developed.
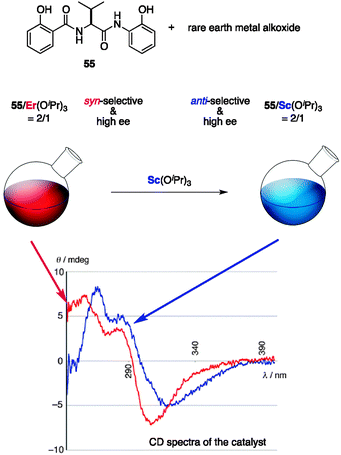 |
| | Fig. 19 CD analysis of 55/Er and 55/Sc, and structural transition from 55/Er to 55/Sc. | |
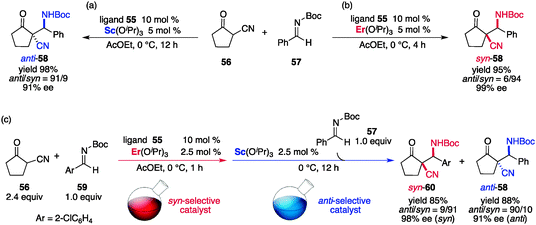 |
| | Scheme 13 (a, b) Catalytic asymmetric Mannich-type reaction of α-cyanoketone 56 and N-Boc-imine 57 with 55/rare earth metal catalyst. (c) Manifestation of diastereoswitching during the course of the reaction. | |
Melchiorre et al. reported a similar in situ stereoswitching catalytic system in which the diastereoselection of an organocatalyst was reversed by the choice of the acidic additive and the reaction medium.70 In the catalytic asymmetric sulfa-Michael reaction71 of benzylthiol (61) and enone 62, a cinchona alkaloid derivative 6372 acted as an effective organocatalyst and its stereoselectivity was enhanced by the concomitant use of 2-fluorobenzoic acid (64) in CHCl3, preferentially affording syn-65 with high enantioselectivity (Scheme 14). The use of diphenyl hydrogen phosphate (DPP, 66) and acetone as a solvent resulted in switching of diastereoselectivity to give anti-65, presumably because the structure of the proposed iminium intermediate (67) derived from the conformationally flexible catalyst 6373 was altered by the counter anion and solvent polarity. By taking advantage of the stronger acidic nature of DPP (66) over 2-fluorobenzoic acid (64), in situ displacement of 64 from the acid–base equilibrium by 66 is possible, thereby altering the diastereoselectivity from syn to anti. Although the addition of a different solvent (acetone) and different reaction temperature were required as well as relatively high catalyst loading, diastereoswitching by the addition of 66 was achieved with high enantioselectivity during the course of the reaction, and the second substrate 68 was preferentially transformed into anti-69.
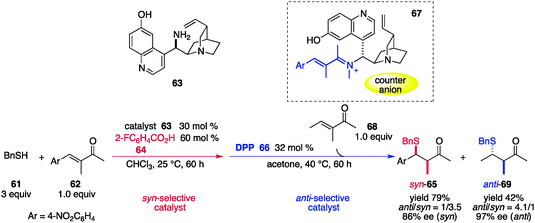 |
| | Scheme 14 Catalytic asymmetric sulfa-Michael reaction of benzylthiol (61) and enone 62, and diastereoswitching during the course of the reaction. | |
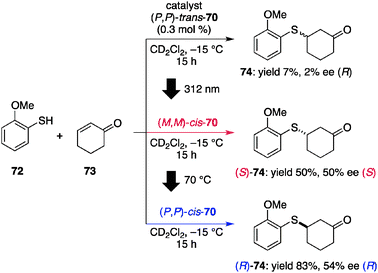
Feringa et al. reported a light-driven rotary motor molecule with four distinct states during a 360° unidirectional cycle, two of which are responsible for catalyzing an asymmetric reaction to give products with opposite chirality (Fig. 20).74 The rotary motor catalyst (P,P)-trans-70 has a central stereogenic indane pair that is connected with P helicity and the pendant functional groups are located in a trans orientation (Fig. 20).75 Each indane holds distinct functional groups, a Brønsted basic 4-(dimethylamino)pyridine (DMAP) unit, and a thiourea unit as a hydrogen-bonding donor. Those two functional groups are anticipated to work together to promote the asymmetric sulfa-Michael reaction71 and the stereochemical outcome will reflect the asymmetric environment of these two functional groups, which is directly linked to the rotational state of catalyst 70. In the reaction of 2-methoxythiophenol (72) and 2-cyclohexen-1-one (73) with 0.3 mol% of (P,P)-trans-70 at −15 °C, a slow reaction occurred and only 7% of the product 74 was obtained in a racemic form after 15 h, probably because of the lack of cooperative activation of the substrates (Scheme 15).76 In contrast, after photoirradiation at 312 nm to rotate the catalyst (P,P)-trans-70 to (M,M)-cis-70, the product (S)-74 was obtained in 50% yield with 50% ee under identical reaction conditions. The cis orientation allows the thioamide and DMAP units to function cooperatively (model 71); the activation of 73 by hydrogen bonding77 with the thiourea and deprotonation of 72 by the DMAP unit as a Brønsted base. (M,M)-cis-70 was fairly stable under reaction conditions (half-life = 120 days at 0 °C), but heating at 70 °C drove the rotational motion to give (P,P)-cis-70, which promoted the same reaction at higher catalytic efficiency (83%) and favored the formation of the enantiomer (R)-74 in 54% ee. Although the enantiomeric excess of this rotational catalyst was moderate, the judicious and iterative control of rotational motion and the exploitation of cooperative catalysis for switching on the catalytic function are intriguing.
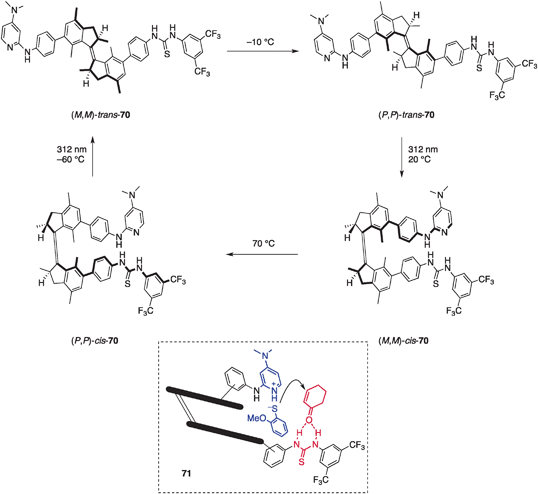 |
| | Fig. 20 The structure of molecular rotor-based multistate catalyst 70. | |
 |
| | Scheme 15 Distinct catalytic efficiency and stereoselectivity produced by each state of the catalyst. | |
5. Future outlook
Impressive and significant advances have led to the development of a number of dynamic multistate catalysts with multiple catalytic functions having adequate spatiotemporal resolution. Various external triggers, such as metal cations, anions, organic molecules/polymers, and photoirradiation/heat may be successfully implemented for timely control of the three-dimensional architecture of multistate catalysts. However, a large gap remains between the function-oriented investigation and reaction-oriented catalyst development. Although catalysts in a general sense need to establish their practical significance for efficient production of particular chemical entities, most of the multistate catalysts presented in this review fail to be distinguished from a practical point of view. Reinforcement of the catalytic efficiency and a broader scope of applicable reactions will be the next big step toward the development of dynamic multistate catalysts that can be practically applied, e.g., a smart catalyst to promote mechanistically orthogonal multiple reactions and otherwise intractable transformations. Furthermore, these catalysts are not only advantageous for their utility in organic synthesis, but can also be applied to a much broader field because these catalysts can produce multiple outputs from an intricate molecular ensemble. With prospective interdisciplinary applications and underutilized switching motifs,78–86 the development of sophisticated dynamic multistate catalysts based on new ideas and efforts will promote progress in this fascinating field.87References
-
(a) Catalysis for Fine Chemical Synthesis, ed. S. M. Roberts, G. Poignant, I. Kozhevnikov, J. Xiao, J. Whittall, T. E. Pickett and E. G. Derouane, John Wiley & Sons, 2004, vol. 1–5 Search PubMedP. W. N. M. van Leeuwen, Homogeneous Catalysis: Understanding the Art, Kluwer, Dordrecht, 2004 Search PubMed.
-
(a) Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 2nd edn. 2000 Search PubMed;
(b) P. J. Walsh and M. C. Kozlowski, Fundamentals of Asymmetric Catalysis, University Science Books, Sausalito, 2009 Search PubMed;
(c) E. J. Corey and L. Kürti, Enantioselective Chemical Synthesis, Direct Book Publishing, Dallas, 2010 Search PubMed.
- Recent reviews on molecular machineries and molecular switches:
(a) V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3349 CrossRef;
(b) W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25 CrossRef CAS;
(c) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72 CrossRef CAS;
(d) V. Balzani, A. Credi, M. Venturi, Molecular Devices and Machines, Wiley, Weinheim, 2nd edn. 2008 Search PubMed;
(e) J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802 RSC;
(f) J. Michl and E. C. H. Sykes, ACS Nano, 2009, 3, 1042 CrossRef CAS;
(g) C. S. Vogelsberg and M. A. Garcia-Garibay, Chem. Soc. Rev., 2012, 41, 1892 RSC.
- Recent reviews on artificial switchable catalysis regulated by photoirradiation and allosteric effector:
(a) L. Kovbasyuk and R. Krämer, Chem. Rev., 2004, 104, 3161 CrossRef CAS;
(b) R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054 CrossRef CAS;
(c) M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114 CrossRef CAS;
(d) A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19 RSC;
(e) Z. Dai, J. Lee and W. Zhang, Molecules, 2012, 17, 1247 Search PubMed.
-
(a) J. Monod, J.-P. Changeux and F. Jacob, J. Mol. Biol., 1963, 6, 306 CrossRef CAS;
(b) D. E. Koshland Jr, in The Enzymes, ed. P. Boyer, Academic Press, New York, 1970, vol. 1, pp. 341 Search PubMed.
-
(a) L. Zhu and E. V. Anslyn, Angew. Chem., Int. Ed., 2006, 45, 3161 CrossRef CAS;
(b) P. Scrimin and L. J. Prins, Chem. Soc. Rev., 2011, 40, 4488 RSC and references cited therein. See also ref. 4c.
- Recent reviews on supramolecular catalysis, see:
(a) Supramolecular Catalysis, ed. P. W. N. M. van Leeuwen, Wiley-VCH, Weinheim, 2008 Search PubMed;
(b) M. Yoshizawa and M. Fujita, Bull. Chem. Soc. Jpn., 2010, 83, 609 CrossRef CAS;
(c) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615 CrossRef CAS.
- Recent selected examples of supramolecular catalysis, see:
(a) C. J. Hastings, M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 6938 CrossRef CAS;
(b) S. Horiuchi, Y. Nishioka, T. Murase and M. Fujita, Chem. Commun., 2010, 46, 3460 RSC;
(c) M. Ikemi, T. Kikuchi, S. Matsumura, K. Shiba, S. Sato and M. Fujita, Chem. Sci., 2010, 1, 68 RSC;
(d) J. S. Mugridge, D. Fiedler and K. N. Raymond, J. Coord. Chem., 2010, 63, 2779 CrossRef CAS;
(e) T. Sawada and M. Fujita, J. Am. Chem. Soc., 2010, 132, 7194 CrossRef CAS;
(f) K. Ohara, M. Kawano, Y. Inokuma and M. Fujita, J. Am. Chem. Soc., 2010, 132, 30–31 CrossRef CAS;
(g) Y. Yamauchi, M. Yoshizawa, M. Akita and M. Fujita, J. Am. Chem. Soc., 2010, 132, 960 CrossRef CAS;
(h) C. Sgarlata, J. S. Mugridge, M. D. Pluth, B. E. F. Tiedemann, V. Zito, G. Arena and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 1005 CrossRef CAS;
(i) J. S. Mugridge, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 1182 CrossRef CAS;
(j) K. Suzuki, K. Takao, S. Sato and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2544 CrossRef CAS;
(k) T. Murase, S. Horiuchi and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2866 CrossRef CAS;
(l) K. Suzuki, S. Sato and M. Fujita, Nat. Chem., 2010, 2, 25–29 CrossRef CAS;
(m) C. Monnereau, P. H. Ramos, A. B. C. Deutman, J. A. A. W. Elemans, R. J. M. Nolte and A. E. Rowan, J. Am. Chem. Soc., 2010, 132, 1529 CrossRef CAS;
(n) S. Sato, O. Morohara, D. Fujita, Y. Yamaguchi, K. Kato and M. Fujita, J. Am. Chem. Soc., 2010, 132, 3670 CrossRef CAS;
(o) T. Kawamichi, Y. Inokuma, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2010, 49, 2375 CrossRef CAS;
(p) Y. Sakata, S. Hiraoka and M. Shionoya, Chem.–Eur. J., 2010, 16, 3318–3325 CrossRef CAS.
-
(a) I. O. Fritsky, R. Ott and R. Krämer, Angew. Chem., Int. Ed., 2000, 39, 3255 CrossRef CAS;
(b) I. O. Fritsky, R. Ott, H. Pritzkow and R. Krämer, Chem.–Eur. J., 2001, 7, 1221 CrossRef CAS;
(c) L. Kovbasyuk, M. Hoppe, H. Pritzkow and R. Krämer, Eur. J. Inorg. Chem., 2001, 1353 CrossRef CAS;
(d) K. P. Strotmeyer, I. O. Fritsky, R. Ott, H. Pritzkow and R. Krämer, Supramol. Chem., 2003, 15, 880;
(e) L. Kovbasyuk, H. Pritzkow, R. Krämer and I. O. Fritsky, Chem. Commun., 2004, 880 RSC.
- D. M. Brown and D. A. Usher, J. Chem. Soc., 1965, 6558 RSC.
- I. O. Fritsky, R. Ott, H. Pritzkow and R. Krämer, Inorg. Chim. Acta, 2003, 346, 111 CrossRef CAS.
- T. Tozawa, S. Tokita and Y. Kubo, Tetrahedron Lett., 2002, 43, 3455 CrossRef CAS.
-
(a) A. Scarso, U. Sceffer, M. Göbel, Q. B. Broxterman, B. Kaptein, F. Formaggio, C. Toniolo and P. Scrimin, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5144 CrossRef CAS;
(b) A. Scarso, G. Zaupa, F. B. Houillon, L. J. Prins and P. Scrimin, J. Org. Chem., 2007, 72, 376 CrossRef CAS.
-
(a) S. Takebayashi, M. Ikeda, M. Takeuchi and S. Shinkai, Chem. Commun., 2004, 420 RSC;
(b) S. Takebayashi, S. Shinakai, M. Ikeda and M. Takeuchi, Org. Biomol. Chem., 2008, 6, 493 RSC.
-
(a) N. C. Gianneschi, P. A. Bertin, S. T. Nguyen, C. A. Mirkin, L. N. Zakharov and A. L. Rheingold, J. Am. Chem. Soc., 2003, 125, 10508 CrossRef CAS;
(b) N. C. Gianneschi, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 1644 CrossRef CAS;
(c) C. G. Oliveri, N. C. Gianneschi, S. T. Nguyen, C. A. Mirkin, C. L. Stern, Z. Wawrzak and M. Pink, J. Am. Chem. Soc., 2006, 128, 16286 CrossRef CAS;
(d) M. S. Masar III., N. C. Gianneschi, C. G. Oliveri, C. L. Stern, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 10149 CrossRef CAS;
(e) H. J. Yoon, J. Heo and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 14182 CrossRef CAS;
(f) H. J. Yoon and C. A. Mirkin, J. Am. Chem. Soc., 2008, 130, 11590 CrossRef CAS.
-
(a) J. R. Farrell, C. A. Mirkin, I. A. Guzei, L. M. Liable-Sands and A. L. Rheingold, Angew. Chem., Int. Ed., 1998, 37, 465 CrossRef CAS;
(b) J. R. Farrell, C. A. Mirkin, I. A. Guzei, L. M. Liable-Sands and A. L. Rheingold, J. Am. Chem. Soc., 1998, 120, 11834 CrossRef CAS;
(c) J. R. Farrell, A. H. Eisenberg, C. A. Mirkin, I. A. Guzei, L. M. Liable-Sands, C. D. Incarvito, A. L. Rheingold and C. L. Stern, Organometallics, 1999, 18, 4856 CrossRef CAS;
(d) B. J. Holliday, J. R. Farrell, C. A. Mirkin, K.-C. Lam and A. L. Rheingold, J. Am. Chem. Soc., 1999, 121, 6316 CrossRef CAS;
(e) F. M. Dixon, A. H. Eisenberg, J. R. Farrell, C. A. Mirkin, L. M. Liable-Sands and A. L. Rheingold, Inorg. Chem., 2000, 39, 3432 CrossRef CAS;
(f) A. H. Eisenberg, F. M. Dixon, C. A. Mirkin, C. L. Stern, C. D. Incarvito and A. L. Rheingold, Organometallics, 2001, 20, 2052 CrossRef CAS;
(g) B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022 CrossRef CAS;
(h) X. Liu, C. L. Stern and C. A. Mirkin, Organometallics, 2002, 21, 1017 CrossRef CAS;
(i) M. V. Ovchinnikov, B. J. Holliday, C. A. Mirkin, L. N. Zakharov and A. L. Rheingold, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4927 CrossRef CAS;
(j) A. H. Eisenberg, M. V. Ovchinnikov and C. A. Mirkin, J. Am. Chem. Soc., 2003, 125, 2836 CrossRef CAS;
(k) M. S. Marsar III., C. A. Mirkin, C. L. Stern, L. N. Zakharov and A. L. Rheingold, Inorg. Chem., 2003, 42, 6851 CrossRef;
(l) M. S. Khoshbin, M. V. Ovchinnikov, C. A. Mirkin, L. N. Zakharov and A. L. Rheingold, Inorg. Chem., 2005, 44, 496 CrossRef CAS;
(m) N. C. Gianneschi, M. S. Marsar III. and C. A. Mirkin, Acc. Chem. Res., 2005, 38, 825 CrossRef CAS;
(n) H. J. Yoon, J. Heo and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 14182 CrossRef CAS;
(o) J. Kuwabara, M. V. Ovchinnikov, C. L. Stern and C. A. Mirkin, Organometallics, 2008, 27, 789 CrossRef CAS;
(p) C. G. Oliveri, P. A. Ulmann, M. J. Wiester and C. A. Mirkin, Acc. Chem. Res., 2008, 41, 1618 CrossRef CAS;
(q) H. J. Yoon and C. A. Mirkin, J. Am. Chem. Soc., 2008, 130, 11590 CrossRef CAS.
-
(a) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853 CrossRef CAS;
(b) K.-M. Park, S.-Y. Park, J. Heo, D. Whang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2002, 124, 2140 CrossRef CAS;
(c) H. Jiang and W. Lin, J. Am. Chem. Soc., 2003, 125, 8084 CrossRef CAS;
(d) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS;
(e) J. W. Sibert, P. B. Forshee and V. Lynch, Inorg. Chem., 2005, 44, 8602 CrossRef CAS.
-
(a) K. B. Hansen, J. L. Leighton and E. N. Jacobsen, J. Am. Chem. Soc., 1996, 118, 10924 CrossRef CAS;
(b) R. G. Konsler, J. Karl and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 10780 CrossRef CAS;
(c) E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421 CrossRef CAS;
(d) J. M. Ready and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 2687 CrossRef CAS;
(e) J. M. Ready and E. N. Jacobsen, Angew. Chem., Int. Ed., 2002, 41, 1374 CrossRef CAS.
- E. O. Fischer, W. Held and F. R. Kreissl, Chem. Ber., 1977, 110, 3842 CrossRef CAS.
- H. Yamashita, Chem. Lett., 1987, 525 CAS.
-
(a) N. C. Gianneschi, S.-H. Cho, S. T. Nguyen and C. A. Mirkin, Angew. Chem., Int. Ed., 2004, 43, 5503 CrossRef CAS;
(b) J. Kuwabara, H. J. Yoon, C. A. Mirkin, A. G. DiPasquale and A. L. Rheingold, Chem. Commun., 2009, 4557 RSC;
(c) H. J. Yoon, J. Kuwabara, J.-H. Kim and C. A. Mirkin, Science, 2010, 330, 66 CrossRef CAS;
(d) C. W. Machan, A. M. Spokoyny, M. R. Jones, A. A. Sarjeant, C. L. Stern and C. A. Mirkin, J. Am. Chem. Soc., 2011, 133, 3023 CrossRef CAS.
-
(a) S. Shinkai, M. Ishihara, K. Ueda and O. Manabe, J. Chem. Soc., Perkin Trans. 2, 1985, 511 RSC;
(b) A. Ueno, I. Suzuki and T. Osa, J. Am. Chem. Soc., 1989, 111, 6391 CrossRef CAS;
(c) R. Corradini, A. Dossena, R. Marchelli, A. Panagia, G. Sartor, M. Saviano, A. Lombardi and V. Pavone, Chem.–Eur. J., 1996, 2, 373 CrossRef CAS;
(d) P. R. Ashton, R. Ballardini, V. Balzani, S. E. Boyd, A. Credi, M. T. Gandolfi, M. Gómez-López, S. Iqbal, D. Philp, J. A. Preece, L. Prodi, H. G. Ricketts, J. F. Stoddart, M. S. Tolley, M. Venturi, A. J. P. White and D. J. Williams, Chem.–Eur. J., 1997, 3, 152 CrossRef CAS;
(e) P. R. Ashton, M. Gómez-López, S. Iqbal, J. A. Preece and J. F. Stoddart, Tetrahedron Lett., 1997, 38, 3635 CrossRef CAS;
(f) M. B. Nielsen, J. G. Hansen and J. Becher, Eur. J. Org. Chem., 1999, 2807;
(g) V. Balzani, P. Ceroni, A. Credi, M. Gómez-López, C. Hamers, J. F. Stoddart and R. Wolf, New J. Chem., 2001, 25, 25 RSC;
(h) Y. Liu, A. H. Flood and J. F. Stoddart, J. Am. Chem. Soc., 2004, 126, 9150 CrossRef CAS;
(i) G. Cooke, P. Woisel, M. Bria, F. Delattre, J. F. Garety, S. G. Hewage, G. Rabani and G. M. Rosair, Org. Lett., 2006, 8, 1423 CrossRef CAS;
(j) T. Muraoka, K. Kinbara and T. Aida, J. Am. Chem. Soc., 2006, 128, 11600 CrossRef CAS;
(k) D.-H. Qu and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 1107 CrossRef CAS;
(l) F. Durola and J. Rebek Jr., Angew. Chem., Int. Ed., 2010, 49, 3189 CrossRef CAS;
(m) O. B. Berryman, A. C. Sather, A. Lledó and J. Rebek Jr., Angew. Chem., Int. Ed., 2011, 50, 9400 CrossRef CAS.
- M. Schmittel, S. De and S. Pramanik, Angew. Chem., Int. Ed., 2012, 51, 3832 CrossRef CAS.
-
(a) M. Schmittel and A. Ganz, Chem. Commun., 1997, 999 RSC;
(b) M. Schmittel, A. Ganz and D. Fenske, Org. Lett., 2002, 4, 2289 CrossRef CAS;
(c) M. Schmittel, V. Kalsani and J. W. Bats, Inorg. Chem., 2005, 44, 4115 CrossRef CAS;
(d) S. De, K. Mahata and M. Schmittel, Chem. Soc. Rev., 2010, 39, 1555 RSC.
-
(a) E. Knoevenagel, Ber. Dtsch. Chem. Ges., 1896, 29, 172 CrossRef CAS;
(b) E. Knoevenagel, Ber. Dtsch. Chem. Ges., 1898, 31, 738 CrossRef CAS;
(c) E. Knoevenagel, Ber. Dtsch. Chem. Ges., 1898, 31, 2585 CrossRef CAS;
(d) E. Knoevenagel, Ber. Dtsch. Chem. Ges., 1898, 31, 2596 CrossRef CAS;
(e) L. F. Tietze and U. Beifuss, Chem. Synth., 1991, 2, 341 Search PubMed.
-
(a) Organic Photochromic and Thermochromic Compounds, ed. J. C. Crano and R. J. Guglielmetti, Kluwer/Plenum, New York, 1999 Search PubMed;
(b) Special Issue: “Photochroism: Memories and Switches”, ed. M. Irie, Chem. Rev., 2000, 100, 1683 Search PubMed;
(c) Molecular Switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, 2001 Search PubMed;
(d) Photochroism: Molecules and Systems, ed. H. Dürr, H. Bouas-Laurent, Elsevier, Amsterdam, 2003 Search PubMed;
(e) S. Hecht, Small, 2005, 1, 26 CrossRef CAS;
(f) M.-M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348 CrossRef CAS;
(g) C.-C. Ko and V. W.-W. Yam, J. Mater. Chem., 2010, 20, 2063 RSC.
- For reviews, see:
(a) G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900 CrossRef CAS;
(b) I. Wilner and B. Wilner, in Molecular Switches, ed. B. L. Feringa, Wiley-VCH, Weinheim, 2001, pp.165–187 Search PubMed.
-
(a) I. Wilner, S. Rubin and A. Riklin, J. Am. Chem. Soc., 1991, 113, 3321 CrossRef CAS;
(b) I. Wilner, Acc. Chem. Res., 1997, 30, 347 CrossRef CAS;
(c) I. Hamachi, T. Hiraoka, Y. Yamada and S. Shinkai, Chem. Lett., 1998, 537 CrossRef CAS;
(d) N. Muranaka, T. Hohsaka and M. Sisido, FEBS Lett., 2002, 510, 10 CrossRef CAS;
(e) J. H. Harvey and D. Trauner, ChemBioChem, 2008, 9, 191 CrossRef CAS;
(f) D. L. Fortin, M. R. Banghart, T. W. Dunn, K. Borges, D. A. Wagenaar, Q. Gaudry, M. H. Karakossian, T. S. Otis, W. B. Kristan and D. Trauner, Nat. Methods, 2008, 5, 331 CAS.
- A comprehensive review on light-gated catalysis, see: R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054 Search PubMed.
-
(a) A. Ueno, K. Takahashi and T. Osa, J. Chem. Soc., Chem. Commun., 1980, 837 RSC;
(b) P. Bortolus and S. Monti, J. Phys. Chem., 1987, 91, 5046 CrossRef CAS;
(c) H. Murakami, A. Kawabuchi, K. Kotoo, M. Kunitake and N. Nakashima, J. Am. Chem. Soc., 1997, 119, 7605 CrossRef CAS;
(d) H. Sugimoto, T. Kimura and S. Inoue, J. Am. Chem. Soc., 1999, 121, 2325 CrossRef CAS;
(e) D. G. Whitten, P. D. Wildes and C. A. DeRosier, J. Am. Chem. Soc., 1972, 94, 7811–23 CrossRef CAS.
- A. Ueno, K. Takahashi and T. Osa, J. Chem. Soc., Chem. Commun., 1981, 94 RSC.
- Pioneering works of the use of azobenzenes as photochromic unit for the construction of molecular switches:
(a) S. Shinkai, T. Ogawa, T. Nakaji, Y. Kusano and O. Manabe, Tetrahedron Lett., 1979, 20, 4569 CrossRef;
(b) A. Ueno, H. Yoshimura, R. Saka and T. Osa, J. Am. Chem. Soc., 1979, 101, 2779 CrossRef CAS;
(c) A. Ueno, R. Saka and T. Osa, Chem. Lett., 1979, 841 CAS;
(d) A. Ueno, R. Saka and T. Osa, Chem. Lett., 1979, 1007 CAS;
(e) A. Ueno, R. Saka and T. Osa, Chem. Lett., 1980, 29 CAS;
(f) S. Shinkai, T. Ogawa, T. Nakaji and O. Manabe, Chem. Commun., 1980, 375 Search PubMed;
(g) S. Shinkai, T. Nakaji, Y. Nishida, T. Ogawa and O. Manabe, J. Am. Chem. Soc., 1980, 102, 5860 CrossRef CAS;
(h) S. Shinkai, T. Nakaji, T. Ogawa, K. Shigematsu and O. Manabe, J. Am. Chem. Soc., 1981, 103, 111 CrossRef;
(i) S. Shinkai, K. Shigematsu, Y. Kusano and O. Manabe, J. Chem. Soc., Perkin Trans. 1, 1981, 3279 RSC;
(j) S. Shinkai, T. Ogawa, Y. Kusano, O. Manabe, K. Kikukawa, T. Goto and T. Matsuda, J. Am. Chem. Soc., 1982, 104, 1960 CrossRef CAS.
-
(a) M. C. Bender and M. Komiyama, Cyclodextrin Chemistry, Springer Verlag, New York, 1977 Search PubMed;
(b) J. Szejtli, Comprehensive Supramolecular Chemistry, ed. J. L. Atwood, J. E. D. Davies, D. D. MacNicol and F. Voegtle, Pergamon, Oxford 1996, vol. 3, pp. 189–203 Search PubMed.
- W.-S. Lee and A. Ueno, Macromol. Rapid Commun., 2001, 22, 448 CrossRef CAS.
-
(a) M. V. Peters, R. S. Stoll, A. Kühn and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 5968 CrossRef CAS;
(b) R. S. Stoll, M. V. Peters, A. Kühn, S. Heiles, R. Goddard, M. Buuhl, C. M. Tiele and S. Hecht, J. Am. Chem. Soc., 2009, 131, 357 CrossRef CAS.
- Irreversible generation of organic bases by photoirradiation, see:
(a) J. F. Cameron and J. M. J. Fréchet, J. Org. Chem., 1990, 55, 5919 CrossRef CAS;
(b) J. F. Cameron and J. M. J. Fréchet, J. Am. Chem. Soc., 1991, 113, 4303 CrossRef CAS;
(c) J. F. Cameron, C. G. Willson and J. M. J. Fréchet, J. Am. Chem. Soc., 1996, 118, 12925 CrossRef CAS.
-
(a) L. Henry, C. R. Hebd. Seances Acad. Sci., 1895, 120, 1265 Search PubMed for a review: ;
(b) F. A. Luzzio, Tetrahedron, 2001, 57, 915 CrossRef CAS.
- E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, Wiley, New York, 1994, p. 740 , and references cited therein Search PubMed.
- k26c-Z (rate constant of 100% 26c-Z) was obtained by extrapolation from the observed rate constant of photostationary state.
- R. S. Stoll and S. Hecht, Org. Lett., 2009, 11, 4790 CrossRef CAS.
-
(a) F. Würthner and J. Rebek Jr., Angew. Chem., Int. Ed., 1995, 34, 446 CrossRef;
(b) F. Würthner and J. Rebek Jr., J. Chem. Soc., Perkin Trans. 1, 1995, 1727 Search PubMed.
-
(a) M. M. Conn, G. Deslongchamps, J. de Mendoza and J. Rebek Jr., J. Am. Chem. Soc., 1993, 115, 3548 CrossRef;
(b) R. J. Pieters and J. Rebek Jr., Recl. Trav. Chim. Pays-Bas, 1993, 112, 330 CAS.
-
(a) R. J. Pieters, I. Huc and J. Rebek Jr., Angew. Chem., Int. Ed., 1994, 33, 1579 CrossRef;
(b) I. Huc, R. J. Pieters and J. Rebek Jr., J. Am. Chem. Soc., 1994, 116, 10296 CrossRef CAS;
(c) R. J. Pieters, I. Huc and J. Rebek Jr., Chem.–Eur. J., 1995, 1, 223;
(d) R. J. Pieters, I. Huc and J. Rebek Jr., Tetrahedron, 1995, 51, 485 CrossRef CAS.
- R. Cacciapaglia, S. Di Stefano and L. Mandolini, J. Am. Chem. Soc., 2003, 125, 2224 CrossRef CAS.
-
(a) R. Cacciapaglia, S. Di Stefano, E. Kelderman and L. Mandolini, Angew. Chem., Int. Ed., 1999, 38, 348 CrossRef CAS;
(b) R. Cacciapaglia, S. Di Stefano and L. Mandolini, J. Org. Chem., 2001, 66, 5926 CrossRef CAS;
(c) R. Cacciapaglia, S. Di Stefano and L. Mandolini, J. Org. Chem., 2002, 67, 521 CrossRef CAS.
- O. B. Berryman, A. C. Sather, A. Lledó and J. Rebek Jr., Angew. Chem., Int. Ed., 2011, 50, 9400 CrossRef CAS.
-
(a) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349 CrossRef CAS;
(b) D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 2746 CrossRef CAS;
(c) S. Bai, H. Yang, P. Wang, J. Gao, B. Li, Q. Yang and C. Li, Chem. Commun., 2010, 46, 8145 RSC;
(d) M. A. Sarmentero, H. Fernandez-Perez, E. Zuidema, C. Bo, A. Vidal-Ferran and P. Ballester, Angew. Chem., Int. Ed., 2010, 49, 7489 CrossRef CAS;
(e) A. Cavarzan, A. Scarso, P. Sgarbossa, G. Strukul and J. N. H. Reek, J. Am. Chem. Soc., 2011, 133, 2848 CrossRef CAS;
(f) C. J. Brown, G. M. Miller, M. W. Johnson, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2011, 133, 11964 CrossRef CAS;
(g) Z. J. Wang, C. J. Brown, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 7358 CrossRef CAS.
- Recent reviews on cavitands, see:
(a) S. M. Biros and J. Rebek Jr., Chem. Soc. Rev., 2007, 36, 93 RSC;
(b) R. J. Hooley and J. Rebek Jr., Chem. Biol., 2009, 16, 255 CrossRef CAS;
(c) J. Rebek Jr., Acc. Chem. Res., 2009, 42, 1660 CrossRef;
(d) O. B. Berryman, H. Dube and J. Rebek Jr., Isr. J. Chem., 2011, 51, 700 CrossRef CAS.
- Y. Wei, S. Han, J. Kim, S. Soh and B. A. Grzybowski, J. Am. Chem. Soc., 2010, 132, 11018 CrossRef CAS.
-
(a) D. Witt, R. Klajn, P. Barski and B. A. Grzybowski, Curr. Org. Chem., 2004, 8, 1763 CrossRef CAS;
(b) R. Klajn, K. J. M. Bishop and B. A. Grzybowski, Proc. Nat. Acad. Sci. U. S. A., 2007, 104, 10305 CrossRef CAS;
(c) R. Klajn, P. J. Wesson, K. J. M. Bishop and B. A. Grzybowski, Angew. Chem., Int. Ed., 2009, 48, 1 CrossRef.
-
(a) A. Corma, C. Gonzalez-Arellano, M. Iglesias and F. Sanchez, Angew. Chem., Int. Ed., 2007, 46, 7820 CrossRef CAS;
(b) P. Raffa, C. Evangelisti, G. Vitulli and P. Salvadori, Tetrahedron Lett., 2008, 49, 3221 CrossRef CAS.
-
(a) S. A. Nikolaev, L. N. Zanaveskin, V. V. Smirnov, V. A. Averyanoy and K. L. Zanaveskin, Russ. Chem. Rev. (Engl. Transl.), 2009, 78, 231 Search PubMed;
(b) T. K. Sau, A. Pal and T. Pal, J. Phys. Chem. B, 2001, 105, 9266 CrossRef CAS.
- D. Sud, T. B. Norsten and N. R. Branda, Angew. Chem., Int. Ed., 2005, 44, 2019 CrossRef.
- H. A. McManus and P. J. Guiry, Chem. Rev., 2004, 104, 4151 CrossRef CAS.
-
(a) T. Sawada and M. Nakada, Adv. Synth. Catal., 2005, 347, 1527 CrossRef CAS;
(b) Z.-H. Xu, S.-N. Zhu, X.-L. Sun, Y. Tang and L.-X. Dai, Chem. Commun., 2007, 1960 RSC.
- P. Dydio, C. Rubay, T. Gadzikwa, M. Lutz and J. N. H. Reek, J. Am. Chem. Soc., 2011, 133, 17176 CrossRef CAS.
- For related studies:
(a) S. J. Lee, S. H. Cho, K. L. Mulfort, D. M. Tiede, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2008, 130, 16828 CrossRef CAS;
(b) M. L. Merlau, M. Mejia, P. del, S. T. Nguyen and J. T. Hupp, Angew. Chem., Int. Ed., 2001, 40, 4239 CrossRef CAS.
- Examples of chirality transduction or enhancement in a static system, see:
(a) Y. Imai, W. Zhang, T. Kida, Y. Nakatsuji and I. Ikeda, Tetrahedron Lett., 1997, 38, 2681 CrossRef CAS;
(b) T. Mizutani, H. Takagi, O. Hara, T. Horiguchi and H. Ogoshi, Tetrahedron Lett., 1997, 38, 1991 CrossRef CAS;
(c) M. T. Reetz and T. Neugebauer, Angew. Chem., Int. Ed., 1999, 38, 179 CrossRef CAS;
(d) C. Bolm and O. Beckmann, Chirality, 2000, 12, 523 CrossRef CAS; K. Mikami and M. Yamanaka, Chem. Rev., 2003, 103, 3369 CrossRef CAS;
(e) J. Clayden, A. Lund, L. Vallverdu and M. Helliwell, Nature, 2004, 431, 966 CrossRef CAS;
(f) A. Alexakis, D. Polet, S. Rosset and S. March, J. Org. Chem., 2004, 69, 5660 CrossRef CAS;
(g) A. Leitner, S. Shekhar, M. J. Pouy and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 15506 CrossRef CAS;
(h) J. Clayden and N. Vassiliou, Org. Biomol. Chem., 2006, 4, 2667 RSC;
(i) M. Schmitkamp, D. Chen, W. Leitner, J. Klankermayer and G. Francio, Chem. Commun., 2007, 4012 RSC;
(j) C. S. Smith and T. V. RajanBabu, Org. Lett., 2008, 10, 1657 CrossRef CAS;
(k) J. Yu, T. V. RajanBabu and J. R. Parquette, J. Am. Chem. Soc., 2008, 130, 7845 CrossRef CAS.
-
(a) N. C. Gianneschi, P. A. Bertin, S. T. Nguyen, C. A. Mirkin, L. N. Zakharov and A. L. Rheingold, J. Am. Chem. Soc., 2003, 125, 10508 CrossRef CAS;
(b) N. C. Gianneschi, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 1644 CrossRef CAS;
(c) M. S. Masar III., N. C. Gianneschi, C. G. Oliveri, C. L. Stern, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 10149 CrossRef CAS;
(d) H. J. Yoon, J. Kuwabara, J.-H. Kim and C. A. Mirkin, Science, 2010, 330, 66 CrossRef CAS;
(e) N. C. Gianneschi, M. S. Masar III. and C. A. Mirkin, Acc. Chem. Res., 2005, 38, 825 CrossRef CAS;
(f) C. G. Oliveri, P. A. Ulmann, M. J. Wiester and C. A. Mirkin, Acc. Chem. Res., 2008, 41, 1618–1629 CrossRef CAS.
-
(a) P. Dydio, T. Zielinski and J. Jurczak, Chem. Commun., 2009, 4560 RSC;
(b) P. Dydio, T. Zielinski and J. Jurczak, Org. Lett., 2010, 12, 1076 CrossRef CAS;
(c) P. Dydio, D. Lichosyt and J. Jurczak, Chem. Soc. Rev., 2011, 40, 2971 RSC.
- S. J. Wezenberg, G. Salassa, E. C. Escudero-Adan, J. Benet-Buchholz and A. W. Kleij, Angew. Chem., Int. Ed., 2011, 50, 713 CrossRef CAS and references cited therein.
-
(a) H. B. Kagan and T. P. Dang, J. Chem. Soc. D, 1971, 481 RSC;
(b) W. S. Knowles, M. J. Sabacky and B. D. Vineyard, J. Chem. Soc., Chem. Commun., 1972, 10 RSC;
(c) A. Miyashita, A. Yasuda, H. Takaya, K. Toriumi, T. Ito, T. Souchi and R. Noyori, J. Am. Chem. Soc., 1980, 102, 7932 CrossRef CAS;
(d) R. Noyori, Asymmetric catalysis in organic synthesis; ed. b. mm, Wiley, New York, 1994, ch. 2 Search PubMed;
(e) J. P. Genet, Acc. Chem. Res., 2003, 36, 908 CrossRef CAS.
-
(a) S. J. Connon, Chem.–Eur. J., 2006, 12, 5418 CrossRef;
(b) S. J. Connon, Synlett, 2009, 354 CAS;
(c) A. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187 RSC;
(d) Y. Sohtome and K. Nagasawa, Synlett, 2010, 1 CAS;
(e) N. Volz and J. Clayden, Angew. Chem., Int. Ed., 2011, 50, 12148 CrossRef CAS.
- P. W. N. M. van Leeuwen, D. Rivillo, M. Raynal and Z. Freixa, J. Am. Chem. Soc., 2011, 133, 18562 CrossRef CAS.
- For reviews, see:
(a) Y. H. Kim, Acc. Chem. Res., 2001, 34, 955 CrossRef CAS;
(b) M. P. Sibi and M. Liu, Curr. Org. Chem., 2001, 5, 719 CAS;
(c) G. Zanoni, F. Castronovo, M. Franzini, G. Vidari and E. Giannini, Chem. Soc. Rev., 2003, 32, 115 RSC;
(d) M. Hayashi and T. Tanaka, Synthesis, 2008, 3361 CAS;
(e) M. Batók, Chem. Rev., 2010, 110, 1663 CrossRef CAS.
- A. Nojiri, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 3779 CrossRef CAS.
- For the utility of amide-based ligand 77, see:
(a) T. Mashiko, K. Hara, D. Tanaka, Y. Fujiwara, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 11342 CrossRef CAS;
(b) T. Mashiko, N. Kumagai and M. Shibasaki, Org. Lett., 2008, 10, 2725 CrossRef CAS;
(c) A. Nojiri, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2008, 130, 5630 CrossRef CAS;
(d) T. Nitabaru, N. Kumagai and M. Shibasaki, Tetrahedron Lett., 2008, 49, 272 CrossRef CAS;
(e) M. Shibasaki, N. Kumagai and T. Mashiko, Curr. Opin. Drug Discovery. Dev., 2009, 12, 862 CAS;
(f) T. Nitabaru, A. Nojiri, M. Kobayashi, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 13860 CrossRef CAS;
(g) Y. Kawato, N. Takahashi, N. Kumagai and M. Shibasaki, Org. Lett., 2010, 12, 1484 CrossRef CAS;
(h) A. Matsuzawa, A. Nojiri, N. Kumagai and M. Shibasaki, Chem.–Eur. J., 2010, 16, 5036 CrossRef CAS;
(i) F. Berhal, S. Takechi, N. Kumagai and M. Shibasaki, Chem.–Eur. J., 2011, 17, 1915 CrossRef CAS;
(j) S. Takechi, N. Kumagai and M. Shibasaki, Tetrahedron Lett., 2011, 52, 2140 CrossRef CAS;
(k) N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 4760 CrossRef CAS;
(l) A. Matsuzawa, T. Mashiko, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 7616 CrossRef CAS;
(m) T. Nitabaru, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2012, 51, 1644 CrossRef CAS;
(n) A. Nojiri, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2012, 51, 2137 CrossRef CAS.
- Other examples of asymmetric catalysis using rare earth metal/amide-based ligand catalysts:
(a) A. Nishida, M. Yamanaka and M. Nakagawa, Tetrahedron Lett., 1999, 40, 1555 CrossRef CAS;
(b) Y. Sudo, D. Shirasaki, S. Harada and A. Nishida, J. Am. Chem. Soc., 2008, 130, 12588 CrossRef CAS.
- For general reviews on catalytic asymmetric Mannich-type reactions, see:
(a) A. Ting and S. E. Schaus, Eur. J. Org. Chem., 2007, 5797 CrossRef CAS;
(b) J. M. Verkade, J. C. van Hemert, P. L. M. Quaedflieg and F. J. T. Rutjes, Chem. Soc. Rev., 2008, 37, 29 RSC;
(c) R. Gómez Arrayás and J. C. Carretero, Chem. Soc. Rev., 2009, 38, 1940 RSC.
- X. Tian, C. Cassani, Y. Liu, A. Moran, A. Urakawa, P. Galzerano, E. Arceo and P. Melchiore, J. Am. Chem. Soc., 2011, 133, 17934 CrossRef CAS.
- D. Enders, K. Lüttgen and A. A. Narin, Synthesis, 2007, 959 CrossRef CAS.
-
(a) W. Chen, W. Du, Y. Duan, Y. Wu, S.-Y. Yang and Y.-C. Chen, Angew. Chem., Int. Ed., 2007, 46, 7667 CrossRef CAS;
(b) G. Bencivenni, P. Galzerano, A. Mazzanti, G. Bartoli and P. Melchiorre, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20642 CrossRef CAS.
-
(a) T. Bürgi and A. Baiker, J. Am. Chem. Soc., 1998, 120, 12920 CrossRef;
(b) G. D. H. Dijkstra, R. M. Kellogg, H. Wynberg, J. S. Svendsen, I. Marko and K. B. Sharpless, J. Am. Chem. Soc., 1989, 111, 8069 CrossRef CAS;
(c) A. Urakawa, D. M. Meier, H. Rüegger and A. Baiker, J. Phys. Chem. A, 2008, 112, 7250 CrossRef CAS.
-
(a) Molecular Motors, ed. M. Schliwa, Wiley-VCH, Weinheim, 2003 Search PubMed;
(b) N. Koumura, R. W. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152 CrossRef CAS;
(c) R. Eelkema, M. M. Pollard, J. Vicario, N. Katsonis, B. Serrano Ramon, C. W. M. Bastiaansen, D. J. Broer and B. L. Feringa, Nature, 2005, 440, 163;
(d) M. M. Pollard, A. Meetsma and B. L. Feringa, Org. Biomol. Chem., 2008, 6, 507 RSC;
(e) J. Wang, A. Kulago, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2010, 132, 4191 CrossRef CAS.
- J. Wang and B. L. Feringa, Science, 2011, 331, 1429 CrossRef CAS.
- Recent reviews on cooperative catalysis, see: Lewis acid/Brønsted base
(a) M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187 CrossRef CAS;
(b) N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 4760 CrossRef CAS Lewis acid/Lewis base: ;
(c) M. Kanai, N. Kato, E. Ichikawa and M. Shibasaki, Synlett, 2005, 1491 CrossRef CAS;
(d) D. H. Paull, C. J. Abraham, M. T. Scerba, E. Alden-Danforth and T. Lectka, Acc. Chem. Res., 2008, 41, 655 CrossRef CAS ; Lewis acid/Brønsted acid and Lewis acid/Lewis acid: ;
(e) H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924 CrossRef CAS;
(f) H. Yamamoto and K. Futatsugi, in Acid Catalysis in Modern Organic Synthesis, ed. H. Yamamoto and K. Ishihara, Wiley-VCH, Weinheim, 2008 Search PubMed.
-
(a) P. R. Schreiner, Chem. Soc. Rev., 2003, 32, 289 RSC;
(b) P. M. Pihko, Angew. Chem., Int. Ed., 2004, 43, 2062 CrossRef CAS;
(c) K. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999 CrossRef CAS;
(d) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS;
(e) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS;
(f) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS;
(g) History and Perspective of Chiral Organic Catalysts, in New Frontiers in Asymmetric Catalysis, ed. G. Lelais and D. W. C. MacMillan, John Wiley & Sons, New Jersey, 2007 Search PubMed;
(h) Brønsted Acids Acid Catalysis, in Modern Organic Synthesis, ed. H. Yamamoto and D. Nakashima, Wiley-VCH, Weinheim, 2008, vol. 1 Search PubMed.
- Modification of coordination numbers:
(a) N. Kano, F. Komatsu and T. Kawashima, J. Am. Chem. Soc., 2001, 123, 10778 CrossRef CAS;
(b) N. Kano, M. Yamamura, F. Komatsu and T. Kawashima, J. Organomet. Chem., 2003, 686, 192 CrossRef CAS;
(c) N. Kano, M. Yamamura and T. Kawashima, J. Am. Chem. Soc., 2004, 126, 6250 CrossRef CAS;
(d) M. Yamamura, N. Kano and T. Kawashima, J. Am. Chem. Soc., 2005, 127, 11954 CrossRef CAS;
(e) N. Kano, F. Komatsu, M. Yamamura and T. Kawashima, J. Am. Chem. Soc., 2006, 128, 7097 CrossRef CAS.
- Modification of Lewis acidity:
(a) N. Kano, J. Yoshino and T. Kawashima, Org. Lett., 2005, 7, 3909 CrossRef CAS;
(b) H. D. Samachetty and N. R. Branda, Pure Appl. Chem., 2006, 78, 2351 CrossRef CAS;
(c) V. Lemieux, M. D. Spantulescu, K. K. Baldridge and N. R. Branda, Angew. Chem., Int. Ed., 2008, 47, 5034 CrossRef CAS.
- Modification of binding affinity:
(a) M. Takeshita, K. Uchida and M. Irie, Chem. Commun., 1996, 1807 RSC;
(b) S. H. Kawai, Tetrahedron Lett., 1998, 39, 4445 CrossRef CAS;
(c) M. Takeshita and M. Irie, J. Org. Chem., 1998, 63, 6643 CrossRef CAS;
(d) A. Mulder, A. Jukovic, F. W. B. van Leeuwen, H. Kooijman, A. L. Spek, J. Huskens and D. N. Reinhoudt, Chem.–Eur. J., 2004, 10, 1114 CrossRef CAS.
- Modification of binding pKa:
(a) S. H. Kawai, S. L. Gilat and J.-M. Lehn, Eur. J. Org. Chem., 1999, 2359 CrossRef CAS;
(b) Y. Odo, K. Matsuda and M. Irie, Chem.–Eur. J., 2006, 12, 4283 CrossRef CAS.
- Modification of Lewis basicity and nucleophilicity:
(a) H. D. Samachetty and N. R. Branda, Chem. Commun., 2005, 2840 RSC;
(b) H. D. Samachetty and N. R. Branda, Tetrahedron, 2008, 64, 8292 CrossRef CAS.
- Modification of electronic nature and reactivity of carbene:
(a) V. Lemieux and N. R. Branda, Org. Lett., 2005, 7, 2969 CrossRef CAS;
(b) V. Lemieux, S. Gauthier and N. R. Branda, Angew. Chem., Int. Ed., 2006, 45, 6820 CrossRef CAS;
(c) D. Sud, T. J. Wigglesworth and N. R. Branda, Angew. Chem., Int. Ed., 2007, 46, 8017 CrossRef CAS;
(d) B. M. Neilson, V. M. Lynch and C. W. Bielawski, Angew. Chem., Int. Ed., 2011, 50, 10322 CrossRef CAS.
- Modification of proton delivery:
(a) F. M. Raymo, S. Giordani, A. J. P. White and D. J. Williams, J. Org. Chem., 2003, 68, 4158 CrossRef CAS;
(b) F. M. Raymo, R. J. Alvarado, S. Giordani and M. A. Cejas, J. Am. Chem. Soc., 2003, 125, 2361 CrossRef CAS;
(c) S. Silvi, A. Arduini, A. Pochini, A. Secchi, M. Tomasulo, F. M. Raymo, M. Baroncini and A. Credi, J. Am. Chem. Soc., 2007, 129, 13378 CrossRef CAS;
(d) S. Silvi, E. C. Constable, C. E. Housecroft, J. E. Beves, E. L. Dunphy, M. Tomasulo, F. M. Raymo and A. Credi, Chem.–Eur. J., 2009, 15, 178 CrossRef CAS;
(e) S. Silvi, E. C. Constable, C. E. Housecroft, J. E. Beves, E. L. Dunphy, M. Tomasulo, F. M. Raymo and A. Credi, Chem. Commun., 2009, 1484 RSC.
- Hydrogen-bonded network and aggregates with photomodulation:
(a) M. S. Vollmer, T. D. Clark, C. Steinem and M. R. Ghadiri, Angew. Chem., Int. Ed., 1999, 38, 1598 CrossRef CAS;
(b) L. N. Lucas, J. van Esch, R. M. Kellog and B. L. Feringa, Chem. Commun., 2001, 759 RSC;
(c) F. Rakotondradany, M. A. Whitehead, A.-M. Lebuis and H. F. Sleiman, Chem.–Eur. J., 2003, 9, 4771 CrossRef CAS;
(d) S. Yagai, T. Nakajima, T. Karatsu, K. Saitow and A. Kitamura, J. Am. Chem. Soc., 2004, 126, 11500 CrossRef CAS;
(e) J. J. D. de Jong, L. N. Lucas, R. M. Kellog, J. van Esch and B. L. Feringa, Science, 2004, 304, 278 CrossRef CAS;
(f) S. Yagai, T. Karatsu and A. Kitamura, Chem.–Eur. J., 2005, 11, 4054 CrossRef CAS;
(g) M. Takeshita, M. Hayashi, S. Kadota, K. H. Mohammed and T. Yamato, Chem. Commun., 2005, 761 RSC;
(h) M. Takeshita, M. Hayashi and T. Miyazaki, Chem. Lett., 2010, 39, 82 CrossRef CAS.
- Hydrogen-bonded complex with photomodulated secondary interaction:
(a) A. Goodman, E. Breinlinger, M. Ober and V. M. Rotello, J. Am. Chem. Soc., 2001, 123, 6213 CrossRef CAS;
(b) T. Okuyama, Y. Yokoyama and Y. Yokoyama, Bull. Chem. Soc. Jpn., 2001, 74, 2181 CrossRef CAS;
(c) M. Herder, M. Pätzel, L. Grubert and S. Hecht, Chem. Commun., 2011, 47, 460 RSC.
- Other on/off type multistate catalysts has been published after evaluation of this manuscript.
(a) V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, Angew. Chem., Int. Ed., 2012, 51, 5166 CrossRef;
(b) D. Wilson and N. R. Branda, Angew. Chem., Int. Ed., 2012, 51, 5431 CrossRef CAS;
(c) T. Imahori, R. Yamaguchi and S. Kurihara, Chem.–Eur. J., 2012, 18, 10802 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.