Catalytic activity of unsupported gold nanoparticles
Yusuke
Mikami
,
Amarajothi
Dhakshinamoorthy
,
Mercedes
Alvaro
and
Hermenegildo
García
*
Instituto Universitario de Tecnología Química CSIC-UPV, Departamento de Química, Univ. Politécnica de Valencia, Av. De los Naranjos s/n, 46022 Valencia, Spain. E-mail: hgarcia@qim.upv.es; Fax: +34 9638-7807
First published on 13th June 2012
Abstract
This article reviews some of the compelling evidence showing that colloidal gold nanoparticles without any solid support exhibit intrinsic catalytic activity for some of the typical gold-catalyzed reactions including CO oxidation, aerobic oxidation of alcohols and diols, borohydride reductions and carbon–carbon cross coupling reaction among other reactions. A critical view of the state-of-the-art indicating open issues such as the role of the nature and concentration of the ligand and the possibility of preparing colloidal samples with preferential crystallographic planes is provided.
 Yusuke Mikami | Yusuke Mikami obtained his PhD at Osaka University in Osaka, Japan, under the guidance of Prof. Kiyotomi Kaneda and Prof. Koichiro Jitsukawa. He was a postdoctoral researcher in Universidad Politecnica de Valencia, Spain. He works in the design of supported metal nanoparticle catalysts for their applications in environmentally-benign organic synthesis. |
 Amarajothi Dhakshinamoorthy | Amarajothi Dhakshinamoorthy received his postgraduate degree in Chemistry from Loyola College (Autonomous), Chennai, India, with two gold medals in 2002. He received a PhD degree under the guidance of Prof. K. Pitchumani from Madurai Kamaraj University, India. Since 2008 he has been working as a postdoctoral fellow in the group of Prof. Hermenegildo Garcia and his current research interests are on developing green and sustainable catalysts based on metal organic frameworks or graphene oxides for aerobic oxidation of hydrocarbons, Lewis-acid catalysis, and alkylation reactions. He has published more than thirty-five papers and holds one international patent. |
 Mercedes Alvaro | Mercedes Alvaro is Full Professor at the Chemistry Department of the Technical University of Valencia. She has co-authored over 150 papers and has supervised 9 theses. Her actual interests include advanced oxidation techniques for water treatment, applied photochemistry for environmental remediation and organic synthesis. |
 Hermenegildo García | Hermenegildo García has been Full Professor at the Technical University of Valencia since 1996 and a staff member of the Instituto de Tecnología Química, a joint center of the Technical University of Valencia and the Spanish National Research Council. He had postdoctoral stays at the University of Reading with Professor Andrew Gilbert and several periods of sabbatical leave in the group of Professor J. C. Scaiano at the University of Ottawa. Prof. Garcia has been active in the field of heterogeneous catalysis working with porous catalysts and nanoparticles, has published over 450 papers and has filed over 25 patents, two of them in industrial exploitation. Prof. Garcia is Doctor Honoris Causa from the University of Bucharest and the recipient of the 2011 Janssen-Cilag award given by the Spanish Royal Society of Chemistry and the 2008 Alpha Gold of the Spanish Society of Glass and Ceramics. |
1 Introduction
The present review deals with the catalytic activity of colloidal gold nanoparticles (Au NPs) dissolved in a liquid phase. To focus the topic of this article it is pertinent to state an explicit definition of the term “colloid”. There are some precedents in which the term colloid has been employed to describe a solid, non-suspensible material constituted by Au NPs sintered together forming large agglomerates with microscopic appearance of foam.1–3 This type of studies constitutes a clear case of heterogeneous catalysis in which a substrate in the liquid phase is in contact with a solid that eventually will settle down by the Earth's gravity. Although no support or metal oxide is contained in the catalyst, which is constituted exclusively of Au, this type of studies are outside the scope of the present review since they are not reporting the catalytic activity of independent Au NPs as they can be present in many supported Au catalysts. Thus, in the present article, we limit ourselves to those precedents in which the catalytic activity of Au NPs that are indefinitely persistent in a liquid phase without any solid support is described and where the presence of two separate phases is not macroscopically observable. The purpose of the present review is to show compelling evidence that colloidal Au NP solutions exhibit intrinsic catalytic activity for aerobic oxidation of alcohols, 1,2-diols and CO as well as borohydride reduction and carbon–carbon coupling among other reactions. This information about the catalytic activity of colloidal Au is of great interest in the area of heterogeneous catalysis by supported Au catalysts because, due to the large influence of the support, there is some misconception that the activity of colloidal Au NPs in the absence of any support is negligible without cooperation of the support.In this way, this review will contribute to establish more exactly the role of the support and Au in the catalytic process. The manuscript has been organized according to the type of ligands used to stabilize the colloids and Table 1 collects the list of reactions that has been included in the present review to illustrate the catalytic activity of colloidal Au solutions discussed throughout the text. A concluding section summarizes some open issues in this area, particularly the role of the ligand in decreasing the activity inherent to the Au NPs and the interplay between colloidal Au and theoretical models.
| Catalyst | Gold precursor | Reducing agent | Stabilizer | Catalysis | Ref. |
|---|---|---|---|---|---|
| PVP: poly(N-vinyl-2-pyrrolidone); PVA: poly(vinyl alcohol); THPC: tetrakishydroxypropylphosphonium chloride; PAMAM: poly(amidoamine). | |||||
| Au:PVP | HAuCl4 | NaBH4 | PVP | Alcohol oxidation | 4 |
| Au:PVP | HAuCl4 | NaBH4 | PVP | Alcohol oxidation | 5 |
| Au:PVP | HAuCl4 | NaBH4 | PVP | Alcohol oxidation | 6 |
| Au:PVP | HAuCl4 | NaBH4 | PVP | Homocoupling of phenylboronic acid | 7 |
| Au:PVP | HAuCl4 | NaBH4 | PVP | Alcohol oxidation | 8 |
| Ionic polymer stabilized Au NPs | HAuCl4 | NaBH4 | Ionic polymer | Reduction of nitrophenol and hydrogenation of cinnamaldehyde | 9 |
| Au(PVA) | HAuCl4 | NaBH4 | PVA | Oxidation of diols | 10 |
| Au(PVA) | NaAuCl4 | NaBH4 | PVA | Oxidation of glycerol | 11 |
| AuTHPC or Aucitrate | Au solution | PVA, THPC, citrate | PVA, THPC, citrate | Oxidation of glycerol | 12 |
| Dendrimer-encapsulated gold nanoparticles | HAuCl4 | NaBH4 | PAMAM dendrimer | CO oxidation | 13 |
| Unsupported gold | HAuCl4 | NaBH4 | Triton X-100 | Reduction of eosin | 14 |
| Colloidal gold | HAuCl4 | Trisodium citrate | Trisodium citrate | Reaction ferricyanide and thiosulphate ions | 15, 16 |
| Polygonal gold nanoparticles | HAuCl4 | Ferric ammonium citrate | None | Reduction of nitrophenols, oxidation of hexoses | 17 |
| Unsupported gold powder | HAuCl4 | NaBH4 | None | Chemiluninescence of luminol | 18 |
| Unsupported gold powder | HAuCl4 | NaBH4 | None | Chemiluninescence of luminol | 19 |
| Au nanowires (GNWs) | HAuCl4 | Oleylamine | Oleylamine | Aerobic oxidation styrene and ethylbenzene | 20 |
| Octylsilane-stabilized colloidal gold | HAuCl4 | NaBH4 | n-Octylsilane | Epoxidation, allylic oxidation | 21 |
| “Naked” gold sol | HAuCl4 | NaBH4 | None | Oxidation of glucose | 22–25 |
| Unsupported gold | HAuCl4 | NaBH4 | Glucose | Oxidative polymerization of aniline | 26 |
| Unsupported gold | HAuCl4 | NaBH4 | Glucose | Oxidative polymerization of pyrrole | 27 |
| Fine gold powder | Evaporating high purity gold metal | None | None | CO oxidation | 28 |
| Gold nanotube membrane | Na3Au(SO3)2 | Electroless Au deposition | None | CO oxidation | 29 |
| “Naked” gold nanoparticles | Au2O3 | H2 | None | Catalytic hydrogen evolution | 30 |
| Thiol-stabilized Au25 nanoclusters | HAuCl4·3H2O | NaBH4 | PhCH2CH2SH | Hydrogenation of benzalacetone | 31 |
| binap-stabilized Au NPs | [(binap)Au2I2] | B2cat2 | binap | Diboration of styrene | 32 |
2 Catalytic activity of polymer-stabilized Au NPs
Due to the instability of small size Au NPs, various polymer stabilizers have been used to prevent their aggregation into inactive bulk Au.33 For example, Au NPs stabilized by polystyrene-based copolymers,34–37 poly(2-aminothiophenol),38 poly(N-vinyl-2-pyrrolidone) (PVP),4–8,39,40 ionic polymers,9,41,42 and ion-exchange resins43,44 exhibited good durability toward agglomeration and catalysis. However, most of these reports employ Au NPs immobilized on insoluble polymer supports, that is, colloidal Au NPs were totally heterogenized and they are outside the scope of this review. In this section, we comment on selected examples of colloidal Au catalysts indefinitely persistent in liquid phase that have been stabilized by a polymer.Tsukuda and co-workers prepared monodisperse Au NPs stabilized by polyvinylpyrrolidone (PVP) (Au:PVP) and investigated their activity for the aerobic oxidation of benzylic alcohols and homocoupling of phenylboronic acid in water.4–7 A Au:PVP-1 catalyst having 1.3 nm of mean diameter was prepared by rapid injection of an aqueous NaBH4 into an aqueous solution of AuCl4−–PVP complex at room temperature. The Au:PVP-1 was treated with Na2SO3 to grow the NPs, leading to the formation of Au:PVP-2 with 9.5 nm average NP size. The catalytic activity of the PVP-stabilized Au NPs was checked for the oxidation of benzylic alcohols in water (Scheme 1).
 | ||
| Scheme 1 | ||
From TEM and optical measurements, Au:PVP-1 was revealed to be stable in size after the aerobic oxidation, but particle growth was observed in some cases. Au NPs having a diameter of 1.3 nm were found to be more active than Pd NPs with a diameter of 1.5 nm prepared by a similar procedure to the Au ones, which is consistent with the result of glucose oxidation using “naked” Au and Pd NPs.22 In addition, from the kinetics experiments, the rate-determining step of the Au:PVP-1-catalyzed oxidation of benzylic alcohol may involve hydrogen atom abstraction by a superoxo-like molecular oxygen species adsorbed on the catalyst. These superoxo-like species are only formed on small Au clusters which lead to the size-specific catalytic activities of Au NPs. Superoxo species (O2˙−) can be bonded onto the surface of Au NPs preferentially to positive Au atoms forming Au–O–O. This raises again a continuous intriguing issue in Au catalysis about the effect of the positive or negative charge density on the activity. It is generally accepted that one of the roles of the solid support is to provide charge density to the gold atoms at the interface between the solid and Au NPs by polarizing the Au atoms in contact with the solid.
Dyson and co-workers synthesized a water-soluble ionic polymer (Scheme 2), poly-(3-((2,4-divinylcyclopentyl)methyl)-1,2-dimethyl-1H-imidazolium methanesulfonate), using ring-opening metathesis polymerization and Au NPs stabilized by the synthesized ionic polymer were evaluated as catalysts for the reduction of p-nitrophenol and the hydrogenation of cinnamaldehyde.9
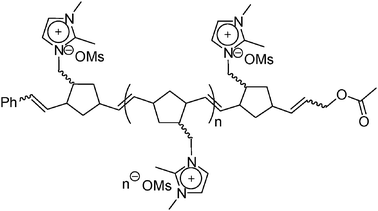 | ||
| Scheme 2 Structure of a water-soluble ionic polymer employed in ref. 9. | ||
Au NPs were prepared in water in the presence of HAuCl4, the polymer, and NaBH4 with various Au/polymer ratios.9 The particle size of the polymer-stabilized Au NPs was confirmed using TEM analysis. The sample with the highest polymer/Au ratio of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 gave a very narrow particle size distribution in the range of 1.8 to 2.8 nm. On the other hand, a broad size distribution from 2 to 12 nm was obtained at lower polymer/Au ratio of 3:1. The Au NPs catalyst having polymer/Au ratio of 50:1 showed the best activity toward reduction of nitrophenol using NaBH4 compared to other Au catalysts with bigger particle sizes (Scheme 3). The reaction mechanism for this nitro reduction is still unclear. But, it has been proposed that it involves an electron transfer from BH4− to Au NPs. We have to remind that NaBH4 is one of the favourite reducing agents for the formation of noble metal NPs and particularly for the formation of Au NPs, the process involving certainly an electron transfer. Other possibility that could be considered is the formation of the stationary concentration of gold hydrides that would be the intermediates effecting the reduction of the nitro group. It would be of interest if in situ spectroscopic techniques could provide some experimental evidence about the reaction mechanism.
:1 gave a very narrow particle size distribution in the range of 1.8 to 2.8 nm. On the other hand, a broad size distribution from 2 to 12 nm was obtained at lower polymer/Au ratio of 3:1. The Au NPs catalyst having polymer/Au ratio of 50:1 showed the best activity toward reduction of nitrophenol using NaBH4 compared to other Au catalysts with bigger particle sizes (Scheme 3). The reaction mechanism for this nitro reduction is still unclear. But, it has been proposed that it involves an electron transfer from BH4− to Au NPs. We have to remind that NaBH4 is one of the favourite reducing agents for the formation of noble metal NPs and particularly for the formation of Au NPs, the process involving certainly an electron transfer. Other possibility that could be considered is the formation of the stationary concentration of gold hydrides that would be the intermediates effecting the reduction of the nitro group. It would be of interest if in situ spectroscopic techniques could provide some experimental evidence about the reaction mechanism.
 | ||
| Scheme 3 | ||
Similar studies of nitrophenol reduction by NaBH4 using Au NPs prepared by HAuCl4 and a radical photoinitiator (Irgacure) have been reported by Scaiano and co-workers.45
De Vos and co-workers have also studied the stability of Au colloids with regard to their ability to promote the aerobic oxidation of 1,2-diols to their corresponding α-hydroxycarboxylates.10,46 Au colloids were prepared from HAuCl4 aqueous solutions by reduction with excess of NaBH4 at 40 °C. The resulting Au NPs were stabilized by poly(vinyl alcohol) (PVA) that was in the solution during the preparation of Au NPs. TEM study of the colloidal Au sample determined that over 60% of the total number of Au NPs had a diameter between 2 and 4 nm. This result is in agreement with the ability of NaBH4 to promote a fast reduction of Au salts leading to the formation of small Au NPs.47 PVA-stabilized Au NPs were found to be active for the aerobic oxidation of 1,2-propanediol, 1,2-hexanediol and 1,2-ocatanediol in tert-butanol and water with an almost complete selectivity to the corresponding α-hydroxycarboxylates. A turnover frequency (TOF) value of 850 h−1 is comparable to those reported for the oxidation of the same substrates for carbon supported Au NPs (Scheme 4).48 Although, the reaction mechanism was not addressed, it can be assumed that oxygen activation will form some kind of superoxide species strongly or weakly bonded to Au NPs depending on the charge density of the clusters in a similar way as proposed for the Au-PVP-1.
 | ||
| Scheme 4 | ||
Although the colloidal Au NPs are as active as those supported on solids, a key point of this study was to determine the stability of the colloidal Au solutions over the time elapsed after their preparation. To check aging stability, oxidation of 1,2-propanediol was promoted by Au colloids that were prepared up to 24 days before their use. Although a certain decrease was observed in the catalytic activity (measured as TOF value) depending on the aging time, it was observed that at least 35% of the original activity was maintained. Another point in the use of colloidal Au as catalyst is the possibility to recover Au after the reaction and reuse for consecutive runs. This point was addressed by using a poly(dimethylsiloxane) (PDMS) membrane to recover the Au sol by nanofiltration, whereby over 99% of Au could be retained by the membrane. Interestingly, no fouling of the membrane was observed. The PDMS membrane allowed the reuse of the Au colloids for four consecutive runs.
In the research field of colloidal Au catalysts, the effects of stabilizers on their catalysis cannot be neglected. Prati and co-workers investigated the catalytic activity and stability of Au colloids in the presence of various protective agents such as PVA, tetrakishydroxypropylphosphonium chloride (THPC) and citrate in the aerobic oxidation of glycerol.11,12 PVA-stabilized Au NPs (AuPVA) were prepared by sodium borohydride reduction of aqueous Au ion solution in the presence of PVA as a stabilizer. THPC- and citrate-stabilized Au NPs (AuTHPC and Aucitrate) were synthesized by reducing the aqueous Au solution, with THPC and citrate as reducing agents and stabilizer. TEM analysis for the three different methods of synthesis of colloidal Au established that the mean diameters of Au NPs increase in the order AuTHPC (2.02 nm) < AuPVA (2.45 nm) < Aucitrate (9.76 nm). After the aging for one week in the absence of light, no significant agglomeration was observed in the case of AuPVA (2.50 nm) and Aucitrate (10.64 nm), whereas drastic increase in size was found for AuTHPC. This result indicated that a bulky stabilizer, PVA, was more effective as a protective agent for small NPs than an electrostatic stabilizer, THPC. A series of Au colloids was tested as catalysts for the oxidation of glycerol in the liquid phase (Scheme 5).
 | ||
| Scheme 5 | ||
The catalytic activity increased with decreasing the particle size, which is in good agreement with previous studies using solid-supported Au catalysts.49 However, a large difference in the activity (TOF of 2478 h−1vs. 715 h−1) was observed for Au colloid samples for which difference in the particle sizes between AuTHPC and AuPVA (2.02 nm vs. 2.45 nm) was small. The authors explained that the lower activity of AuPVA compared to AuTHPC arises from the limited accessibility of the substrate to Au when the stabilizer was PVA due to steric congestion caused by these macromolecules. Overall, these catalytic data nicely illustrate that the catalytic activity of Au colloid must depend largely on the nature of the stabilizer.
Dendrimers are among the preferred ligands for Au13,50 and other metal51 NPs. One study using the fifth generation of poly(amidoamine) (PAMAM) dendrimers and effecting the formation of the Au NPs by reduction of HAuCl4 with a ten-fold excess of NaBH4 has shown that the resulting composite colloids are active to promote CO oxidation.50 TEM images of the material showed that the average Au NP size is 2.2 nm.
Using eosin dye as probe molecule to study the catalytic behavior of Au NPs in reverse micelles, an interesting observation of the influence of the average particle size on the performance of Au NPs has been observed.14 The reverse micelles system of the water–cyclohexane mixture was obtained using Triton X-100 (TX-100, see Scheme 6) as surfactant.
 | ||
| Scheme 6 Structure of Triton X-100. | ||
Formation of Au NPs of various sizes ranging from 10 to 46 nm in average diameter was achieved by a two step procedure in which firstly Au NPs seeds were formed by UV irradiation of an aqueous solution of HAuCl4 containing TX-100. Au NP seeds were grown by reduction of HAuCl4 with ascorbic acid leading to mostly spherical particles. The catalytic experiments were performed with keeping constant the amount of Au and the reaction was monitored by photospectrometry. The important point of the study was not only that the unsupported colloidal Au acts as a catalyst for the BH4− eosin reduction, but also that the catalytic activity of the NPs exhibits two different size regimes. Thus, when the specific reaction rate of eosin disappearance divided by the unit area of the catalyst is plotted against the particle size, two straight lines were obtained. The shift between the two regimes occurred around 15 nm. These experimental results indicate that besides the particle diameter some other factors must control the catalytic activity of the colloidal Au. However, no suggestions about this intriguing additional factor were provided. It could be worth studying if changes in the particle morphology and/or formation of preferential crystal planes upon growth of the NPs are responsible for the shift of the catalytic activity. In fact, a topic of much current interest in Au catalysis is to find evidence to support theoretical calculations that predict remarkable catalytic differences depending on the crystallographic planes of Au crystallites exposed in the model to the substrates.52 This aspect as well as the morphology of the Au NPs has been neglected in most of the studies, mainly due to the difficulty in the preparation of well-defined samples with predominant crystal planes. It could happen that the procedure based on the two-step Au NP formation used in the present study could be a suitable protocol to prepare colloidal Au with defined crystal structures. Besides sample preparation, the difficulty of this type of studies focused on characterization of the activity of specific crystal planes arises from the required TEM characterization to prove the structure and orientation of the nanocrystals.
3 Citrate-stabilized Au NPs
An early example of citrate-stabilized Au NPs was reported by Freund and co-workers who studied the activity of Au for the reaction between ferricyanide and thiosulfate (Scheme 7).15,16 | ||
| Scheme 7 | ||
The citrate-stabilized Au NPs were prepared in accordance with the method reported by Freund and co-workers.15 The aqueous mixture of HAuCl4 and citrate was stirred at reflux temperature at different citrate concentrations, yielding the citrate-stabilized Au NPs with various mean particle sizes. The mean diameter of the particle was increased by decreasing the concentration of citrate. The obtained Au NPs were stable even after 4 weeks. Spectroscopic analysis showed that the obtained Au NPs were prolate spheroids. The catalytic activity of the Au catalysts was investigated in the reaction between ferricyanide and thiosulfate ions. It was observed that the catalytic rates increased with increasing concentration of the catalyst linearly. From the kinetic study, the authors claimed that this reaction occurred on the surface of Au NPs and was not diffusion controlled reaction.
One important issue in heterogeneous Au catalysis, besides the assessment of the intrinsic activity of Au colloids that is the central topic of this review, is the specific catalytic activity of Au atoms in different crystallographic planes or at the edges and corners of a NP. Theoretical calculations using Au clusters clearly predicted that the activity of the unsaturated Au atoms at corners and edges should be higher than those in other positions, and also that unsaturated sites should exhibit increasing catalytic activity as the coordination number of Au is lower. This issue is of high importance since the preparation of highly active Au catalysts should be based on the control not only of the NP dimensions but also of the morphology of the Au crystallites. The issue of preferential catalytic activity by certain Au crystal planes has been largely neglected and poorly studied due to the difficulty in preparation of well-defined samples. In this context, it has been possible to prepare polygonal or spherical Au NPs of average edge ranging from 100 to 158 nm that were used as catalyst for NaBH4 reduction of nitrophenol to aminophenol and for the aerobic oxidation of three different D-hexoses.17 Apparently, the cation accompanying the citrate anion plays a key role in controlling the polygonal (ferric ammonium citrate) or spherical (triammonium citrate) morphology of the Au NPs resulting from the reduction. TEM studies clearly showed the preferential growth of the Au(111) lattice planes although the thickness distribution of the crystallites was not determined. The catalytic activity of these prismatic Au NPs was compared with that of spherical Au NPs that were prepared by triammonium citrate reduction of HAuCl4. Importantly, it was found that the catalytic activity of polygonal Au NPs can be 1000 times higher than that of spherical Au NPs for nitrophenol reduction.53
Similarly, Au NPs of different sizes ranging from 6 to 57 nm prepared by the conventional citrate reduction method (larger than 15 nm of Au NP diameter) or the borohydride method (6 nm diameter) are effective to promote the chemiluminescence from luminol.54,55 Previous studies in the literature have shown that supported Au catalysts are able to promote the chemiluminescence of luminol using hydrogen peroxide and ferricyanide as reagents.19,56,57 This reaction requires the generation of hydroxyl radicals by reduction of hydrogen peroxide that subsequently attack luminol to form 3-aminophthalate in its singlet excited state that relaxes to the corresponding ground state by emitting one photon. Safavi and co-workers have shown that unsupported colloidal Au can also be used as catalyst for this reaction.18 Furthermore, colloidal Au was also able to form in situ hydrogen peroxide by promoting the aerobic oxidation of hydrazine. The overall tandem reactions catalyzed by Au NPs responsible for luminol chemiluminescence are presented in Scheme 8.
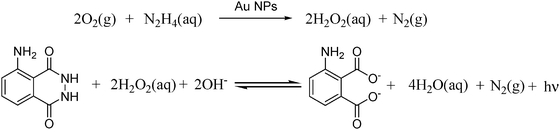 | ||
| Scheme 8 | ||
By measuring the chemiluminescence using a flow injection system and monitoring the emission intensity of a cell over time, it was concluded that colloidal Au NPs were efficient in catalyzing the tandem reactions and their activity depends on average particle sizes. It was observed that the most efficient sample was that in which the average particle size was 15 nm and beyond this value the catalytic activity decreases as the average diameter of the NPs increases. However, it is important to know that chemiluminescence intensity may not reflect properly the catalytic activity of the Au samples since the efficiency of Au catalysts in generating the singlet excited state of aminophthalate can be counter balanced by the quenching of this excited state by Au NPs, a process that has been found to be more favorable for small sized NPs.19,56 Thus, it may happen that Au NPs smaller than 15 nm in size can be very efficient as catalysts for the formation of aminophthalate but this catalytic activity could be masked by subsequent inhibition of the chemiluminescence emission due to the efficient quenching of the aminophthalate excited state by small Au NPs. In this sense, it is advisable to confirm chemiluminescence activity data with other analytical techniques that are not influenced by this quenching effect.
4 Au NPs having other stabilizers
Aerobic oxidation is one reaction type for which supported Au has been found among the best catalytic systems.58 The remarkable influence of the catalytic activity of supported Au depending on the support and the fact that this catalytic activity decreases and eventually disappears when the average Au particle size is above 20 nm are widely accepted as some of the main reasons why colloidal Au should be devoid of any catalytic activity. For this reason, it is important in this review to highlight those reports in which various nanoscopic Au materials have been used as oxidation catalysts to specifically address the key issue of intrinsic activity of unsupported Au.Au nanowires (NWs) have been prepared by reduction of HAuCl4 in oleylamine at 80 °C.59 The NWs exhibit a remarkably narrow diameter size distribution centered in 1.4 nm as it was already reported for this protocol based on oleylamine.59 The aerobic oxidation at benzylic positions by Au NWs under atmospheric pressure has been reported.20 This sample of Au NWs has been tested for the aerobic oxidation of styrene at 100 °C. A mixture of benzaldehyde, acetophenone and styrene oxide was obtained (Scheme 9). However, it was observed that the TEM image of Au NWs after usage exhibits the presence of dark spots that could correspond to Au NPs. It may happen that Au NPs, probably coordinated with the ligand, are the species that are formed from the NWs and migrate to the liquid phase and act as the real catalytic species. This point should have been addressed by careful survey of the species present in the liquid phase and probably also by conventional leaching experiments based on hot filtration at the reaction temperature.
 | ||
| Scheme 9 | ||
A second reaction that has also been performed using Au NWs as catalysts was the benzylic oxidation of aromatic hydrocarbons20 (Scheme 10). The experimental results have been complemented by DFT calculations of a possible reaction mechanism for ethylbenzene oxidation. This theoretical study concluded that the reaction starts with the endothermic dissociation of O2 on the Au NWs (reaction energy; −0.11 eV, activation barrier; 1.01 eV). It was estimated that the O2 dissociation is facilitated by the surface roughness characteristic of Au NWs compared to the models in which a flat Au(111) surface is considered. Subsequently, ethylbenzene is activated by hydrogen abstraction of surface bound oxygen atoms leading to the generation of phenylethyl radicals and surface bound hydroxy groups, phenylethyl radicals would be trapped by molecular oxygen, triggering a peroxyl oxidation chain mechanism. The key point is again that no support is needed in the mechanism and that the Au colloid with defined morphology exhibits catalytic activity for the aerobic oxidation of benzylic functions.
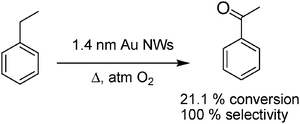 | ||
| Scheme 10 | ||
Octylsilane-stabilized colloidal Au NPs of particle sizes below 2.1 nm were found to be highly efficient for the aerobic oxidation of trans-stilbene to the corresponding epoxide and cyclohexene to 2-cyclohexen-1-one using tert-butyl hydrogen peroxide (TBHP) as a radical initiator.21 The colloidal Au particles were prepared by the two-phase method in which a transfer agent that will act also as a ligand is used to move HAuCl4 from the aqueous to the organic phase. In the present case, Thieuleux, Caps and co-workers prepared three similar samples with small particle size differing in the phase transfer agent used. Besides, the conventional tetraoctylammonium ion, two other neutral ligands, namely n-octylsilane and octylthiol, were also used. It was found that the three Au colloids exhibited high catalytic activity for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond epoxidation (trans-stilbene, epoxide yield between 40–90%) or allylic oxidation of cyclohexene, conversion between 40%. As a result of comparison of the activity between a commercially available TiO2-supported Au (Au/TiO2, 1.5 wt% Au, 3.5 nm particle diameter) and Au colloids, the initial reaction rate observed for the colloidal Au solutions was always higher than that obtained with the supported catalyst. This fact was attributed to the lack of mass transfer limitations in the case of solubilized colloidal Au, and to the higher dispersion of Au atoms in the Au colloid. The important point is that these experiments really showed that unsupported Au NPs can activate molecular oxygen from the air without the assistance of any support and can have an intrinsic activity even higher than conventional supported Au catalysts. However, as expected in view of the previous comments, it was observed that tetraoctylammonium stabilized colloids rapidly deactivate, a fact that was ascribed to the agglomeration of poorly stabilized Au NPs. In line with this rationalization, the slower deactivation of octylthiol capped Au NPs was interpreted as due to a better stabilization of the Au colloid arising from the stronger Au–ligand interaction. It remains, however, to be understood why the most active colloidal sample was not the one with the smallest particle size (6 nm) and if capping agents strongly bonded to the Au surface decrease significantly the catalytic activity of the colloid.
C double bond epoxidation (trans-stilbene, epoxide yield between 40–90%) or allylic oxidation of cyclohexene, conversion between 40%. As a result of comparison of the activity between a commercially available TiO2-supported Au (Au/TiO2, 1.5 wt% Au, 3.5 nm particle diameter) and Au colloids, the initial reaction rate observed for the colloidal Au solutions was always higher than that obtained with the supported catalyst. This fact was attributed to the lack of mass transfer limitations in the case of solubilized colloidal Au, and to the higher dispersion of Au atoms in the Au colloid. The important point is that these experiments really showed that unsupported Au NPs can activate molecular oxygen from the air without the assistance of any support and can have an intrinsic activity even higher than conventional supported Au catalysts. However, as expected in view of the previous comments, it was observed that tetraoctylammonium stabilized colloids rapidly deactivate, a fact that was ascribed to the agglomeration of poorly stabilized Au NPs. In line with this rationalization, the slower deactivation of octylthiol capped Au NPs was interpreted as due to a better stabilization of the Au colloid arising from the stronger Au–ligand interaction. It remains, however, to be understood why the most active colloidal sample was not the one with the smallest particle size (6 nm) and if capping agents strongly bonded to the Au surface decrease significantly the catalytic activity of the colloid.
5 “Naked” Au NPs
Small particles of gold differ from the bulk as they contain edge atoms that have a low coordination number60 and can adopt binding geometries which lead to a more reactive electronic structure.61 Thus, the main reason of the catalytic properties of gold nanoparticles lies at least partly in their geometric structure.62 Truly naked Au NPs are probably impossible to prepare except maybe in the gas phase. A certain number of reports have addressed the formation and reactivity of Au clusters in the vacuum chamber of a mass spectrometer.63 The atomic (geometric) and electronic structure of gas-phase and supported clusters and particularly gold clusters62,64–66 and their chemical reactivity67 on the size of the clusters are among the foremost challenges of modern cluster science.68 Early studies have shown that the reactivity of small gas-phase gold clusters (Aun in the range of up to 30 atoms) depends on the exact number of atoms and the charge state of the clusters, with cluster anions with an even n showing enhanced reactivity.69 Further, it has been shown theoretically that the reactivity toward oxygen of gold cluster anions originates from their propensity to transfer electronic charge into the antibonding 2π* orbital of molecular oxygen, thus weakening the O–O bond which becomes activated into a superoxo- or peroxo-like state. A similar influence of charge density occurs upon binding of CO to charged (gas-phase or supported) gold clusters.70 Studies in the gas phase are limited by the conditions of high vacuum required for the preparation and of limited value in understanding the chemical reactivity of Au colloids in condensed media.In a very interesting study trying to address the catalytic activity intrinsically due to Au, in the absence of any support, Rossi and co-workers reported the aerobic oxidation of glucose in aqueous media under strongly basic conditions catalyzed by naked Au NPs.22–25 It was found that unsupported Au colloids exhibited comparable initial activity to that of Au NPs supported on active carbon. During the course of the reaction unsupported Au colloids undergo considerable deactivation, losing most of the initial activity of the fresh sample. Interestingly, if during the reaction active carbon is added to the reaction medium, then the activity of Au colloid was preserved for longer time.
The above results were interpreted considering that Au colloid has intrinsic activity to promote the aerobic oxidation of glucose in aqueous solution even in the absence of any support. But due to the tendency of Au NPs to grow and agglomerate, these Au NPs are not stable and undergo deactivation. According to this, the role of active carbon will be just to stabilize the size of the Au NPs impeding their growth and agglomeration without introducing any relevant catalytic activity. In fact, based on these observations, it has been proposed that in contrast to the case in which Au NPs are supported on metal oxides, particularly those in which the metal can exhibit two redox states, those reactions catalyzed by activated carbon supported Au should correspond to reactions and conditions in which Au NPs alone should be also catalytically active. It would be important to check the validity of this guideline and to delineate which reactions can or cannot be promoted by colloidal Au.
The above colloidal Au catalyst using a large excess of glucose as a stabilizer was also applicable for the oxidative polymerization.26,27 In the case of the synthesis of polyaniline from aniline,26 a comparison of the catalytic activity of colloidal Au with other solid-supported catalysts was provided. Because of the short catalytic life of colloidal Au in oxidation reactions,22 unsupported Au NPs were less active than the solid catalyst of Au/TiO2.
Fine Au powder of around 70 nm average particle size has been found to be active to promote the low temperature CO oxidation to CO2.28 Previous studies with Au powder of much larger particle size (ranging from 500 to 800 nm) have shown that these large Au particles are inactive for the same process.71 The authors checked the different impurity content for both Au samples and concluded that it was not relevant and that the different catalytic behavior should be attributed to the large difference in the particle size.
Moreover, this fine Au powder was also found to be more reactive with oxygen and that it can retain some oxygen (probably as Au oxide) during thermal pretreatments if calcination is carried out in the presence of oxygen. In addition, the powdered Au was also found to be more efficient in promoting CO oxidation than a single Au(110) crystal. This better performance of fine powdered Au over single Au crystals was attributed to the presence of a larger number of Au atoms in edges and defects to interact with the substrate in powdered samples than in the perfect Au(110) crystals.72 However, in spite of catalytic activity of nanoparticulate Au powder, it is worth commenting that this unsupported particulate Au sample exhibits lower performance than that of TiO2 supported Au. The rate constant of CO oxidation per unit surface area of Au NPs was higher by two orders of magnitude for Au/TiO2 than for Au powder.
Trying to address the intrinsic catalytic activity of Au in the absence of any support that could cooperate in the reaction mechanism, Dumesic and co-workers have studied the catalytic activity of Au nanotubes partially embedded in a porous inert polycarbonate membrane for the oxidation of CO.29 Au nanotubes were prepared by electroless deposition of Au within the pores of a 10 μm thick polycarbonate membrane containing 220 nm diameter pores. Scanning electron microscopy (SEM) analysis revealed that the Au nanotubes exhibit a high level of surface roughness (around 53 nm) that could be related to the sintering of individual Au NPs. Au nanotubes partially embedded into the polycarbonate membrane are contacted on one side by a gas stream containing CO and O2 and, on the other, an aqueous solution at the desired pH and containing or not H2O2. The most remarkable conclusion from this work is the occurrence of a promotional effect of basic water or base treatment on the activity of Au nanotubes. It was claimed that negatively charged hydroxyl groups formed by heterolytic dissociation of liquid water can be reactive against CO, favoring the oxidation of CO to CO2.73 In addition, a small amount of hydrogen (0.05 μmol min−1 per gram of Au) was detected when the Au nanotubes came into contact with gaseous CO and liquid water. Therefore, it was concluded that the presence of base favors the presence of these hydroxy groups.
Another mechanistic detail is that the presence of oxygen in the system generates a small amount of hydrogen peroxide (1.5 μmol min−1 per gram of Au). In this regard, if the liquid phase in contact with the Au nanotubes contains some hydrogen peroxide as a reagent instead of molecular oxygen, then, a much higher catalytic activity of the Au membrane is observed. Again, what is clear from the previous study is that Au nanotubes without the assistance of any support exhibit activity for the CO oxidation of CO2. The mechanistic details suggest that this activity for CO oxidation is related to the ability of the Au nanotubes to promote electron transfer reactions. This electron transfer process becomes facilitated under basic conditions or when an oxidizing reagent (hydrogen peroxide) is present in the reaction mixture. It has been claimed that one of the major roles of the support in many reactions catalyzed by Au is to provide surface-bound hydroxyl groups. Apparently, these hydroxyl groups do not need to be necessarily associated to the support and they can be supplied by liquid water particularly at high pH values. Under these favorable conditions, it was estimated that the rates of CO oxidation by oxygen on Au nanotubes are similar to those rates measured using Au NPs supported on oxide supports. Estimation of TOF for CO oxidation by oxygen in liquid water at high pH values gives about 0.04 s−1 which lies in the range of TOF values reported for supported Au catalysts that can vary from 0.01 to 0.5 s−1.74,75
As noted in the previous section, colloidal Au NPs have been prepared by the reduction of Au salts using borohydride or citrate as a reducing agent and the stabilizers such as polymers or citrates were needed to prevent their agglomeration. However, these foreign stabilizers might affect the intrinsic catalytic properties of the obtained Au NPs. To investigate the characteristics of naked Au NPs without addition of any foreign stabilizers, Meisel and co-workers prepared Au NPs using Au oxide (Au2O3) as a starting material by H2 reduction (Scheme 11). The preparation procedure for naked Au NPs30 consisted in treating commercially available Au2O3 with 1.5 atm of ultrapure molecular hydrogen at 70 °C in deionized water, which leads to the formation of Au NPs of 50 nm in mean diameter. The obtained Au NPs can be employed as active redox catalysts for hydrogen evolution from isopropanol oxidation.
 | ||
| Scheme 11 | ||
Scaiano and co-workers76–78 studied the thermal decomposition of dicumyl peroxide into the alcohol and ketone promoted by photochemical excitation on the surface plasmon band (SPB) of unsupported Au NPs (Scheme 12).
 | ||
| Scheme 12 | ||
The target was to establish the local temperature on the surface of Au NPs that results upon laser excitation at its surface plasmon band and subsequent relaxation. Cumyl peroxide decomposition was used as molecular thermometer. The synthesis of unsupported Au NPs was conducted using Irgacure-2959 (1-[4-(2-hydroxyethoxy)phenyl]-2-hydroxy-2-methyl-1-propane-1-one) as a precursor of a reducing agent. Irgacure-2959 yields ketyl radicals upon 350 nm excitation and the obtained radicals work as reductants for Au3+ to Au0 (Scheme 13).
 | ||
| Scheme 13 | ||
The colloidal Au NPs solutions were stable in water in spite of the absence of polymers or any other typical Au ligand. The shape of Au NPs was mainly spherical and the mean diameter was 12.7 nm. The photocatalytic decomposition of dicumyl peroxide in the presence of unsupported Au NPs was performed by using a laser-drop technique.79 This photochemical irradiation consists in using a needle to form a drop of the solution and once the drop is formed a laser flash is fired to excite the Au NP SPB. Complete conversion of dicumyl peroxide was achieved and the main reaction products were 2-phenyl-2-propanol and acetophenone.
6 Ligand-protected well-defined Au NPs
Various organic ligands such as phosphine and thiol have been used as stabilizers for the synthesis of precise number of Au clusters.80–88 For example, the phosphine ligand has been employed to prepare Au55 clusters.89–92 It has been reported that phosphine-stabilized Au NPs could be immobilized on solid supports such as TiO2 and SiO2 with keeping their NPs structure and exhibited catalytic activity.Thiolate-protected gold clusters have recently become an intensively studied field. Their optical, magnetic and electrochemical properties differ widely from bigger NPs that exhibit surface plasmon resonances.93 For small Au clusters, Garzon was the first to point out that thiolate ligation can cause significant structural distortion of the Au core.94 This could be an indication that the catalytic activity of these clusters should be strongly influenced by the interaction with the thiol ligand.
In 2007, Kornberg95 achieved a breakthrough in the research of Au:SR systems by elucidating the structure of Au102(SR)44 using single-crystal X-ray diffraction. Subsequently, Murray96 and Jin97 experimentally determined the structure of Au25(SR)18, while Hakkinen98 theoretically predicted the structure of this cluster. It would be of interest to determine the catalytic activity of defined Au clusters.
Recently, Tsukuda and coworkers prepared Au:SC18H37 clusters by a reaction between C18H37SH and mixtures of PVP-stabilized Aun clusters (n = 34, 42, 58).99 The Au:SC18H37 samples thus prepared were subjected to recycling size-exclusion chromatography to afford a Au:SC18H37 cluster (core mass: 11 kDa) accompanied by Au38(SC18H37)24. On the basis of the MALDI mass spectra recorded at a low laser power, it was concluded that the 11 kDa Au:SC18H37 cluster is a mixture of Au54(SC18H37)30 and Au55(SC18H37)31. The stability of these clusters could not be explained by the classical structure model, in which the thiolates are bound to the Au54 and Au55 cores. Further, it was proposed that Au54(SC18H37)30 and Au55(SC18H37)31 are composed of a Au37 core whose surface is completely protected by –SR–[Au–SR–]x (x = 1 and 2) oligomers.99 One of the interests of these studies would be to use well characterized Au clusters in catalysis in a way that the relative activity of these clusters could be determined. The long term objective will be to assess the optimal structure of Au size to achieve the maximum efficiency in the catalytic activity of Au by preparing materials in which only the site with maximum activity is present.
Further, Murray and co-workers have shown how to obtain atomic metal core structures of subnanometer clusters Au13[PPh3]4[S(CH2)11CH3]2Cl2 and Au13[PPh3]4[S(CH2)11CH3]4. These species have been characterized by electron microscopy and X-ray absorption spectroscopy. The number of gold atoms in the cores of these two clusters was determined quantitatively using high-angle annular dark field scanning transmission electron microscopy. In addition, multiple scattering-path analyses of extended X-ray absorption fine structure spectra suggested that the Au metal cores of each of these complexes adopt relaxed quasi icosahedral structure. Data from microscopy and spectroscopy studies extended to larger thiolate-protected gold clusters showing a broader distribution in nanoparticle core sizes (183 ± 116 Au atoms) which revealed a bulklike fcc geometry.100 Recently, one-phase size-focusing approach has been used to prepare gold clusters protected by chiral camphorthiolate, Au25-(CamS)18, as a major product and characterized by circular dichroism and mass spectrometry.101
These comments on reports describing ligand protected Au clusters show that it is nowadays possible to prepare well defined Au species whose catalytic activity could be tested for different reactions. Even, if they are not stable under the reactions conditions, the initial catalytic activity combined with theoretical studies will give hints about the optimal geometry of Au NPs to promote catalytic reactions.
Jin and co-workers employed thiol-stabilized Au25 nanoclusters as catalysts for the selective hydrogenation of α,β-unsaturated carbonyl compounds into unsaturated alcohols.31 Various thiol-stabilized Au clusters have been prepared using a two-phase method.102 The catalytic hydrogenation of benzalacetone was performed with H2 at atmospheric pressure selectively to unsaturated alcohol with 22% conversion in 3 h using Au25(SC2H4Ph)18 as catalyst (Scheme 14).
 | ||
| Scheme 14 | ||
However, complete selectivity towards the unsaturated alcohols was obtained when the conversion was below 50%. The effect of thiol ligand was investigated by using alkyl thiol as stabilizer instead of aryl thiol. Significant differences in catalytic activity and product selectivity were not observed. The Au25(SC2H4Ph)18 catalyst could be immobilized on various oxide supports such as Fe2O3, TiO2 and SiO2 and the supported Au25 cluster catalysts showed comparable activity and selectivity to the homogeneous ones. Therefore, this comparison between Au colloidal solutions and heterogeneous catalysts provides again a nice example illustrating that the catalytic activity of Au containing solids can also be observed for the unsupported Au NPs. Further studies describing the catalytic activity of well defined Au clusters of a defined number of Au atoms with different ligands will be very useful and could delineate the intrinsic catalytic of Au NPs as catalysts. On the other hand, it should be commented that optimization of the catalytic sites for one substrate cannot necessarily mean that the catalyst has been optimized for all the substrates. Depending on the structure of the α,β-unsaturated ketone, it may happen that different Au clusters exhibit different order of relative catalytic activity depending on the interaction with the substrate.
Fernández and co-workers reported catalytic diboration of styrene using binap-stabilized Au NPs (Scheme 15).32
 | ||
| Scheme 15 | ||
The Au NPs were formed in situ during the catalytic reaction by employing [(binap)Au2Cl2] and [(binap)Au2I2] as catalyst precursors in the diboration of styrene. This reaction was performed in THF at room temperature in the presence of base. For example, [(binap)Au2I2] gave complete conversion of styrene to a diborated compound with 99% selectivity in 2 h and almost no conversion in the absence of base. The in situ reduction of [(binap)Au2I2] by a diboronate reagent (bis(4-methylcatecholate)diborane; B2cat) was confirmed by XRD and TEM measurements. The in situ formed Au NPs were highly stable and could be reused in subsequent catalytic runs. When (R)-binap was used as the ligand for Au, asymmetric induction was not observed. Another example that has been recently reported is Sonogashira coupling of phenylacetylene and iodobenzene promoted by Au complexes.103 The observed induction period could be attributed to the formation of Au NPs generated during the course of the reaction.
Overall, this study provides compelling evidence of a phenomenon that is probably more general in many reactions catalyzed by, supposedly, homogeneous AuI and AuIII salts and complexes. Considering that many Au complexes and salts exhibit low stability and hence the reaction condition sometimes requires the use of reducing agents and heating, it could likely be that the Au salts or ligands do not survive the reaction conditions and very early they decompose to give Au NPs that really act as active catalytic species.
Interestingly, Aoshima and co-workers have prepared stable and durable Au nanoclusters of less than 4 nm using thermosensitive vinyl ether star polymers obtained through living cationic polymerization.104 The resulting clusters effectively catalyzed the aerobic oxidation of benzyl alcohol in the presence of potassium hydroxide in water at 32 °C and observed 99% of benzoic acid. The clusters were easily separated from the reaction mixture by utilizing their thermosensitive nature, allowing reusing for six times without any decrease in its activity. Even after sixth use, the catalyst was able to promote the formation of 99% of benzoic acid at final time.104
7 Summary and outlook
The present review has commented examples from the literature that clearly prove the intrinsic activity of unsupported colloidal Au NPs for some of the typical reactions promoted by heterogeneous catalysts based on supported Au NPs, such as low temperature CO oxidation and aerobic alcohol oxidation. In these two reactions, the activity of Au colloids could be comparable or even surpass those of conventional supported Au NP catalysts. In other cases, such as BH4− reductions that have less industrial impact due to the high cost of the reagent compared to molecular hydrogen, the activity of colloidal Au has also been proved, although comparison with supported Au catalysts has not been provided. In this context, it necessary to expand further the range of reactions catalyzed by colloidal Au to those that have been carried out with supported Au and to provide the comparison of the activity between supported and unsupported Au catalysts.With regard to the activity of colloidal Au, an issue that has been not sufficiently addressed is the role of the ligand used to stabilize the NPs from the particle growth and the influence of its concentration on the catalytic activity. It would be interesting to clearly demonstrate whether the ligand plays any role in the catalytic reactions enhancing or disfavoring the intrinsic activity of Au NPs besides avoiding particle aggregation. In this regard, it has been frequently assumed that strong Au–ligand interactions occurring in thiols and other sulfur-containing ligands or in phosphines must decrease considerably the catalytic activity of the NPs. However, it could be that a compromise between activity and reusability requires an optimal concentration of some strongly coordinating ligands that may cause a decrease in its initial reaction rate but allow longer durability. In addition, it would be convenient to provide examples in which this assumption is quantitatively established by measuring TOF or other kinetic parameters. Also from an applied point of view it is of much interest to exploit the possibility to find ligands that can allow the reuse and recovery of the catalyst or to make possible performing continuous flow reactions with colloidal Au NPs.
Finally, considering that Au colloids offer the advantage with respect to any supported Au catalyst that can be prepared following highly reliable experimental protocols under a variety of conditions including solvents and reducing agents, it could be interesting to prepare samples in which characterization convincingly shows that the surface of the Au NP is constituted preferentially by well-defined crystallographic planes with narrow average particle size. This type of well-defined samples can be very close to theoretical models and could be used to validate predictions that some catalytic reactions are better promoted by certain crystallographic planes.105 Due to the simplicity of preparation of unsupported Au and the possibility of obtaining them in small average particle size, Au colloids can be extremely useful to prove and validate the preference of certain catalytic reactions for certain orientations of the crystals. Also, formation, characterization and study of the catalytic activity of well defined Au clusters will be of great interest to understand the optimal geometry of active sites. These experimental studies should be complemented by theoretical calculations of the transition states and reaction mechanisms.
It can be anticipated that the research area of colloidal Au will grow still and will become important for the development of Au catalysis in particular for modeling and testing theoretical concepts.
Acknowledgements
Financial support by the Spanish Ministry of Science and Innovation (CTQ2009-15865, CTQ2010-18671 and Consolider Multicat) is gratefully acknowledged.References
- M. Haruta, ChemPhysChem, 2007, 8, 1911 CrossRef CAS.
- V. Zielasek, B. Jürgens, C. Schulz, J. Biener, M. M. Biener, A. V. Hamza and M. Bäumer, Angew. Chem., Int. Ed., 2006, 45, 8241 CrossRef CAS.
- C. Xu, J. Su, X. Xu, P. Liu, H. Zhao, F. Tian and Y. Ding, J. Am. Chem. Soc., 2007, 129, 42 CrossRef CAS.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374 CrossRef CAS.
- H. Tsunoyama, N. Ichikuni, H. Sakurai and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 7086 CrossRef CAS.
- H. Tsunoyama, T. Tsukuda and H. Sakurai, Chem. Lett., 2007, 36, 212 CrossRef CAS.
- H. Tsunoyama, H. Sakurai, N. Ichikuni, Y. Negishi and T. Tsukuda, Langmuir, 2004, 20, 11293 CrossRef CAS.
- W. Hou, N. A. Dehm and R. W. J. Scott, J. Catal., 2008, 253, 22 CrossRef CAS.
- I. Biondi, G. Laurenczy and P. J. Dyson, Inorg. Chem., 2011, 50, 8038 CrossRef CAS.
- P. G. N. Mertens, M. Bulut, L. E. M. Gevers, I. F. J. Vankelecom, P. A. Jacobs and D. E. De Vos, Catal. Lett., 2005, 102, 57 CrossRef CAS.
- L. Prati, P. Spontoni and A. Gaiassi, Top. Catal., 2009, 52, 288 CrossRef CAS.
- A. Villa, D. Wang, D. S. Su and L. Prati, ChemCatChem, 2009, 1, 510 CrossRef CAS.
- K. Hayakawa, T. Yoshimura and K. Esumi, Langmuir, 2003, 19, 5517 CrossRef CAS.
- T. K. Sau, A. Pal and T. Pal, J. Phys. Chem. B, 2001, 105, 9266 CrossRef CAS.
- P. L. Freund and M. Spiro, J. Phys. Chem. A, 1985, 89, 1074 CrossRef CAS.
- P. L. Freund and M. Spiro, J. Chem. Soc., Faraday Trans., 1986, 82, 2277 RSC.
- M. H. Rashid and T. K. Mandal, Adv. Funct. Mater., 2008, 18, 2261 CrossRef CAS.
- A. Safavi, G. Absalan and F. Bamdad, Anal. Chim. Acta, 2008, 610, 243 CrossRef CAS.
- C. Duan, H. Cui, Z. Zhang, B. Liu, J. Guo and W. Wang, J. Phys. Chem. C, 2007, 111, 4561 CAS.
- L. Hu, X. Cao, J. Yang, M. Li, H. Hong, Q. Xu, J. Ge, L. Wang, J. Lu and L. Chen, Chem. Commun., 2011, 47, 1303 RSC.
- M. Boualleg, K. Guillois, B. Istria, L. Burel, L. Veyre, J.-M. Basset, C. Thieuleux and V. Caps, Chem. Commun., 2010, 46, 5361 RSC.
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812 CrossRef CAS.
- P. Beltrame, M. Comotti, C. Della Pina and M. Rossi, Appl. Catal., A, 2006, 297, 1 CrossRef CAS.
- M. Comotti, C. Della Pina, E. Falletta and M. Rossi, Adv. Synth. Catal., 2006, 348, 313 CrossRef CAS.
- M. Comotti, C. Della Pina and M. Rossi, J. Mol. Catal. A, 2006, 252, 89 CrossRef.
- Z. Chen, C. Dellapina, E. Falletta, M. Lofaro, M. Pasta, M. Rossi and N. Santo, J. Catal., 2008, 259, 1 CrossRef CAS.
- C. D. Pina, E. Falletta, M. Lo Faro, M. Pasta and M. Rossi, Gold Bull., 2009, 42, 27 CrossRef.
- Y. Iizuka, T. Tode, T. Takao, K. Yatsu, T. Takeuchi, S. Tsubota and M. Haruta, J. Catal., 1999, 187, 50 CrossRef CAS.
- M. A. Sanchez-Castillo, C. Couto, W. B. Kim and J. A. Dumesic, Angew. Chem., Int. Ed., 2004, 43, 1140 CrossRef CAS.
- G. Merga, N. Saucedo, L. C. Cass, J. Puthussery and D. Meisel, J. Phys. Chem. C, 2010, 114, 14811 CAS.
- Y. Zhu, H. Qian, B. A. Drake and R. Jin, Angew. Chem., Int. Ed., 2010, 49, 1295 CrossRef CAS.
- J. Ramírez, M. Sanaú and E. Fernández, Angew. Chem., Int. Ed., 2008, 47, 5194 CrossRef.
- T. Ishida and M. Haruta, Angew. Chem., Int. Ed., 2007, 46, 7154 CrossRef CAS.
- H. Miyamura, M. Morita, T. Inasaki and S. Kobayashi, Bull. Chem. Soc. Jpn., 2011, 84, 588 CrossRef CAS.
- H. Miyamura, T. Yasukawa and S. Kobayashi, Green Chem., 2010, 12, 776 RSC.
- C. Lucchesi, T. Inasaki, H. Miyamura, R. Matsubara and S. Kobayashi, Adv. Synth. Catal., 2008, 350, 1996 CrossRef CAS.
- H. Miyamura, R. Matsubara, Y. Miyazaki and S. Kobayashi, Angew. Chem., Int. Ed., 2007, 46, 4151 CrossRef CAS.
- J. Han, Y. Liu and R. Guo, J. Am. Chem. Soc., 2009, 131, 2060 CrossRef CAS.
- H. Tsunoyama, H. Sakurai and T. Tsukuda, Chem. Phys. Lett., 2006, 429, 528 CrossRef CAS.
- P. G. N. Mertens, P. Vandezande, X. Ye, H. Poelman, I. F. J. Vankelecom and D. E. De Vos, Appl. Catal., A, 2009, 355, 176 CrossRef CAS.
- D. Zhao, Z. Fei, W. H. Ang and P. J. Dyson, Small, 2006, 2, 879 CrossRef CAS.
- C. Shan, F. Li, F. Yuan, G. Yang, L. Niu and Q. Zhang, Nanotechnology, 2008, 19, 285601 CrossRef.
- F. Shi and Y. Deng, J. Catal., 2002, 211, 518 Search PubMed.
- F. Shi, Q. Zhang, Y. Ma, Y. He and Y. Deng, J. Am. Chem. Soc., 2005, 127, 4182 CrossRef CAS.
- G. L. Hallett-Tapley, C. L. Crites, M. González-Béjar, K. L. McGilvray, J. C. Netto-Ferreira and J. C. Scaiano, J. Photochem. Photobiol., A, 2011, 224, 8 CrossRef CAS.
- P. G. N. Mertens, I. F. J. Vankelecom, P. A. Jacobs and D. E. De Vos, Gold Bull., 2005, 38, 157 CrossRef CAS.
- F. Porta, L. Prati, M. Rossi and G. Scari, J. Catal., 2002, 211, 464 CAS.
- S. Carrettin, P. McMorn, P. Johnston, K. Griffin and G. J. Hutchings, Chem. Commun., 2002, 696 RSC.
- N. Dimitratos, J. A. Lopez-Sanchez, D. Lennon, F. Porta, L. Prati and A. Villa, Catal. Lett., 2006, 108, 147 CrossRef CAS.
- P. Kracke, T. Haas, H. Saltsburg and M. Flytzani-Stephanopoulos, J. Phys. Chem. C, 2010, 114, 16401 CAS.
- L. M. Bronstein and Z. B. Shifrina, Chem. Rev., 2011, 111, 5301 CrossRef CAS.
- M. Boronat, F. Illas and A. Corma, J. Phys. Chem. A, 2009, 113, 3750 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- G. Frens, Nature (London) Phys. Sci., 1973, 241, 20 CAS.
- K. R. Brown, A. P. Fox and M. J. Natan, J. Am. Chem. Soc., 1996, 118, 1154 CrossRef CAS.
- Z. F. Zhang, H. Cui, C. Z. Lai and L. J. Liu, Anal. Chem., 2005, 77, 3324 CrossRef CAS.
- H. Cui, Z. F. Zhang and M. J. Shi, J. Phys. Chem. B, 2005, 109, 3099 CrossRef CAS.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096 RSC.
- Z. Huo, C.-K. Tsung, W. Huang, X. Zhang and P. Yang, Nano Lett., 2008, 8, 2041 CrossRef CAS.
- C. Lemire, R. Meyer, S. Shaikhutdinov and H.-J. Freund, Angew. Chem., Int. Ed., 2004, 43, 118 CrossRef.
- G. Mills, M. S. Gordon and H. Metiu, J. Chem. Phys., 2003, 118, 4198 CrossRef CAS.
- K. J. Taylor, C. L. Pettiettehall, O. Cheshnovsky and R. E. Smalley, J. Chem. Phys., 1992, 96, 3319 CrossRef CAS.
- W. T. Wallace and R. L. Whetten, J. Am. Chem. Soc., 2002, 124, 7499 CrossRef CAS.
- H. Hakkinen and U. Landman, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, R2278 CrossRef.
- F. Furche, R. Ahlrichs, P. Weis, C. Jacob, S. Gilb, T. Bierweiler and M. M. Kappes, J. Chem. Phys., 2002, 117, 6982 CrossRef CAS.
- H. Hakkinen, B. Yoon, U. Landman, X. Li, H.-J. Zhai and L.-S. Wang, J. Phys. Chem. A, 2003, 107, 6168 CrossRef.
- B. Yoon, P. Koskinen, B. Huber, O. Kostko, B. von Issendorff, H. Hakkinen, M. Moseler and U. Landman, ChemPhysChem, 2007, 8, 157 CrossRef CAS.
- X. Xing, B. Yoon, U. Landman and J. H. Parks, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 165423 CrossRef.
- D. M. Cox, R. Brickman, K. Creegan and A. Kaldor, Mater. Res. Soc. Symp. Proc., 1991, 206, 43 CrossRef CAS.
- B. Yoon, H. Hakkinen, U. Landman, A. S. Worz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403 CrossRef CAS.
- W. C. Ketchie, Y. Fang, M. S. Wong, M. Murayama and R. J. Davis, J. Catal., 2007, 250, 94 CrossRef CAS.
- M. Boronat, P. Concepcion, A. A. Corma, S. Gonzalez, F. Illas and P. Serna, J. Am. Chem. Soc., 2007, 129, 16230 CrossRef CAS.
- S. K. Desai and M. Neurock, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 075420 CrossRef.
- H. H. Kung, M. C. Kung and C. K. Costello, J. Catal., 2003, 216, 425 CrossRef CAS.
- M. Haruta, Stud. Surf. Sci. Catal., 2003, 145, 31 CrossRef CAS.
- C. Fasciani, C. J. B. Alejo, M. Grenier, J. C. Netto-Ferreira and J. C. Scaiano, Org. Lett., 2001, 13, 204 CrossRef.
- N. L. Pacioni, M. González-Béjar, E. Alarcón, K. L. McGilvray and J. C. Scaiano, J. Am. Chem. Soc., 2010, 132, 6298 CrossRef CAS.
- M. L. Marin, K. L. McGilvray and J. C. Scaiano, J. Am. Chem. Soc., 2008, 130, 16572 CrossRef CAS.
- J. T. Banks and J. C. Scaiano, J. Am. Chem. Soc., 1993, 115, 6409 CrossRef CAS.
- W. W. Weare, S. M. Reed, M. G. Warner and J. E. Hutchison, J. Am. Chem. Soc., 2000, 122, 12890 CrossRef CAS.
- Z. Wu, J. Suhan and R. Jin, J. Mater. Chem., 2009, 19, 622 RSC.
- O. Uzun, Y. Hu, A. Verma, S. Chen, A. Centrone and F. Stellacci, Chem. Commun., 2008, 196 RSC.
- A. Ghosh, S. Basak, B. H. Wunsch, R. Kumar and F. Stellacci, Angew. Chem., Int. Ed., 2011, 50, 790 CrossRef.
- W. J. Sommer and M. Weck, Langmuir, 2007, 23, 11991 CrossRef CAS.
- B. Y. Zhu, H. Qian, M. Zhu and R. Jin, Adv. Mater., 2010, 22, 1915 CrossRef.
- C. Briggs, T. B. Norsten and V. M. Rotello, Chem. Commun., 2002, 1890 RSC.
- O. Lopez-Acevedo, K. A. Kacprzak, J. Akola and H. Häkkinen, Nat. Chem., 2010, 2, 329 CrossRef CAS.
- G. Schmid, Chem. Rev., 1992, 92, 1709 CrossRef CAS.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nat. Chem., 2008, 454, 981 CAS.
- H.-G. Boyen, G. Kästle, F. Weigl, B. Koslowski, C. Dietrich, P. Ziemann, J. P. Spatz, S. Riethmüller, C. Hartmann, M. Möller, G. Schmid, M. G. Garnier and P. Oelhafen, Science, 2002, 297, 1533 CrossRef CAS.
- Y. Pei, N. Shao, Y. Gao and X. C. Zeng, ACS Nano, 2010, 4, 2009 CrossRef CAS.
- G. Schmid, Chem. Soc. Rev., 2008, 37, 1909 RSC.
- R. Jin, Nanoscale, 2010, 2, 343 RSC.
- I. L. Garzon, K. Michaelian, M. R. Beltran, A. Posada-Amarillas, P. Ordejon, E. Artacho, D. Sanchez-Portal and J. M. Soler, Phys. Rev. Lett., 1998, 81, 1600 CrossRef CAS.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430 CrossRef CAS.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754 CrossRef CAS.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883 CrossRef CAS.
- J. Akola, M. Walter, R. L. Whetten, H. Hakkinen and H. Gronbeck, J. Am. Chem. Soc., 2008, 130, 3756 CrossRef CAS.
- R. Tsunoyama, H. Tsunoyama, P. Pannopard, J. Limtrakul and T. Tsukuda, J. Phys. Chem. C, 2010, 114, 16004 CAS.
- L. D. Menard, H. Xu, S.-P. Gao, R. D. Twesten, A. S. Harper, Y. Song, G. Wang, A. D. Douglas, J. C. Yang, A. I. Frenkel, R. W. Murray and R. G. Nuzzo, J. Phys. Chem. B, 2006, 110, 14564 CrossRef CAS.
- S. Knoppe, N. Kothalawala, V. R. Jupally, A. Dass and T. Burgi, Chem. Commun., 2012, 48, 4630 RSC.
- M. Zhu, E. Lanni, N. Garg, M. E. Bier and R. Jin, J. Am. Chem. Soc., 2008, 130, 1138 CrossRef CAS.
- A. Corma, R. Juárez, M. Boronat, F. Sánchez, M. Iglesias and H. García, Chem. Commun., 2011, 47, 1446 RSC.
- S. Kanaoka, N. Yagi, Y. Fukuyama, S. Aoshima, H. Tsunoyama, T. Tsukuda and H. Sakurai, J. Am. Chem. Soc., 2007, 129, 12060 CrossRef CAS.
- M. Boronat and A. Corma, Dalton Trans., 2010, 39, 8538 RSC.
| This journal is © The Royal Society of Chemistry 2013 |