Energy transfer cassettes based on organic fluorophores: construction and applications in ratiometric sensing
Jiangli
Fan
,
Mingming
Hu
,
Peng
Zhan
and
Xiaojun
Peng
*
State Key Laboratory of Fine Chemicals, Faculty of Chemical, Environmental and Biological Science and Technology, Dalian University of Technology, Dalian, China. E-mail: pengxj@dlut.edu.cn; Fax: +86-411-8498-6306; Tel: +86-411-8498-6327
First published on 11th October 2012
Abstract
This tutorial review presents some recent developments in the construction and applications of cassettes based on resonance energy transfer between fluorescent dyes in the visible and infrared region. We focused on the contributions of different connections between the energy donor and acceptor according to the “through-space” and “through-bond” methods, and emphasised their applications in ratiometric sensing for the detection of ions and small molecules.
 Jiangli Fan | Jiangli Fan received her PhD from the Dalian University of Technology (China) in 2005. In 2010, she attended the University of South Carolina as a visiting scholar. She is currently an associate professor at the State Key Laboratory of Fine Chemicals, Dalian University of Technology. Her research is focused on fluorescent dye-based probes and their biological applications. |
 Mingming Hu | Mingming Hu received her Bachelor of Engineering degree from the Dalian Jiaotong University (China) in 2007. She is now a PhD candidate under the supervision of Prof. Xiaojun Peng and Associate Prof. Jiangli Fan in the Dalian University of Technology (China). Her research interests include fluorescent molecular systems and their applications in small molecule (ion) recognition. |
 Peng Zhan | Peng Zhan obtained his Bachelor of Science degree from the Anhui University of Science and Technology (China) in 2010. And he will receive the Master degree of Applied Chemistry in 2013 under the supervision of Associate Prof. Jiangli Fan in the Dalian University of Technology (China). His research is focused on the designing and application of TBET-based fluorescent probes. |
 Xiaojun Peng | Xiaojun Peng received his PhD from the Dalian University of Technology in 1990. After completing postdoctoral research in Nankai University, he worked at the Dalian University of Technology in 1992. In 2001 and 2002, he attended the Stockholm University and Northwestern University as a visiting scholar. He is currently the director of the State Key Laboratory of Fine Chemicals of China at the Dalian University of Technology, where he is also a professor. His research interests cover fluorescent dyes for the bio-imaging, biolabeling, and photochemistry of supramolecules. |
1. Introduction
Small molecular organic dyes, such as BODIPY, rhodamine, fluorescein, coumarin and cyanine derivatives have been widely used in bioimaging, biosensing, medical diagnosis, and environmental detection.1 However, the undesirable photophysical properties of various fluorophores still constrain the full potential of their applications. For instance, all these bright organic dyes usually have the serious disadvantage of very small Stokes shifts (typically less than 25 nm), which can lead to serious self-quenching and fluorescence detection errors because of excitation backscattering effects.On the other hand, measuring fluorescence by an increase of the fluorescence intensity without much shift of either excitation or emission wavelength can be influenced by many factors, such as the localization of the probe, changes of environment around the probe (pH, polarity, temperature, and so forth), emission collection efficiency, effective cell thickness in the optical beam, and changes in the excitation intensity. To reduce the influence of such factors, ratiometric measurement is utilized, namely, simultaneous recording of the fluorescence intensities at two wavelengths and calculation of their ratio.2 Besides internal charge transfer (ICT) mechanism using a single fluorophore to get ratiometric changes, the exploration of multifluorophores with energy donor–acceptor architectures can achieve large pseudo-Stokes shifts. It can also afford simultaneous recording ratio signals of two emission intensities at different wavelengths, which could provide a built-in correction for the environmental effects and supply a facile method for visualizing complex biological processes at the molecular level, such as DNA detection, protein labelling, nucleic acid regulation, and other biomarkers. Consequently, studying the construction of energy donor–acceptor architectures, or “energy transfer cassettes” is very interesting and significant to many chemists, biologists, environmentalists, and medical scientists.
Resonance energy transfer (RET) or electronic energy transfer (EET), is a mechanism describing energy transfer between two chromophores (Scheme 1). When both chromophores are fluorescent, the term “fluorescence resonance energy transfer” (FRET) is often used instead, although the energy is not always transferred by fluorescence. This process occurs when: (1) a donor has a high quantum yield; (2) there is a substantial overlap of the donor emission spectrum with the acceptor absorption; (3) there is an appropriate alignment of the absorption and emission transition moments and their separation vector and (4) the donor–acceptor distances are in proximity, typically less than 10 nm through non-radiative dipole–dipole coupling.3
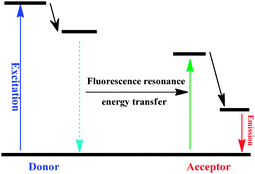 | ||
| Scheme 1 The mechanism of the fluorescence resonance energy transfer process. | ||
Normally, dyes based on FRET processes are usually linked by a non-conjugated spacer, and the energy transfer occurs through space (Scheme 2a). Since Stryer and Haugland took FRET as a spectroscopic ruler,4 FRET has been applied to the analysis of DNA structures, nucleic acid regulation, protein structure, function analysis, and immunoassays.5,6 Although the pseudo-Stokes shifts of FRET-based energy cassettes are larger than the Stokes shifts of either the donor or acceptor dyes, they are still limited by the requirement that the donor emission must have strong overlap with the acceptor absorption. This requirement essentially restricts the pseudo-Stokes shifts as well as the emission shifts (the emission wavelength shift between the donor and acceptor) of FRET-based systems.
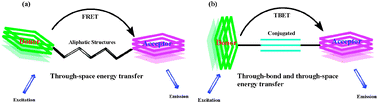 | ||
| Scheme 2 (a) Through-space and (b) through-bond energy transfer cassettes. | ||
The dyes based on TBET (through-bond energy transfer) are the ones which have a donor connected to an acceptor via electronically conjugated linkers. In particular on the interchromophore bridge conformation and configuration (Scheme 2b). Earlier, a systematic study of these phenomena was undertaken by Verhoeven and co-workers.7–9 If the through-bond energy transfer is fast relative to non-radiative decay pathways, then, unlike standard FRET-based cassettes, the TBET process is apparently not constrained by the requirement of spectrum overlap. If so, multiplexing with through-bond energy transfer cassettes would not necessarily involve loss of sensitivity at high resolutions.
Requirements for good through-bond energy transfer cassettes for labeling biological systems include:10 (1) donor components with strong absorbance at the excitation wavelength; (2) acceptor components that fluoresce strongly; (3) functional groups that allow attachment linkers. The linker must prevent the donor and acceptor fragments from becoming planar. Because if they did, the system would behave as a single conjugated dye. Conversely, the linker must allow through-bond energy transfer from the donor to the acceptor, that is rapid relative to non-radiative decay processes.
Though the excited energy transfer has been applied to quantum dots,11,12 nanoparticles,13,14 and protein-DNA complexes,15,16 in this tutorial review, we focused on the organic cassettes of energy transfer based on traditional FRET (through-space energy transfer) and TBET (through-bond energy transfer) developed since the year 2000, and their applications in ratiometric detection of ions, molecules, and other biomarkers. In the aspect of FRET, we classified the systems by different fluorophore skeletons, stressed the function of BODIPY, rhodamine, fluorescein, coumarin, and other fluorophores in the construction of the energy transfer systems. We hope to provide a general overview of the design and applications of these multifluorophores at detecting ions, small molecules, and other biomarkers.
2. Through-space energy transfer cassettes (FRET)
2.1 FRET system with BODIPY skeleton
Boron dipyrromethene (BODIPY) dyes have received much attention due to their outstanding properties, such as large absorption extinction coefficients, sharp fluorescence emissions, high fluorescence quantum yields, and high photophysical stability. However, the absorption and emission wavelengths for classical BODIPY chromophore centre at 470–530 nm. Extension of the π conjugation through introduction of aryl, vinyl, styryl, and other substituents in the 3- or 5-carbon chain can move the BODIPY emission to a longer wavelength. This appropriate structural adjustment allows BODIPY derivatives to be energy donors or acceptors in different energy transfer systems. Connected by triazole, which has good biocompatibility, alkyl chain, or some other proper linkers to justify the distance between donor and acceptor, FRET systems with a BODIPY skeleton have been developed in recent years.F1 was designed by Akkaya and co-workers (Fig. 1).17 In F1, excitation at 526 nm produced an emission peak centered at 618 nm of the PDI (perylenediimides) core. And the quantum yield of emission from the BODIPY units decreased to less than 0.01. The efficiency of energy transfer was estimated as 99%, which yielded a Förster critical radius of 47 Å. This antenna system was the first demonstration of the efficiency of energy transfer in a BODIPY–PDI bichromophoric system via very efficient click chemistry; it appeared to be highly promising for the design and synthesis of similar dendritic structures.
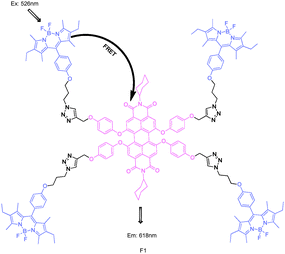 | ||
| Fig. 1 Structure of BODIPY derivative FRET cassette F1. | ||
Soon afterwards, through increasing interchromophoric distance, the Akkaya group synthesized a series of BODIPY dyads (Fig. 2).18 With the same substituent group at the R-position, boron to boron distances in dyads F2b (n = 0), F3b (n = 2), and F4b (n = 4) were estimated at 11.9, 25.5, and 39.1 Å; and the energy transfer efficiencies were calculated as 99.9%, 94% and 85%, decreasing with increasing boron to boron distances. In chemosensor F4c, the metal receptor was an electron donor and also part of the π-system of the chromophore, Hg2+ binding caused a blue shift in the absorption spectrum. In THF, after addition of Hg2+ to the solution of 1 mM F4c, a small decrease in the intensity at 540 nm was indicative of larger EET, the corresponding intensity at 600 nm increased.
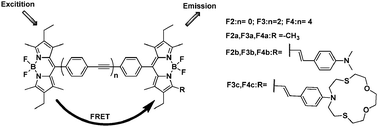 | ||
| Fig. 2 Fluorescent structure series of F2–F4 based on FRET. | ||
As mentioned above, extending the π conjugation in the 3- or 5-carbon chain can shift the BODIPY emission to a longer wavelength. Akkaya et al. reported an energy transfer cassette based entirely on differentially functionalized BODIPY dyes, F5a–c (Fig. 3).19 The absorption spectra showed the composite band at 520 nm arising from the donor, and the band at 650 nm originating from the acceptor. When the donor chromophore was excited at 525 nm, a strong emission from the acceptor at 670 nm was received. Since large absorption cross-sections in the visible region were accessible in this manner, the energy transfer systems were capable of elaborate and efficient energy transfer from donor BODIPY units to the distyryl-BODIPY acceptor.
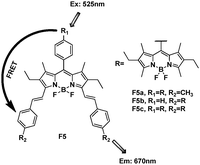 | ||
| Fig. 3 Structure of BODIPY derivative FRET cassette F5a–c. | ||
Based on the efficient syntheses of BODIPY especially for the large π-conjugation BODIPY with long wavelength, a light-harvesting system containing three kinds of BODIPY fluorophores (yellow-green, pink, and purple BODIPY with the absorption maximum at 501, 568, and 647 nm, respectively) was synthesized through click chemistry by Xiao and Qian (Fig. 4).20 Upon excitation of F6 at λex = 490 nm (exclusive absorption of the yellow-green BODIPY unit) or λex 560 nm (exclusive absorption of the pink BODIPY unit), fluorescence emission of the central purple BODIPY dye at 660 nm was observed. Its good photostability, solubility, and efficient energy transfer (99%) within it made compound F6 promising as a light-harvesting system.
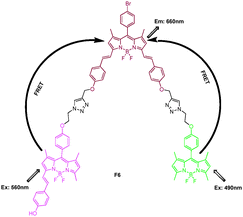 | ||
| Fig. 4 Structure of BODIPY derivative FRET cassette F6. | ||
On the basis of previous work, Akkaya et al. connected the BODIPY donor and large conjugated distyryl-BODIPY acceptor by the click reaction, and introduced the ion ligand to report a FRET Hg2+ sensor with large Stokes shifts and red to near-IR emissions (Fig. 5).21 In the absence of any metal ions, there were two clearly identifiable peaks in the visible region, one around 500 nm and the other one centered at 690 nm. Upon excitation at 480 nm, the free chemosensor showed two emission peaks at 518 and 725 nm. Other metal ions were essentially ineffective, but the addition of Hg2+ ions resulted in a blue shift in the emission together with a large increase in intensity. These were a large pseudo-Stokes shift (approximately 300–250 nm) and multiple ratiometric measurement opportunities for the detection.
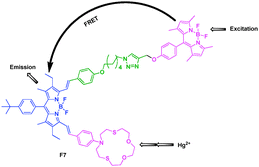 | ||
| Fig. 5 Fluorescent probe F7 for Hg2+ based on FRET. | ||
Zhao and James et al. calculated the FRET process by DFT calculations firstly. They synthesized a FRET fluorescent molecular probe for detection of Cys/Hcy (Fig. 6).22 The energy donor of a parent BODIPY was connected with the acceptor styryl-BODIPY through Suzuki cross-coupling reaction. For the free probe F8, the lack of fluorescence was attributed to the dark excited state S1, which was due to the electron sink effect of the 2,4-dinitrobenzensulfonyl(DNBS) moiety; the fluorescence OFF–ON switch was triggered by cleavage of the DNBS unit from the fluorophore by thiols. Then excited at 505 nm, a red emission at 590 nm was switched on, producing a pseudo-Stokes shift of up to 77 nm.
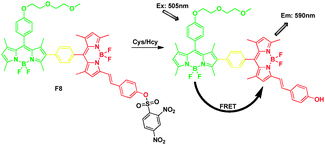 | ||
| Fig. 6 FRET chemosensor F8 for detection of thiol. | ||
It should be pointed out that there was no direct π-conjugation between the donor and acceptor because the phenyl ring between these two units took up a perpendicular geometry to each other. This connection could be helpful to improve the energy transfer through-bond and through-space simultaneously, and increase the energy transfer efficiency.
A molecular dyad F9 and triad F10, comprised of BODIPY (BDP), covalently linked to its structural analog and near-IR emitting sensitizer, BF2-chelated tetraarylazadipyrromethane (ADP), have been synthesized (Fig. 7).23 The ADP (or Aza-BODIPY), which has a nitrogen in the meso-position of the macrocycle, have attracted increased attention because of large fluorescence quantum yields above λ = 700 nm.24 The BDP and ADP entities were separated by about 14 Å with a dihedral angle between the fluorophores of around 70°. The excitation transfer from the singlet BDP to ADP was envisioned due to good spectral overlap of the BDP emission and ADP absorption spectra. In toluene, when excited at λ = 505 nm, the emission of BDP at 517 nm was quenched, with the appearance of new band at 674 nm for F9 and 663 nm for F10, respectively. The energy transfer efficiency of the triad F10 was better than that of dyad F9.
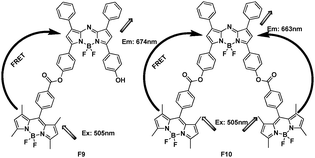 | ||
| Fig. 7 FRET systems F9 and F10 based on BODIPY. | ||
The major absorption and fluorescence emission positions of subphthalocyanines (ca. 560–570 nm) are complementary with those of BODIPY (ca. 500–520 nm) and distyryl BODIPY (ca. 630–680 nm). Therefore, proper conjugates should absorb over a broad range in the visible region and have a good spectral overlap between the donor emission and the acceptor absorption. A BODIPY and a distyryl BODIPY moiety were connected axially with subphthalocyanine substitution by Ng et al., respectively (Fig. 8).25 Both systems exhibited highly efficient photoinduced energy transfer processes, either from the excited BODIPY 470 nm to the subphthalocyanine core 570 nm (F11) or from the excited subphthalocyanine 515 nm to the distyryl BODIPY unit 653 nm (F12). The energy transfer quantum yield ΦENT of F11 and F12 were both found to be 0.98.
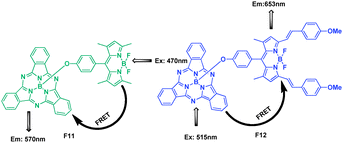 | ||
| Fig. 8 Structure of FRET cassettes F11, F12. | ||
2.2 FRET system with rhodamine skeleton
Rhodamine dyes also have excellent photophysical properties similar to those of BODIPY dyes. However, their absorption and emission spectra shift to a longer wavelength (ca. 580 nm), thus in the FRET systems, the rhodamine fluorophore often acts as the energy acceptor. Furthermore, rhodamine is a convenient platform to construct colorimetric “naked eye” and fluorescence “OFF–ON” probes taking advantage of the opening of the spirolactam ring. The extent of spectrum overlap and the distance of energy transfer are regulated by the behaviour and detection of analytes. All these points make rhodamine derivatives a good choice for recognition sites and signal switches in FRET systems.Most of the following examples were based on analyte promoted spiro-opening of rhodamine. Some other chemosodimeters were due to special reactions with the FRET process.
Based on the spirolactam of rhodamine through induced hydrazide-8-hydroxylquinoline, a flexible multi-chelating site of Cr3+, Li et al.26 synthesized a FRET probe F13 (Fig. 9). F13 displayed an absorption band centered at ca. 380 nm and yellow fluorescence centered at 544 nm in ethanol–water (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). Addition of Cr3+ to the solution caused an obvious enhancement of the absorption band centered at 568 nm, corresponding to a change in color from weak yellow to red–orange. Importantly, when F13 was excited at 405 nm, the intensity of the fluorescent peak at 544 nm gradually decreased and that of a new fluorescent band centered at 592 nm gradually increased. In the presence of Cr3+, the ratio of emission intensities at I592/I544 varied from 0.77 to 5.63, corresponding to a 7.6-fold enhancement.
:1, v/v). Addition of Cr3+ to the solution caused an obvious enhancement of the absorption band centered at 568 nm, corresponding to a change in color from weak yellow to red–orange. Importantly, when F13 was excited at 405 nm, the intensity of the fluorescent peak at 544 nm gradually decreased and that of a new fluorescent band centered at 592 nm gradually increased. In the presence of Cr3+, the ratio of emission intensities at I592/I544 varied from 0.77 to 5.63, corresponding to a 7.6-fold enhancement.
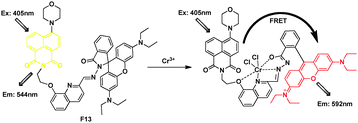 | ||
| Fig. 9 Fluorescent probe F13 for Cr3+ detection based on FRET. | ||
Through connecting a naphthalimine derivative directly with rhodamine spirolactam, F14 was synthesized as a ratiometric sensor for Hg2+/Cr3+ based on the resonance energy transfer process (Fig. 10).27 In the absence of ions, the system F14, was FRET-OFF, when excited with 455 nm, an emission from the donor at 533 nm appeared; while along with the addition of Cr3+/Hg2+, the FRET process happened, a new emission of rhodamine at 583 nm increased, and showed a steady decrease in emission intensity at 533 nm. Furthermore, confocal laser microscopy studies confirmed that the reagent F14 could also be used as an imaging probe for detecting the uptake of these ions in A431 cells.
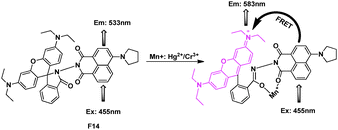 | ||
| Fig. 10 FRET fluorescent probe F14 for detection of Hg2+ and Cr3+. | ||
In pH 7.0, Tris-CH3CN (1:9, v/v) buffer, the emission of dansyl was at 507 nm, which overlaps with the absorption of rhodamine part. Yoon, Park and Kim synthesized a series of rhodamine-dansyl fluorophores incorporated into a tren spacer as Cu2+ FRET sensors, F15a–c (Fig. 11).28 Among them, F15a showed the best FRET efficiency through dansyl emission to rhodamine absorption for Cu2+, due to the addition of a sulfonamide group which presumably assists in trapping Cu2+. When Cu2+ was added into the excited F15a at 420 nm, it showed an emission band at approximately 580 nm, which was the region of rhodamine. The fluorescence ratio at I574/I507 showed a 13-fold increase in the case of the Cu2+ complexation.
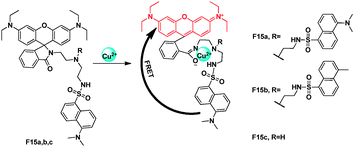 | ||
| Fig. 11 FRET chemosensors F15a,b,c for Cu2+ detection. | ||
N-Dansyl thiophene derivative of rhodamine was utilized successfully for detection of Hg2+ in aqueous solution (Fig. 12).29 On excitation of the CH3CN–H2O solution of F16 at 340 nm, a relatively weak and very broad emission band was observed, which could be assigned to the dansyl unit based emission. After bonding with Hg2+, a new emission of rhodamine was observed. The ratio of acceptor and donor emission intensity (I555/I483), in the absence and presence of different concentration of Hg2+, varied from 0.83 to 22.46, and this 27-fold enhancement was attributed to the FRET process.
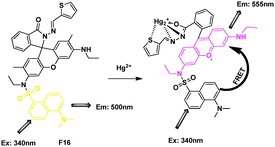 | ||
| Fig. 12 FRET chemosensor F16 for Hg2+ detection. | ||
For the coumarin glutathione-relative receptor and rhodamine glucose-relative receptor, both of them displayed high sensitivity but poor selectivity toward Cr3+ in aqueous solution. However, combining the two receptors as one system could detect Cr3+ in aqueous buffer with excellent selectivity and sensitivity. Duan's group reported a simple FRET-based approach to ratiometric fluorescence sensing of Cr3+ in aqueous solution using glutathione and glucose as building blocks, it was inspired to the binding motifs of Cr3+ in glucose tolerance factors (GTF) (Fig. 13).30 Selectivity and competition experiments showed that this system featured high sensitivity (detection limit ≤0.1 ppm) and excellent selectivity over other metal ions in buffer solution. Confocal fluorescence microscopy experiments had established the utility of the approach in monitoring Cr3+ within living cells.
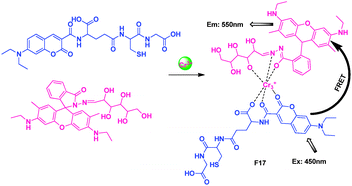 | ||
| Fig. 13 Intermolecular FRET sensor F17 for Cr3+ detection. | ||
F18 was a ratiometric fluorescent thiol probe (Fig. 14).31 The intramolecular energy transfer efficiency from the BODIPY donor to the rhodamine acceptor in F18 was calculated to be 98.4%. When F18 was titrated with increasing concentrations of Cys in potassium phosphate buffer, the nucleophilic thiol group of Cys attacked the electrophilic carbon of the thioester group leading to the cleavage of the FRET dyad. Since the distance was infinite after the thiol-induced cleavage of the FRET dyad, the FRET should be switched off in the presence of thiol. So the intensity at 590 nm gradually decreased with the concomitant growth of a new emission peak at 510 nm, indicating that the FRET was OFF. The fluorescent intensity ratios at 510 and 590 nm (I510/I590) exhibited a drastic change from 0.09 to 24.82 with a 275-fold enhancement in the emission ratios.
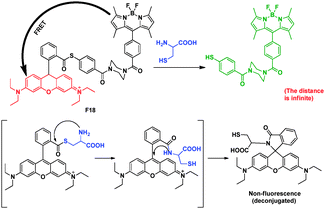 | ||
| Fig. 14 FRET chemosensor F18 for detection of thiol. | ||
Lin's group employed a coumarin–rhodamine platform to act as a FRET-based molecular strategy for the construction of small-molecule dual-excitation ratiometric probes (Fig. 15).32–35 A piperazine moiety was chosen as the rigid linker, which may avoid the likely static fluorescence quenching. Furthermore, the piperazine linker renders a suitable distance between the energy donor (coumarin) and acceptor (rhodamine). Introducing different recognition groups into this system can achieve the detection of different analytes.
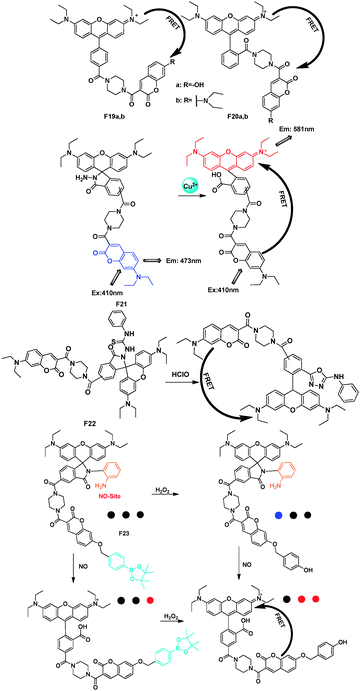 | ||
| Fig. 15 Rhodamine–coumarin FRET platforms F19–F23 and their applications. | ||
For the FRET pairs F19 and F20,32 when excited at the coumarin donor absorption, the emission of the coumarin donors at around 400–520 nm was nearly completely quenched. Concomitantly, the emission of the energy-acceptors of F19–F20 at around 586–592 nm displayed a marked fluorescence enhancement. These cassettes could also have further applications as pH probes. Similarly, based on the spiro-ring of rhodamine, the FRET platform was also developed as a fluorescent chemodosimeter for Cu2+33 and HOCl34 as F21 and F22. And for F23, the first single-fluorescent molecule, can respond to H2O2, NO, and H2O2/NO with three different sets of fluorescence signals.35
Xiao and Qian presented a BODIPY-rhodamine FRET system F24 with a rigid and conjugated phenyl-ethynyl-phenyl group as the linker, which could act as a ratiometric and intracellular Hg2+ sensor (Fig. 16).36 A leuco-rhodamine derivative was chosen as a sensitive and selective chemosensor for Hg2+ ions. A highly efficient ring-opening reaction induced by Hg2+ can change the emission maximum of the system from 514 nm (the characteristic peak of BODIPY) to 589 nm (the characteristic peak of rhodamine). The ratio of the emission intensities at 589 nm and 514 nm (I589/I514) became constant when the amount of Hg2+ added reached 60 ppb with a FRET efficiency of 99%.
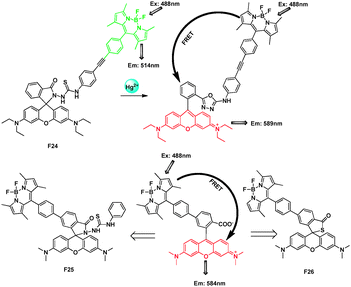 | ||
| Fig. 16 FRET cassettes F24–26 based on BODIPY and rhodamine fluorophores for detecting Hg2+. | ||
Later, the authors connected a BODIPY donor to the 5′-position of a tetramethylrhodamine acceptor through the short and rigid biphenyl spacer. Importantly, this partly conjugated nature was also good for high-efficiency FRET through bonds. To illustrate its value as a versatile platform, they introduced typical Hg2+ receptors and developed two ratiometric sensors, F25 and F26, with different spirocyclic receptors that recognized Hg2+ on different reaction mechanisms.37 The spirocyclic receptors induced on the remaining 2′-carboxyl group can recognize the corresponding cations without influence from/to other parts of the molecule (spacer or donor) for their separation from each other with over 99% FRET efficiency. It's very important for designing FRET sensors for different analytes.
The authors also thought the connection through a rigid and conjugated spacer facilitated the through-bond energy transfer, which will be discussed in detail in our part 3, cassettes based on “energy transfer through bond” and their applications.
Lin et al. described the first FRET-based ratiometric fluorescent gold probe F27 which is capable of quantitative determination of AuNPs (Fig. 17).13 The thio-amide-phenyl-substituted alkyne on the rhodamine platform was selectively promoted by Au3+ over Hg2+ ions in aqueous ethanol to afford 5-ketohiazole. The spiro-ring opening induced by Au3+ made the state of cassettes from FRET-OFF to FRET-ON. The ratiometric probe exhibited an excellent linearity between the emission ratio (I594/I514) and the Au3+ concentration in the range from 5.0 × 10−7 to 1.5 × 10−5 M with a detection limit of 3.9 × 10−7 M.
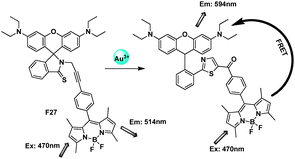 | ||
| Fig. 17 Fluorescent probe F27 for Au3+ based on FRET. | ||
Also modulated by a specific metal ion induced ring-opening process of the rhodamine spirolactam, Zhang et al. described a ratiometric fluorescent cellular imaging probe for Zn2+ (Fig. 18).38 Fluorescein was chosen as the energy donor because its fluorescence spectrum matches well with the absorption spectrum of rhodamine. The high selectivity for Zn2+ over other ions of the probe might be ascribed to the chemical structure of containing sulfur, oxygen, and nitrogen atoms in a special arrangement, and the fluorescein moiety might also play an important role for its selectivity for Zn2+ through electronic or steric hindrance effect. The probe exhibited a stable response of fluorescence intensity ratio at I590/I518 for Zn2+ over a concentration range from 2.0 × 10−7 to 2.0 × 10−5 M.
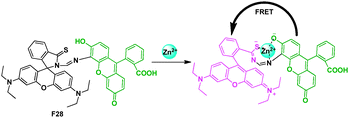 | ||
| Fig. 18 Fluorescent probe F28 for Zn2+ detection based on FRET. | ||
2.3 FRET system with fluorescein skeleton
Fluorescein dyes and their analogues, being introduced into photophysical, biochemical, and biomedical research within the recent years, appeared to be a very promising group of fluorescent dyes owing to their high quantum yield and photostability. It was anticipated that, fluorescein would be appropriate as such an acceptor, because it has two conformations (lactone form and quinoid form) with distinctly different absorption properties. The fluorescence of this quinoid molecule is high, and excitation occurs at ca. 494 nm and emission at ca. 521 nm. The 2′,7′-fluorophore coupled with other organic group makes it suitable for developing in FRET systems. Importantly, fluorescein compounds were non-fluorescent when the phenolic hydroxyl group was caged, while strongly green fluorescent when the hydroxy group is deprotonated.Nagano's group developed fluorescent probes with a 3′-O,6′-O-protected fluorescein (Fig. 19).39 As there was no spectral overlap integral between the coumarin emission and the leuco fluorescein, the fluorescein moiety cannot accept the excitation energy of the donor moiety and the donor fluorescence can be observed. After cleavage of the protective groups by hydrolytic enzymes, the fluorescein moiety showed a strong absorption in the coumarin emission region, and then acceptor fluorescence due the FRET was observed. Compound F30 with the flexible ethylene linker exhibited weaker fluorescence and longer wavelength of the absorption maxima compared to F29a and F29b with the rigid cyclohexane linker. And F29a and F29b were practically useful for the ratiometric measurement of PTP (protein tyrosine phosphatase) activity.
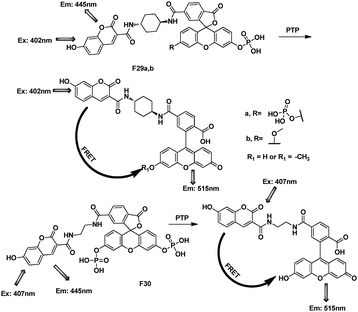 | ||
| Fig. 19 FRET cassettes of F29–30 for detection of protein PTP. | ||
Similarly, cassette F31, comprised of a coumarin donor and a boronate-protected fluorescein acceptor linked by a cyclohexyl spacer was designed by Chang's group (Fig. 20).40 Based on the cleavage of the protective groups of fluorescein, F31 features good selectivity for H2O2 over other ROS in water solution.
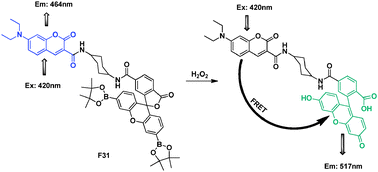 | ||
| Fig. 20 FRET chemosensor F31 for detection of H2O2. | ||
Guo et al. reported a 4-(N,N-dimethylamino)benzamide-fluorescein FRET “OFF–ON” system F32 as a ratiometric fluorescence probe for CN− (Fig. 21).41 In DMF–water (9:1, v/v) system, the nucleophilic attack by cyanide toward the activated hydrazone functionality, followed by fast proton transfer of the acidic phenol proton to the developing nitrogen anion of this compound, would trigger ring-opening in the fluorescein spirolactone unit to generate a long-wavelength fluorescein monoanion fluorophore. The emission intensities at 550 nm and 450 nm (I550/I450) became constant when the amount of CN− added reached 26 equiv., and the emission intensity ratio, I550/I450, varied from 0.60 to 16.6.
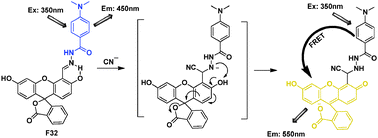 | ||
| Fig. 21 Fluorescent probe F32 for CN− based on FRET. | ||
Based on coumarin–fluorescein FRET cassette, Ojida and Hamichi reported the design of ratiometric chemosensors F33a and F33b for nucleoside polyphosphates such as ATP that were based on binding-induced modulation of FRET coupled with a turn-on fluorescence-sensing mechanism (Fig. 22).42 The FRET efficiencies of the ATP bound complexes of F33a and F33b were calculated as 76% and 83%, respectively.
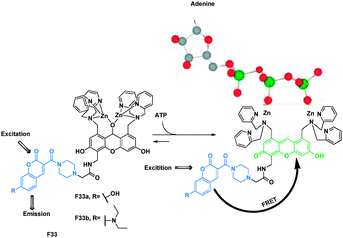 | ||
| Fig. 22 Binding mechanisms of FRET sensors F33a–b with ATP. | ||
If the fluorophores were introduced intramolecularly with a flexible linker, they should form ground-state dye-to-dye close contact in an aqueous environment and the fluorescence should be quenched. Thus, Nagano et al. chose the phenyl moiety as an appropriately cleavable linker to prevent dye-to-dye close contact in an aqueous environment and design a fluorescent ratiometric probe F34 to detect phosphodiesterase I activity (Fig. 23).43 In F34, coumarin was designed as the donor and fluorescein as the acceptor. Before detection, the energy transfer efficiency 94%, after the hydrolysis of the phosphodiester group by a phosphodiesterase, FRET no longer took place.
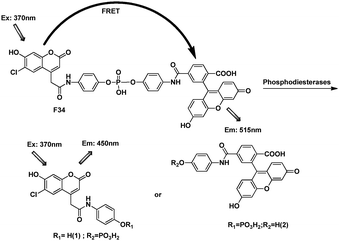 | ||
| Fig. 23 FRET cassette of F34. | ||
2.4 FRET system with coumarin skeleton
Coumarin is a benzopyrone that occurs naturally in many plants and essential oils. Due to their good photochemical and photobiological properties, the coumarin derivatives have attracted much attention recently, especially for applications in pharmacology and medicine. The emission of typical coumarin is relatively short, in the FRET systems, the coumarin fluorophore often acts as the energy donor.Through introduction of different electron donors in the 7-positon, the derivative of coumarin such as coumarin-343, display broader region of green–yellow.
FRET is widely used to monitor association and dynamic processes of biological molecules. There are only a few systems featuring FRET across mechanically interlocked components. Rebek et al. reported a rotaxane with FRET labels (Fig. 24).44 The relevant structural feature of the rotaxane F35 was a macrocycle bearing a fluorophore and the axle bearing another fluorophore at one end. Coumarin 2 was the donor on the axle and coumarin 343 was the acceptor on the macrocycle. The Förster distances of the dyes were typically around 20–80 Å; a distance that mostly exceeds the length of the axle. Accordingly, the system maintained FRET as long as the rotaxane was intact.
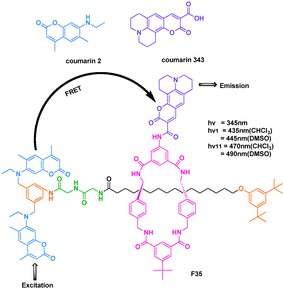 | ||
| Fig. 24 FRET process of cassette F35. | ||
In contrast to these intramolecular energy transfer process, a FRET approach towards potential detection of phosgene, which was based on a selective chemical reaction between phosgene (or triphosgene as a simulant) and donor and acceptor fluorophores was present as F36 by Rudkevich et al. (Fig. 25).45 This reaction covalently brought them together within the appropriate Förster distance ca. 20 Å.
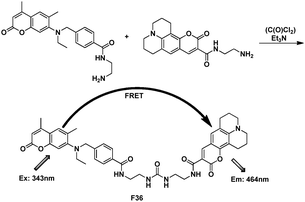 | ||
| Fig. 25 FRET chemosensor F36. | ||
Dendrimer F37 was the first FRET based organic nonionic material that transformed UV and visible light directly to NIR radiation (Fig. 26).46 The emission of coumarin 2 overlaps well with the higher level excited-state absorption of the core perylenebis(dicarboximide), thus creating a pathway for FRET to occur from coumarin 2 to the core derivative. When F37 was excited at the donor λmax (λex = 345 nm), the donor emission was completely quenched. This energy transfer efficiency was 99%.
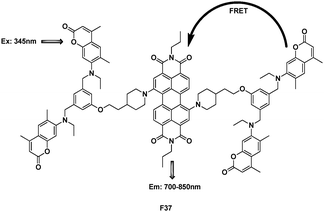 | ||
| Fig. 26 Structure of dendrimer FRET cassette F37. | ||
This system could potentially be applied as the gain medium of solid-state organic-based lasers or as a component of high value photovoltaic (PV) materials, where destructive high energy UV radiation would be converted to useful low energy NIR radiation.
Lin's group designed and synthesized the compound F38, based on a tetrakis (4-hydrosy-phenyl)porphyrin–coumarin scaffold, as a ratiometric fluorescent probe for thiols (Fig. 27).47 For the free F38, the fluorescence of the coumarin dye was quenched due to the FRET from the coumarin donor to the porphyrin acceptor, in good agreement with the overlap between the coumarin emission with the Soret and Q1 band of the porphyrin absorption at 421 and 500 nm. Upon addition of Cys, the disulfide bond was cleaved, and the distance of the coumarin and porphyrin dyes became infinite. The FRET was turned OFF. Surprisingly, the porphyrin emission kept constant in the presence of thiols. The authors considered that maybe it was because the strong overlap between the coumarin donor emission peak and the minor emission band of the porphyrin acceptor elicited the singlet-singlet annihilation process.
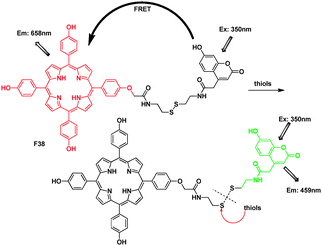 | ||
| Fig. 27 FRET chemosensor F38 for thiols. | ||
The red-emitting iridium complex [(btp)2Ir(acac)] BTP was significantly quenched by dissolved oxygen in solution. Using this property, Tobita et al. reported a ratiometric molecular sensor for monitoring oxygen levels in living cells and tissues (Fig. 28).48 They selected coumarin-343 as the fluorescent donor group and the iridium complex BTP as the phosphorescent acceptor group, and connected by a tetraproline linker to spatially separate these groups. When F39 was excited at 405 nm, the electronic energy was partially transferred from donor to the acceptor moiety by singlet energy transfer, resulting in both blue fluorescence of coumarin-343 and red phosphorescence of BTP. Under aerated conditions, the phosphorescence of BTP was almost entirely quenched. It could also found that the fluorescence and phosphorescence exhibit reasonably good spectral separation, which was favorable for ratiometric measurements at above 610 vs. 480 nm.
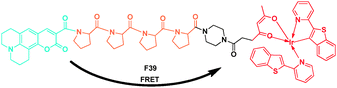 | ||
| Fig. 28 FRET probe F39 for monitoring oxygen levels. | ||
F40 was based on photo-induced electron transfer (PET) and binding-induced modulation of fluorescence resonance energy transfer (FRET) mechanisms for ratiometric detection of H+ (Fig. 29).49 This ratiometric chemosensor was constructed by introduction of a pH-insensitive coumarin fluorophore as an FRET donor into a pH-sensitive amoni-naphthalimide derivative as the FRET acceptor. The sensor exhibited clear dual-mission signal changes in blue and green spectral windows upon pH changes. The pH sensor was applied for not only measuring cellular pH, but also for visualizing stimulus-responsive changes of intracellular pH values. F40 was applied to visualized intracellular pH values of three cell lines, CP-A, J774A.1 and Hela.
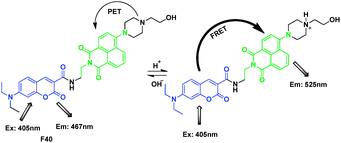 | ||
| Fig. 29 Fluorescent probe F40 for H+ based on FRET. | ||
2.5 FRET system with other fluorophore skeleton
Besides the several categories of organic fluorophores mentioned above, there are still some other fluorescent dyes with attractive photophysical properties, which were used in the energy transfer systems. Compounds such as “squaraine”, which is an important fluorescence “Turn-OFF” type chemodosimeter for the recognition of nucleophiles. One of the smallest available fluorophore, “dansyl group”, which is absorbance in near UV-region, fluorescence band in visible region. And “naphthalimide” fluorophore which often acts as an energy donor because it has strong emission in the visible range and its broad emission (450–650 nm) covers a part of the acceptor's absorption band.In addition, the development of two-photon technology and metal–organic complexes, opened up a new prospect for the traditional energy transfer system.
Xi's group designed and synthesized F41 with total separation of their emission spectra to study base-catalyzed hydrolysis and pig liver esterase (PLE) activity, the widely used esterase in industry (Fig. 30).50 1-Naphthylacetic moiety was selected as the fluorescent donor and a dansyl group as the acceptor. The FRET efficiency of free F41 was larger than 90%. Both donor and acceptor were located on opposite sides in a single molecule with an ester bond as a linker. Compound F41 displayed a large shift (from 500 to 338 nm) in its emission spectrum after the base-catalyzed cleavage of ester bond within two fluorophores.
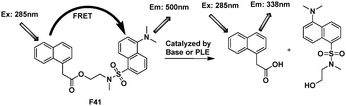 | ||
| Fig. 30 FRET cassettes F41. | ||
After conjugating 8-amino-quinoline to spiropyran backbone via a CH2 spacer at the C8′ carbon, the resultant sensor F42, possessed a tridentate “ONN” binding site for Zn2+, and provided an opportunity to construct a FRET relay between the two fluorophoric subunits present in the system (Fig. 31).51 The probe successfully exhibited a fluorescence “Turn-ON” and ratiometric response for Zn2+ in ethanol solution with high selectivity.
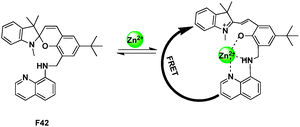 | ||
| Fig. 31 FRET chemosensor F42 for Zn2+ detection. | ||
A N-(pyrenylmethyl)naphtho-azacrown-5 F43 was synthesized as an “ON–OFF” fluorescent chemosensor for Cu2+ by Yoon and Kim (Fig. 32).52 Excited at 240 nm corresponding to the absorption of naphthalene unit (energy donor) of F43, emission at 380 nm from pyrene unit (energy acceptor) was observed, indicating that intramolecular fluorescence resonance energy transfer (FRET-ON) occurred in F43. However, when Cu2+ was added to a solution of F43 the fluorescence of pyrene was strongly quenched (FRET-OFF) whereas that of naphthalene group was revived. The calculated molecular HOMO and LUMOs orbitals suggested the excited-sated interactions leading to FRET from naphthalene to pyrene in F43, but no electron density changes in the F43-Cu2+ complex.
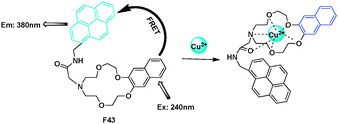 | ||
| Fig. 32 FRET chemosensor F43 for Cu2+ detection. | ||
Squaraine is an important fluorescence “Turn-OFF” type chemodosimeter for the recognition of nucleophiles. Due to significant electron deficiency, squaraine can be readily attacked by nucleophilic compounds, to generate colorless and non-fluorescent products. FRET cassette-type sensors, F44, which contain two naphthalimide donors and a squaraine acceptor, have been developed on the concept of switching off FRET through removing the spectral overlap by the analyte-induced decompositions of the acceptor chromophore (Fig. 33).53 In F44a, a short ethylene group was employed for higher energy transfer efficiency. However, the short distance between donor and acceptor may facilitate PET. Because of its poor solubility in polar solvents, the F− recognition experiment was carried out in CH2Cl2. A longer and more flexible ester chain (diethyoxyl group) was used as the spacer in F44b, which improves the solubility and facilitates reactive recognitions in polar solvents. F44b could detect F− and CN− in different systems based on FRET.
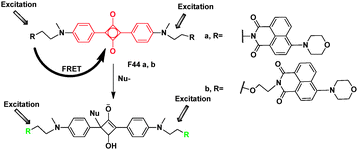 | ||
| Fig. 33 FRET chemosensor F44 for detection of F− and CN−. | ||
Fréchet and Prasad presented a bichromophoric system that was capable of undergoing efficient FRET following either two-photon (IR laser) or single-photon (UV-visible) excitation (Fig. 34).54 The donor chromophore had a large TPA cross-section in the 800 nm region (6700 GM ± 15%), thus for F45, there were bands at 320 and 405 nm arising from the two-photon absorbing chromophore, and the bands at 270 and 540 nm arising from the Nile red moiety. Irradiation of F45 at 405 nm resulted in emission mainly from the Nile red chromophore at 595 nm, with an efficient energy transfer (>99%). Changing the local laser intensity by varying sample position resulted in changes in emission intensity (at 595 nm) that followed a quadratic dependence, indicating that the emission from compounds F45 and Nile red was due to two-photon excitation.
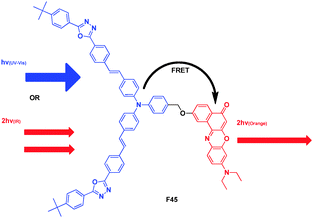 | ||
| Fig. 34 Structure of FRET cassette F45. | ||
3. Cassettes based on “energy transfer through bond” and their applications
Conventional fluorescence detection approaches have the problem that the dyes which emit at longer wavelengths absorb at the excitation wavelength less effectively, so their fluorescence intensities are diminished.10 To solve this problem, the technology of fluorescence resonance energy transfer has been developed. Compared to the limitation of overlap between the wavelengths in through-space transfer, the through-bond energy transfer is fast relative to non-radiative decay pathways, which is apparently not constrained by these requirements. Actually, the appropriate connection between the donor and acceptor could ensure that the energy transfer happens through-space and through-bond simultaneously, which would improve the energy transfer efficiency. Through the conjugated linker, which prevents the donor and acceptor fragments from being planar, the “energy transfer through-bond” cassettes enable greater freedom for the selection of fluorophores. It can produce a larger Stokes shifts, and develop the emission further into the IR wavelength region.Burgess et al. designed cassettes T1–T8, in which the BODIPY fragments acted as acceptors while anthracene derivatives served as donors (Fig. 35).55 Energy transfer efficiencies in such donor–acceptor cassette systems must vary with the relative orientation of the two components, and with the structure of the linkers that attach them. This study was designed to probe these issues for the special case in which the linkers between the donor and the acceptor fragments are conjugated. The electrochemical experiment indicated increasing conjugation in the series T5 < T7 and T1 < T8 < T6. From the X-ray crystallographic studies, the twist between the donor and acceptor planes was more rigidly enforced in T5 than in T7. The authors found that T6 and T8 were not truly cassettes, since they were fully conjugated systems without an internal twist to break that conjugation. Compounds T5 and T7 revealed the favored conformations of the donor and acceptor fragments, at least in the solid state. Fluorescence emission spectra of the cassettes showed maxima varying between 514 and 647 nm.
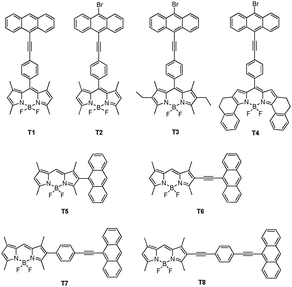 | ||
| Fig. 35 TBET cassettes T1–T8. | ||
To improve the water-compatible fluorescent imaging probes with tunable photonic properties that can be excited at a single wavelength, Burgess et al. synthesized bichromophoric cassettes T9a–c consisting of a BODIPY donor and a cyanine acceptor (Fig. 36).56 Upon excitation of the BODIPY moiety at 488 nm, the excitation energy was transferred through an acetylene bridge to the cyanine dye acceptor, which emitted light at approximately 600, 700 and 800 nm, with a “pseudo-Stokes” shifts between 86 and 290 nm. Encapsulated cassette T9a permeated Clone 9 rat liver cells, where it localized in the mitochondria and fluoresced through the acceptor part, i.e., red light around 590 nm.
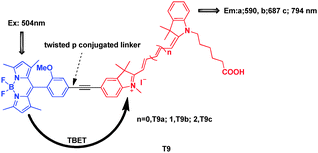 | ||
| Fig. 36 TBET cassettes T9. | ||
Modifications to the BODIPY core can lead to dyes that emit above 600 nm. This makes the derivatives the energy acceptor in the cassettes. Burgess's group reported a series of aza-BODIPY dyes, and picked one to couple with a fluorescein-alkyne derivative to give the energy transfer cassettes T10a and T10b (Fig. 37).57 The absorption and fluorescence characteristics of T10a and T10b were highly dependent on the pH and solvent media. If they can be modified to increase solubility in aqueous media, they will have great potential application as biomolecular probes.
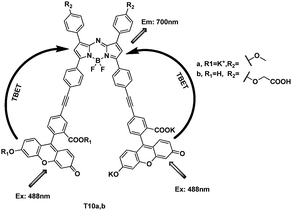 | ||
| Fig. 37 TBET cassettes T10a and T10b. | ||
The same research group chose Nile red, which has emissions in the 630–650 nm region, longer than cellular autofluorescence, as the acceptors, reported the syntheses and photophysical properties of dyads T11, T12a,b, T13a,b (Fig. 38).58 In EtOH, their fluorescence spectra showed efficient energy transfer without significant “leakage” of emission from the donor fragment. Their quantum efficiencies when excited at the donor absorption maxima, were determined to be in the range of 0.31–0.41. The energy transfer efficiencies were calculated to be 77–97%. The apparent Stokes shifts for the dyads ranged from 120 to 150 nm.
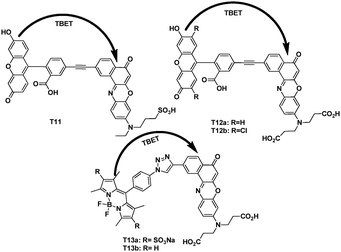 | ||
| Fig. 38 TBET cassettes T11, T12a,b and T13a,b. | ||
Shen et al. reported two TBET cassettes that exhibit large pseudo-Stokes shift (up to 400 nm) and high efficiency (up to 99%) (Fig. 39).59 From the X-ray of T14a, the meso-thiophene was virtually orthogonal to the indacene plane. The quinoline moiety and the indacene plane were nearly planar. When excited with quinoline wavelength, strong emission of the acceptor BODIPY appeared because of the energy transfer. Selective binding Fe3+ with the donor entity significantly suppresses the excitation energy transfer and resulting in fluorescence quenching in aqueous solution. When added to Fe3+, and excited at 334 nm, the fluorescence intensity from the BODIPY of T14a at 523 nm and T14b at 732 nm decreased remarkably, whereas excitation of the BODIPY, the emission band from the BODIPY parts still remain.
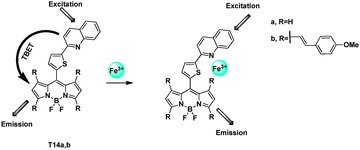 | ||
| Fig. 39 TBET chemosensors T14a,b for Fe3+ detection. | ||
Guo et al. designed energy transfer cassettes composed of coumarin and BODIPY. They have longer wavelength fluorescence emission; large pseudo-Stokes shifts (up to 410 nm) and efficient energy transfer efficiencies (up to 98.6%) (Fig. 40).60 When cassettes F15a–d in ethanol were excited at 340 nm, the emission of the coumarin energy donor around 408 nm was almost completely quenched. The characteristic emission bands of the acceptor BODIPYs (512, 658, 750 and 632 nm, respectively for F15a–d) appeared. Because of PET process of cassette F15c, with decreasing the pH value from 7.5 to 1.5, the emission at 750 nm gradually decreased; concomitantly, the emission at 632 nm was drastically enhanced due to the inhibition of the ICT and PET processes upon protonation of the basic dimethyl amino groups. The change of fluorescence intensity at 632 nm vs. pH variation was over 1460-fold.
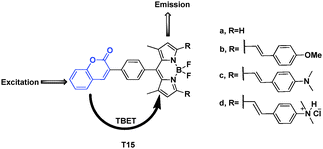 | ||
| Fig. 40 Fluorescent probe series of T15a–d based on TBET. | ||
Under the condensation of coumarin aldehydes with 3-(diethyl-amino) phenol, followed by oxidation with chloranil, Lin et al. synthesized TBET cassettes T16a–e (Fig. 41)61 Upon excitation of the cassettes (5 μM; phosphate buffer–MeOH 3:2, v/v) in the coumarin absorption band, only the characteristic emission band of the acceptor rhodamine (around 582 nm) was observed, thus indicated that the energy transfer efficiencies were nearly perfect (>99%). T16e was a ratiometric fluorescent TBET pH probe. This probe showed a large pseudo-Stokes shift as well as a dramatic change of the ratios of emission intensities at 587 and 465 nm (I587/I465) from 46.5 at pH 3.0 to 0.2 at pH 8.6 because of a large emission shift.
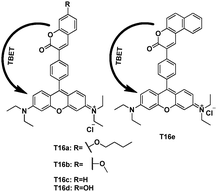 | ||
| Fig. 41 TBET cassettes T16, and T16e as pH probe. | ||
It's worth mentioning that the aryl group is a wonderful linker in TBET systems. Although this concept is not proposed in these papers, the cassettes of Fig. 3, and Fig. 16 may also have the energy transfer through space and bond simultaneously.
The donor fluorophore was connected directly to the rhodamine by a conjunction linker. Manoj Kumar et al. synthesized naphthalimide appended rhodamine based fluorescent chemosensor T17 which showed through-bond energy transfer in the presence of Hg2+ ions in mixed aqueous solution (Fig. 42).62 The fluorescence spectrum of compound T17 in the absence of mercury ions, exhibited a very weak emission at 472 nm attributed to the naphthalimide moiety when excited at 360 nm. After addition of Hg2+, the fluorescence appears at 578 nm with 407-fold enhancement. The detection limit for Hg2+ was 2 × 10−6 mol L−1.
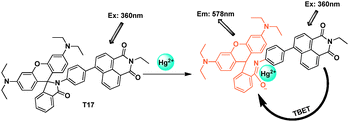 | ||
| Fig. 42 TBET chemosensor T17 for detection of Hg2+. | ||
Also by conjugate connection through an aryl group, a pentaquinone-rhodamine TBET system was designed by Bhalla and Kumar et al. (Fig. 43).63 In this cassette, the rhodamine was the acceptor, and the mechanism was based on the Hg2+ induced spiro-ring opening. In THF–H2O (9.5:0.5, v/v), after addition of Hg2+, when excited at 360 nm, it could get fluorescence enhancement at 582 nm.
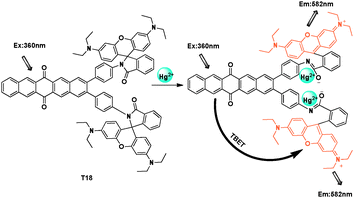 | ||
| Fig. 43 TBET chemosensor T18 for Hg2+ detection. | ||
Kevin Burgess et al. reported a simple design concept for through-bond conjugated dye cassettes and structures that could be useful for multiplexing in many areas of molecular biology and biotechnology (Fig. 44).64 Absorption spectra of T19a–d show maxima characteristics of the donor and acceptor components at 488 and 514 nm. It indicated that compounds were behaving as cassettes and not as planar, totally conjugated dyes. Excitation of the cassettes at 488 or 514 nm produced fluorescence characteristics of only the acceptor component at 538, 582, 603, and 616 nm, respectively.
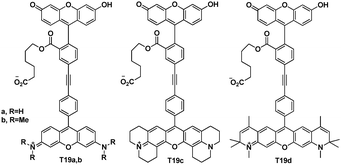 | ||
| Fig. 44 TBET cassettes T19a–d. | ||
Based on the studies of cassettes T19a–d, the Burgess group demonstrated the preliminary application of these dyes in single-color sequencing experiments.65
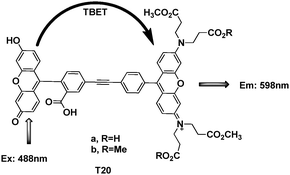
In order to label the proteins outside the cell, Kevin Burgess et al. synthesized some TBET fluorophores (Fig. 45)66 based on their previous studies. Under pH 8 buffer conditions, there was about 80% energy transfer efficiency for compound T20a. After coupling with an illustrative recombinant protein, mouse acyl-coenzyme A binding protein (ACBP), the binding and CD data indicated that labeling ACBP with cassette T20b did not impede its natural binding function or cause it to misfold in vitro. From the fluorescence image in COS-7 cells, it showed ACBP-1 colocalizes around the nuclei when the cells were excited at 488 nm (to excite the donor) and a filter at 598 nm (acceptor fluorescence) was applied.
Forming discrete probes for biotechnology that emit at longer wavelengths are generally more useful. Kevin Burgess's group synthesized chemically activated cassettes T21 and T22 based on fluorescein and nile red (Fig. 46).67 With considerable experimentation, a couple of organic-soluble luminol-fluorescein/nile red based cassettes were synthesised. The dipole–dipole energy-transfer efficiency was 39% and 42% for T21 and T22, respectively. Simple experiments in vitro showed that cassettes T21 and T22 could be activated through treatment with peroxidase under physiological conditions.
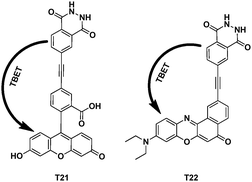 | ||
| Fig. 46 TBET cassettes T21 and T22. | ||
4. Conclusion
In this tutorial review, we focused on the recent major contributions regarding fluorescence resonance energy transfer since 2000. They include the systems of BODIPY–BODIPY, BODIPY–cyanine, BODIPY–rhodamine, rhodamine–coumarin, rhodamine–fluorescein and so on. The traditional energy transfer through-space was connecting the donor chromophore and acceptor fluorophore through a proper linker, for example triazole from click reaction,20 quinoline,26 piperazine,32 and others.49 So Akkaya et al. reported some BODIPY-based FRET cassettes successfully.14–16,18 Nagano et al. synthesized ratiometric fluorescent probes based on fluorescein.39,43 Lin's group developed a series of FRET chemosensors based on coumarin,13,34,35,47 and applied these ratiometric detectors into living cells. Kim et al. used the FRET-based system to detect ions successfully.28,52 Xiao and Qian,20,36,37 also did very excellent work in this field. But these FRET-based cassettes are still limited by the requirement that the donor emission must have a strong overlap with the acceptor absorption, which essentially restricts the design of FRET-based systems. If the donors are connected to acceptors via electronically conjugated linkers, which prevent the donor and acceptor fragments from being planar, energy transfer in such systems occurs through-bond and through-space simultaneously.68 Unlike standard FRET-based cassettes, TBET is apparently not constrained by the overlap requirement and this enables greater freedom for the selection of fluorophores. Thus a serious of brilliant TBET cassettes were reported by Burgess group10,56,66,67 to develop this new energy transfer method between fluorophores. Actually, the above-mentioned sensor molecules indicate that switching by either “through-space” or “through-bond” is feasible for practical use such as ions detection, protein labelling, and organelles localization.Since there are important advantages of fluorescent ratiometric measurement at two wavelengths, these cassettes based on FRET and TBET mechanisms will become useful tools for bioimaging, biosensing, medical diagnosis and environmental detection. We consider that even more wonderful cassettes should be developed to satisfy different applications.
Acknowledgements
This work was supported by the NSF of China (21136002, 21076032 and 20923006), the National Basic Research Program of China (2009CB723706 and 2013CB733702).Notes and references
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS.
- R. Y. Tsien and A. T. Harootunian, Cell Calcium, 1990, 11, 93–109 CrossRef CAS.
- C. G. dos Remedios and P. D. J. Moens, J. Struct. Biol., 1995, 115, 175–185 CrossRef CAS.
- L. Stryer and R. P. Haugland, Proc. Natl. Acad. Sci. U. S. A., 1967, 58, 719–726 CrossRef CAS.
- J. Szöllosi, S. Damjanovich and L. Mátyus, Cytometry, 1998, 34, 159–179 CrossRef.
- R. B. Sekar and A. Periasamy, J. Cell Biol., 2003, 160, 629–633 CrossRef CAS.
- H. Oevering, M. N. Paddon-Row, M. Heppener, A. M. Oliver, E. Cotsaris, J. W. Verhoeven and N. S. Hush, J. Am. Chem. Soc., 1987, 109, 3258–3269 CrossRef CAS.
- M. R. Roest, J. M. Lawson, M. N. Paddon-Row and J. W. Verhoeven, Chem. Phys. Lett., 1994, 230, 536–542 CrossRef CAS.
- M. Antolovich, P. J. Keyte, A. M. Oliver, M. N. Paddon-Row, J. Kroon, J. W. Verhoeven, S. A. Jonker and J. M. Warman, J. Phys. Chem., 1991, 95, 1933–1941 CrossRef CAS.
- G. S. Jiao, L. H. Thoresen and K. Burgess, J. Am. Chem. Soc., 2003, 125, 14668–14669 CrossRef CAS.
- L. F. Shi, V. De Paoli, N. Rosenzweig and Z. Rosenzweig, J. Am. Chem. Soc., 2006, 128, 10378–10379 CrossRef CAS.
- E. Oh, M. Y. Hong, D. Lee, S. H. Nam, H. C. Yoon and H. S. Kim, J. Am. Chem. Soc., 2005, 127, 3270–3271 CrossRef CAS.
- X. Cao, W. Lin and Y. Ding, Chem.–Eur. J., 2011, 17, 9066–9069 CrossRef CAS.
- G. Chen, F. Song, J. Wang, Z. Yang, S. Sun, J. Fan, X. Qiang, X. Wang, B. Dou and X. Peng, Chem. Commun., 2012, 48, 2949–2951 RSC.
- K. S. Sanju, P. P. Neelakandan and D. Ramaiah, Chem. Commun., 2011, 47, 1288–1290 RSC.
- S. Wang, B. S. Gaylord and G. C. Bazan, J. Am. Chem. Soc., 2004, 126, 5446–5451 CrossRef CAS.
- M. D. Yilmaz, O. A. Bozdemir and E. U. Akkaya, Org. Lett., 2006, 8, 2871–2873 CrossRef CAS.
- A. Coskun and E. U. Akkaya, J. Am. Chem. Soc., 2006, 128, 14474–14475 CrossRef CAS.
- G. Barin, M. D. Yilmaz and E. U. Akkaya, Tetrahedron Lett., 2009, 50, 1738–1740 CrossRef CAS.
- X. L. Zhang, Y. Xiao and X. H. Qian, Org. Lett., 2008, 10, 29–32 CrossRef CAS.
- S. Atilgan, T. Ozdemir and E. U. Akkaya, Org. Lett., 2010, 12, 4792–4795 CrossRef CAS.
- J. Shao, H. Sun, H. Guo, S. Ji, J. Zhao, W. Wu, X. Yuan, C. Zhang and T. D. James, Chem. Sci., 2012, 3, 1049–1061 RSC.
- M. E. El-Khouly, A. N. Amin, M. E. Zandler, S. Fukuzumi and F. D'Souza, Chem.–Eur. J., 2012, 18, 5239–5247 CrossRef CAS.
- P. Batat, M. Cantuel, G. Jonusau skas, L. Scarpantonio, A. Palma, D. F. O'Shea and N. D. McClenaghan, J. Phys. Chem. A, 2011, 115, 14034–14039 CrossRef CAS.
- J. Y. Liu, H. S. Yeung, W. Xu, X. Y. Li and D. K. P. Ng, Org. Lett., 2008, 10, 5421–5424 CrossRef CAS.
- F. Y. Li, Z. G. Zhou, M. X. Yu, H. Yang, K. W. Huang, T. Yi and C. H. Huang, Chem. Commun., 2008, 3387–3389 Search PubMed.
- P. Mahato, S. Saha, E. Suresh, R. Di Liddo, P. P. Parnigotto, M. T. Conconi, M. K. Kesharwani, B. Ganguly and A. Das, Inorg. Chem., 2012, 51, 1769–1777 CrossRef CAS.
- S. Yoon, M. H. Lee, H. J. Kim, N. Park and J. S. Kim, Org. Lett., 2008, 10, 213–216 CrossRef.
- M. Suresh, S. Mishra, S. K. Mishra, E. Suresh, A. K. Mandal, A. Shrivastav and A. Das, Org. Lett., 2009, 11, 2740–2743 CrossRef CAS.
- C. Y. Duan, X. Y. Hu, X. L. Zhang, G. J. He and C. He, Tetrahedron, 2011, 67, 1091–1095 CrossRef.
- W. Y. Lin, L. L. Long, B. B. Chen, W. S. Gao and L. Yuan, Chem. Commun., 2011, 47, 893–895 RSC.
- L. Yuan, W. Lin, Z. Cao, J. Wang and B. Chen, Chem.–Eur. J., 2012, 18, 1247–1255 CrossRef CAS.
- L. Yuan, W. Lin, B. Chen and Y. Xie, Org. Lett., 2011, 14, 432–435 CrossRef.
- L. Yuan, W. Lin, Y. Xie, B. Chen and J. Song, Chem.–Eur. J., 2012, 18, 2700–2706 CrossRef CAS.
- L. Yuan, W. Lin, Y. Xie, B. Chen and S. Zhu, J. Am. Chem. Soc., 2011, 134, 1305–1315 CrossRef.
- Y. Xiao, X. L. Zhang and X. H. Qian, Angew. Chem., Int. Ed., 2008, 47, 8025–8029 CrossRef.
- Y. Xiao, H. B. Yu, H. Y. Guo and X. H. Qian, Chem.–Eur. J., 2011, 17, 3179–3191 CrossRef.
- Z. X. Han, X. B. Zhang, L. Zhuo, Y. J. Gong, X. Y. Wu, J. Zhen, C. M. He, L. X. Jian, Z. Jing, G. L. Shen and R. Q. Yu, Anal. Chem., 2010, 82, 3108–3113 CrossRef CAS.
- H. Takakusa, K. Kikuchi, Y. Urano, H. Kojima and T. Nagano, Chem.–Eur. J., 2003, 9, 1479–1485 CrossRef CAS.
- C. J. Chang, A. E. Albers and V. S. Okreglak, J. Am. Chem. Soc., 2006, 128, 9640–9641 CrossRef.
- W. Guo, X. Lv, J. Liu, Y. L. Liu, Y. Zhao, M. L. Chen and P. Wang, Org. Biomol. Chem., 2011, 9, 4954–4958 Search PubMed.
- A. Ojida, Y. Kurishita, T. Kohira and I. Hamachi, J. Am. Chem. Soc., 2010, 132, 13290–13299 CrossRef.
- T. Nagano, H. Takakusa, K. Kikuchi, Y. Urano, S. Sakamoto and K. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 1653–1657 CrossRef.
- H. Onagi and J. Rebek, Chem. Commun., 2005, 4604–4606 RSC.
- D. M. Rudkevich and H. X. Zhang, Chem. Commun., 2007, 1238–1239 Search PubMed.
- J. M. Serin, D. W. Brousmiche and J. M. J. Frechet, J. Am. Chem. Soc., 2002, 124, 11848–11849 CrossRef CAS.
- X. Cao, W. Lin and Q. Yu, J. Org. Chem., 2011, 76, 7423–7430 CrossRef CAS.
- T. Yoshihara, Y. Yamaguchi, M. Hosaka, T. Takeuchi and S. Tobita, Angew. Chem., Int. Ed., 2012, 51, 4148–4151 CrossRef CAS.
- X. Zhou, F. Su, H. Lu, P. Senechal-Willis, Y. Tian, R. H. Johnson and D. R. Meldrum, Biomaterials, 2012, 33, 171–180 CrossRef CAS.
- Z. Xi, L. Yi, L. Cao and L. L. Liu, Tetrahedron, 2008, 64, 8947–8951 CrossRef.
- W. H. Chan, J. F. Zhu, H. Yuan and A. W. M. Lee, Tetrahedron Lett., 2010, 51, 3550–3554 CrossRef.
- S. Yoon, H. J. Kim, S. Y. Park and J. S. Kim, Tetrahedron, 2008, 64, 1294–1300 CrossRef.
- Y. Xiao, H. B. Yu and M. Y. Fu, Phys. Chem. Chem. Phys., 2010, 12, 7386–7391 RSC.
- D. W. Brousmiche, J. M. Serin, J. M. J. Frechet, G. S. He, T. C. Lin, S. J. Chung and P. N. Prasad, J. Am. Chem. Soc., 2003, 125, 1448–1449 CrossRef CAS.
- C. W. Wan, A. Burghart, J. Chen, F. Bergstrom, L. B. A. Johansson, M. F. Wolford, T. G. Kim, M. R. Topp, R. M. Hochstrasser and K. Burgess, Chem.–Eur. J., 2003, 9, 4430–4441 CrossRef CAS.
- Y. Ueno, J. Jose, A. Loudet, C. Perez-Bolivar, P. Anzenbacher and K. Burgess, J. Am. Chem. Soc., 2011, 133, 51–55 CrossRef CAS.
- A. Loudet, R. Bandichhor, L. Wu and K. Burgess, Tetrahedron, 2008, 64, 3642–3654 CrossRef CAS.
- J. Jose, Y. Ueno, J. C. Castro, L. L. Li and K. Burgess, Tetrahedron Lett., 2009, 50, 6442–6445 CrossRef CAS.
- X. Qu, Q. Liu, X. Ji, H. Chen, Z. Zhou and Z. Shen, Chem. Commun., 2012, 48, 4600–4602 RSC.
- Y. Zhao, Y. Zhang, X. Lv, Y. Liu, M. Chen, P. Wang, J. Liu and W. Guo, J. Mater. Chem., 2011, 21, 13168–13171 RSC.
- W. Lin, L. Yuan, Z. Cao, Y. Feng and J. Song, Angew. Chem., Int. Ed., 2010, 49, 375–379 CrossRef CAS.
- M. Kumar, N. Kumar, V. Bhalla, H. Singh, P. R. Sharma and T. Kaur, Org. Lett., 2011, 13, 1422–1425 CrossRef CAS.
- V. Bhalla, Roopa, M. Kumar, P. R. Sharma and T. Kaur, Inorg. Chem., 2012, 51, 2150–2156 CrossRef CAS.
- G.-S. Jiao, L. H. Thoresen and K. Burgess, J. Am. Chem. Soc., 2003, 125, 14668–14669 CrossRef CAS.
- G. S. Jiao, L. H. Thoresen, T. G. Kim, W. C. Haaland, F. Gao, M. R. Topp, R. M. Hochstrasser, M. L. Metzker and K. Burgess, Chem.–Eur. J., 2006, 12, 7816–7826 CrossRef CAS.
- R. Bandichhor, A. D. Petrescu, A. Vespa, A. B. Kier, F. Schroeder and K. Burgess, J. Am. Chem. Soc., 2006, 128, 10688–10689 CrossRef CAS.
- J. Y. Han, J. Jose, E. Mei and K. Burgess, Angew. Chem., Int. Ed., 2007, 46, 1684–1687 CrossRef CAS.
- S. Speiser, Chem. Rev., 1996, 96, 1953–1976 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |