Molecular daisy chains
Jürgen
Rotzler
a and
Marcel
Mayor
*ab
aUniversity of Basel, Department of Chemistry, St. Johannsring 19, CH-4056 Basel, Switzerland. E-mail: marcel.mayor@unibas.ch; Fax: +41 61 267 1016; Tel: +41 61 267 1006
bKarlsruhe Institute of Technology (KIT), Institute of Nanotechnology, P. O. Box 3640, D-76021, Karlsruhe, Germany
First published on 17th September 2012
Abstract
This tutorial review summarizes the progress made towards mechanically interlocked daisy chains. Such materials can be seen as a further development in polymer science, where the conventional covalent interlinking bonds are replaced by supramolecular binding concepts. Materials in which the mechanical bond is an integral part of the polymeric backbone are expected to possess unique macroscopic properties and are therefore the synthetic aim in an ever growing research community. After introducing general considerations about daisy chains, the most common analytic methods to get insight into the aggregation behaviour of such self-complementary monomers are presented. Cyclodextrins/aromatic rods, crown ethers/cationic rods and pillararenes/alkyl chains are systems used to achieve daisy chain-like molecular arrays. By comparison of the reported systems, conclusions about an improved structural design are drawn.
 Jürgen Rotzler | Jürgen Rotzler was born in Lörrach (Germany) in 1982. He studied chemistry at the University of Basel (Switzerland) where he received his Master of Science in 2008 and his PhD in 2012 by working on torsion angle restricted biphenyl based push-pull systems and on a new assembling motif for molecular daisy chains with special emphasis on applications in molecular electronics under the supervision of Prof. Marcel Mayor. Currently he is a PostDoc in the group of Prof. James L. Leighton at Columbia University in New York working on polyketide natural product synthesis. |
 Marcel Mayor | Marcel Mayor was born in Zürich (Switzerland) in 1965. He studied chemistry at the University of Berne (Switzerland) where he received his PhD in 1995 supervised by Professor Rolf Scheffold and Professor Lorenz Walder. After working together with Professor Jean-Marie Lehn in the Laboratoire de Chimie Supramoléculaire of the University Louis Pasteur in Strasbourg as a SNF post-doctoral fellow and at the Collège de France in Paris (France) as Maître de Conférences invité, he founded his own research group in the Institute of Nanotechnology of the Karlsruhe Institute of Technology (KIT, Germany) in 1998. After defending his habilitation at the University Louis Pasteur in Strasbourg in 2002, he became Professor of Chemistry at the University of Basel (Switzerland) in 2004. His current research interests are supramolecular chemistry, molecular electronics, nanoscale architectures, functional materials and hybrid materials. |
Introduction
In the sense of materials science, one of the most amazing examples where the knowledge of structure–property relationships has been used to design matter with distinct properties is the production of polymers. The properties of polymers are directly linked to their molecular composition. The nature of the monomeric structural units and the correct position within the macromolecule define the macroscopic properties and the function.1 Therefore these macroscopic properties can be tailored by altering the chemical character of the monomers using well established synthetic methods.2,3 Chemical aspects that have to be taken into account include the knowledge of the properties of individual structural units (monomers), three-dimensional aggregates (solid state structure, physical properties), solubility and bulk properties like crystallinity, melting temperature, glass transition, etc.2 But to design macromolecules with distinct properties, not only chemical aspects but also architectural aspects have to be considered, resulting in polymer topologies like linear copolymers, branched graft/comb polymers or dendrimers, as well as polymeric network molecules.4 The ease to create a large diversity of polymers with tailored properties led to the replacement of traditional materials in daily life by synthetic organic polymers.Mechanically interlocked polymers
A special case of polymers which attracted considerable attention in the last few decades are mechanically bonded macromolecules/polymers (Fig. 1).5–8 In mechanically interlocked polymers the covalent bonds of traditional polymers are replaced by mechanical bonds often supported by non-covalent interactions of the subunits like for example hydrogen bonds, charge transfer and hydrophobic interactions.9 In some cases dramatic changes in their macroscopic properties are triggered by these non-covalent interactions.10,11 Polyrotaxanes, polycatenanes, daisy chains and mechanically interlocked dendrimers are members of this family of non-covalently linked polymers or oligomers.10,12–14 Encircling of the polymeric backbone by macrocycles having a hydrophobic interior and a hydrophilic exterior, like e.g. cyclodextrins, makes them water soluble in contrast to the parent hydrophobic polymer.7,15 Furthermore an improved shielding of the parent polymer caused by cyclic molecules can result in an increased stability against bleaching or oxidation or even in enhanced conductivities.12![Mechanically interlocked macromolecules as a further development towards new polymeric materials including poly[n]rotaxanes, poly[n]catenanes and daisy chains.](/image/article/2013/CS/c2cs35217f/c2cs35217f-f1.gif) | ||
| Fig. 1 Mechanically interlocked macromolecules as a further development towards new polymeric materials including poly[n]rotaxanes, poly[n]catenanes and daisy chains. | ||
Maybe the most important changes in macroscopic properties of mechanically interlocked macromolecules are the alteration of viscosity,16,17 phase behavior18 and exterior functionality12 which lead to materials which are easier processable compared to most of the parent polymers. The unique properties of such systems not only resulted in improvement of already existing processing techniques in polymer science but also in advances in molecular electronics,19 drug delivery,20 sensing21 and tissue engineering22 to name just a few. The synthesis of defined mechanically bonded macromolecules is the key towards new applications in all areas of research to benefit the requirement of ever more high-performing plastics in today’s society. However, low overall yields and laborious, multi-step synthetic routes towards mechanically interlocked molecules, as well as the lack of control over the polymerization process are issues which have to be addressed before mechanically interlocked macromolecules will find their way towards applications.
Molecular daisy chains
A special case of mechanically interlocked macromolecules are daisy chains which potentially allow alteration of macroscopic properties by switching mechanisms on a molecular scale. In its original sense a daisy chain is a daisy (flower) garland where the stems are interlinked to form a chain or a chaplet (Fig. 2). The term daisy chaining is widely used in computer technology for the serial connection of hardware components leading to bus-systems. | ||
| Fig. 2 Left: original daisy chain composed of flowers; middle: schematic representation of a molecular cyclic daisy chain; right: acyclic daisy chain. | ||
A molecular daisy chain is an array of identical molecules that consist of both a linear thread (guest) and a threadable macrocycle (host) which are covalently bound together – in principle nothing else than coupling of a rotaxane thread to the encircling macrocycle. Threading of the macrocycle by a linear section of another component mediated by intermolecular recognition – rather than intramolecular recognition (preventing “head-tail-biting”) – leads to the formation of either cyclic or acyclic molecular arrays, so-called daisy chains. Introduction of a stopper unit prevents dethreading of these supramolecularly assembled daisy chains and therefore converts the supramolecular binding into a mechanical bond, leading to mechanically interlocked daisy chains. The term daisy chain for such supramolecular assemblies was first introduced by Stoddart.23 The monomeric units are named in the literature plerotopic, hermaphroditic, heteroditopic or self-complementary.
In contrast to polyrotaxanes, in which an already existing polymer is encircled with macrocycles to alter the macroscopic properties of the parent polymer, in systems like polycatenanes or daisy chains the mechanical bond is an integral part of the polymer chain.5,13 Furthermore polyrotaxanes are composed of interlocked macrocycles which exhibit only a motional freedom of the cycle, whereas daisy chains and polycatenanes can alter the length of the polymer providing something like “elongation adaptability” (Fig. 3).24 In principle self-complementary, plerotopic daisy chain monomers allow the formation of long mechanically bonded polymeric structures which exhibit a linear longitudinal mobility.5 These integrated mechanical bonds can potentially lead to new polymers with entirely modified properties like very large modulus loss, low activation energy for the viscous flow and rapid stress relaxation.8,24 By careful design of the recognition sites and incorporation of different guest motifs in one tail of the plerotopic molecules a by external input triggered intramolecular motion in daisy chains can be achieved which results in a controlled elongation mobility by switching mechanisms and therefore to potential formation of molecular actuators or even molecular muscles.5,25,26
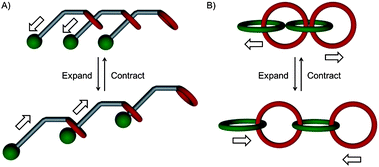 | ||
| Fig. 3 Possible internal elongation mobility of (A) daisy chains and (B) polycatenanes.24 | ||
The fundamental question in the preparation of daisy chains is whether the covalently interlinked hosts and guests form acyclic long oligomers or cyclic structures (Fig. 2).23 By analysis of the stability of these two options the cyclized systems seem to be thermodynamically more stable than acyclic systems. For example in the smallest cyclic version consisting of two monomers where the guest of one monomer is complexed by the host of the second monomer and vice versa ([c2]daisy chain) in one supermolecule two stabilizing host–guest interactions are present in contradiction to the acyclic version where for two stabilizing interactions three monomers are needed (Fig. 4). Additionally in acyclic assemblies always one rod and one tail are frustrated leading to unfavorable cohesive and dispersion forces. Furthermore the formation of long acyclic daisy chains is an entropically unfavored process which can only be overcome if a high monomer concentration is used together with strong binding affinities.
![Stabilizing host–guest interactions in daisy chains. (A) smallest possible cyclic daisy chain ([c2]daisy chain) where two monomers are stabilized by two supramolecular interactions; (B) acyclic trimeric daisy chain ([a3]daisy chain) where three monomers are stabilized by two supramolecular interactions.](/image/article/2013/CS/c2cs35217f/c2cs35217f-f4.gif) | ||
| Fig. 4 Stabilizing host–guest interactions in daisy chains. (A) smallest possible cyclic daisy chain ([c2]daisy chain) where two monomers are stabilized by two supramolecular interactions; (B) acyclic trimeric daisy chain ([a3]daisy chain) where three monomers are stabilized by two supramolecular interactions. | ||
Despite examples where anion-templating27 and metal-templating26,28 were used to construct daisy chains the synthetic efforts towards polymeric mechanically interlinked daisy chains were mainly focused on host–guest supramolecular interactions. Two binding motifs are mainly reported in various publications which are the formation of inclusion complexes by hydrophobic guests into the hydrophobic cavity of cyclodextrins and complexation of dialkylammonium centers by crown ethers. The hydrophobic effect in polar solvents in the cyclodextrin motifs causes strong association in contrast to the crown ether-cation motif leaving more space for synthetic manipulations and therefore to more design possibilities.
In this article the deployed recognition concepts are reviewed, also with special emphasis on the analytical methods used to investigate the aggregation behavior of ditopic monomers. By analysis of the reported examples, conclusions about the molecular design of the monomeric subunit are drawn potentially guiding towards long acyclic daisy chains.
Analytical methods to investigate the aggregation behavior of self-complementary monomers
A particular issue in the research area of mechanically interlocked molecules and especially of daisy chains is the analysis of the formed aggregates in solution. In principle various aggregates can be formed upon intermolecular threading of the molecular rod into a cavity of the covalently attached macrocyclic moiety.29 Supramolecularly bonded daisy chain monomers can potentially form two different dimer structures excluding aggregation of the ring components and the molecular rods like in micelles (Fig. 5). The formation of [c2]daisy chains ([c2]HH) is well known in the literature and is often a thermodynamically stable aggregate preventing formation of longer oligomers.5 Also the aggregation of two hydrophobic rods ([a2]TT) is possible but it is expected that such a binding is weaker than the inclusion of the rod into the hosts cavity and therefore causes, because of the reversible aggregation, no interruption of oligomerization. Formation of an acyclic head–tail dimer ([a2]HT) can be seen as the first propagation step towards polymerization. Nevertheless each possible n-mer of the propagating chain has the possibility to form, because of the expected reversible binding, its cyclic analogs. The hypothetical formation of this variety of aggregates makes a detailed analysis difficult. Therefore the most convenient methods used are summarized with a special emphasis on qualitative outcomes.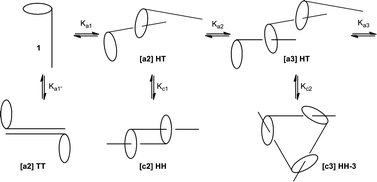 | ||
| Fig. 5 Possible aggregates formed by daisy chain monomers.5 | ||
Optical methods
Absorption measurements are particularly interesting for daisy chains composed of UV active molecular rods and macrocycles with different electronic nature. Upon complexation of for example an electron deficient guest into the electron rich cavity mediated by donor–acceptor interactions a charge transfer band can be observed.30–33 Furthermore a change of intensity in UV bands with temperature can be assigned to a shift of the equilibrium since the entropic contribution is decreased.33 For instance in cases where solvent molecules are released from the cavity upon complexation to the bulk media the change in entropy ΔS will be negative.34 Thus a decrease in temperature will shift the equilibrium to the associated form. Additionally titration experiments allow for plotting λmax against the concentration. According to Lambert–Beer’s law the dependence should be linear, what means that a deviation from linearity can be assigned to oligomer formation.30,35 Moreover encircling of the linear component can lead to a change of the conformation and with it to a different electronic signature. For example an oligophenylene rod can be forced into a planar alignment what should result in a change in conjugation and therefore to changes in the UV/Vis spectra.19The same arguments as for UV/Vis spectroscopy are valid in fluorescence spectra of daisy chains. The shielding effect of the ring component leads to a decreased quantum yield of the emission. Thus the intensity of the emission bands decrease in the complexed form what results in a nonlinear plot of the emission intensity against the concentration.36–38 Deviations from linearity in such plots can be directly assigned to critical aggregation concentrations. Fluorescence is also strongly related to the electronic character of the emitting molecule and therefore sensitive to changes in conformation or to the influence of the macrocyclic component. Furthermore formation of a daisy chain can potentially result in the perfect alignment of two chromophores causing excimer formation.39 Concentration dependent fluorescence titration allows for estimation of the association constant which is a measure for the strength of the inclusion complex.36,40
NMR
Very powerful methods for the characterization of aggregates formed by daisy chain monomers – which can also be seen as A–B type monomers, keeping to the nomenclature of polymer science – are NMR techniques. Two different cases of aggregates have to be considered: (i) dynamic aggregates with a fast exchange on the NMR time scale which show averaged signals in the spectra; (ii) aggregates with a slow exchange on the NMR time scale which show individual signals for monomers and aggregates. In the first case the observed averaged chemical shifts can be expressed as the sum of the chemical shifts of each individual aggregate present in solution, normalized by its molar fraction (eqn (1)).41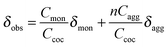 | (1) |
| δobs = δaggp + δmon (1 – p) | (2) |
| n = 1/(1 − p) | (3) |
One drawback of the evaluation of the aggregation number n using Carother’s equation is the assumption that cyclic species do not contribute to the consumption of host and guest sites.44 Therefore this method is not suitable for systems which tend to form small cyclic species. Other equations used to characterize the self-assemblies formed by daisy chain monomers are based on an equilibrium where n monomers form a single aggregate consisting out of n repeat units (eqn (4)). Ka represents the association constant.
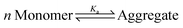 | (4) |
From eqn (1) and Ka = Cagg/Cmonn, eqn (5) can be obtained.37,39,47,48
ln((δmon − δobs)Ctot) = n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln((δobs − δagg)Ctot) + ln Ka + ln n − (n − 1) ln (δmon − δagg) ln((δobs − δagg)Ctot) + ln Ka + ln n − (n − 1) ln (δmon − δagg) | (5) |
Plots of ln[Ctot(|δobs − δmon|)] vs. ln[Ctot(|δagg − δobs|)] give a straight line from which the slope and the intercept can be calculated yielding n and Ka, respectively. By plotting δobs against the total concentration and extrapolating to zero monomer concentration the chemical shift of the monomer (δmon) can be estimated graphically. Extrapolation of the concentration to infinity yields the chemical shift of the aggregate (δagg).48
In the case of slow exchange on the NMR time scale individual signals for monomer and aggregate can be observed in the 1H-NMR spectra. Therefore the aggregation number n and the association constant Ka can be calculated from eqn (6)
| Ka = ([M]0 − [M])/n [M]n | (6) |
Besides the evaluation of these important parameters like n, Ka and critical aggregation concentration, 1H-NMR titration can show a significant signal broadening at high concentrations. This behavior can be seen as evidence for the formation of long oligomeric species since their presence influence the viscosity of the solution dramatically, leading to broader signals.30
The threading of the molecular rod into the cavity of the macrocyclic moiety can be demonstrated by 2D-NOESY or ROESY-NMR. Observation of cross peaks of protons inside the cavity and protons assigned to the thread is indicative for the inclusion of the rod into the host and thus for the formation of daisy chains.39,51,52 Furthermore structural information can be obtained by 2D-NMR making it to a very powerful tool to analyze the aggregation behavior of self-complementary ditopic monomers.
Polymer techniques
Several standard techniques used in polymer science to characterize high mass compounds were adopted to investigate daisy chain oligomers, among them dynamic light scattering (DLS),35,53 viscosimetry,35,44,46,51 differential scanning calorimetry,44 vapor pressure osmometry (VPO),51 size exclusion chromatography32,35 and diffusion ordered (DOSY) NMR.33,46,51,54 All these techniques are based on the increasing hydrodynamic radius with increasing aggregation number. DLS, DOSY-NMR and size exclusion can be used to determine the diffusion coefficient at various concentrations. Thus the hydrodynamic radius and with it the size of the supramolecular oligomers, as well as transitions in aggregation (critical aggregation concentrations) can be estimated. Furthermore comparison of the obtained data with molecular modeling can provide hints for the spatial arrangement of the daisy chain. Smaller cyclic daisy chains have a smaller hydrodynamic radius and therefore a lower viscosity than higher oligomers. Hence concentration dependent viscosity measurements will display a nonlinear increase in viscosity with increasing concentration upon formation of longer daisy chains. Another important parameter in polymer science is the glass transition temperature Tg. Differential scanning calorimetry can be used to estimate Tg. An increasing Tg with concentration points to a higher extent of self-organization. Especially long oligomers are expected to have an amorphous nature. Size exclusion chromatography is an ideal method for thermodynamically and kinetically stable daisy chains. There the formed aggregates are separated by their hydrodynamic radius. Especially the possibility to use a multi-angle light scattering photometer as detector allows for direct evaluation of the weight averaged molar mass of the eluted compound by Zimm plot analysis. The molar mass of individual daisy chain aggregates can also be estimated by vapor pressure osmometry. This method is based on the lower vapor pressure of solutions containing solute compared to the vapor pressure of pure solvent. In a Wheatstone bridge (two thermistors, one thermistor is covered by pure solvent and one with a solution of the molecule of choice in a defined concentration) the difference in vapor pressure is compensated by condensation of pure solvent, that saturates the gas phase, on the droplet of the solution. The increasing vapor pressure of the solution droplet leads to an increase in temperature. Thus the difference in temperature is measured which is proportional to the number of particles dissolved in solution. If sample concentrations are known the molecular mass can be determined.Additionally all conventional methods for mass determination are suitable to characterize the size of the daisy chain, but it has to be mentioned that conventional mass spectrometry is measured in the gas phase. MS methods based on solution therefore require an evaporation of solvent which results in an increase in concentration during this process which potentially results in higher aggregates.
Cyclodextrin based daisy chains
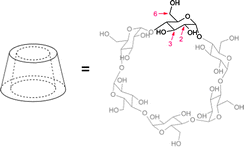
Hirotsu, Fujita and Tabushi were the first who studied the formation of daisy chains in 1982 although indirect evidences were reported before.55 They substituted β-cyclodextrins with a tert-butylthio group (1). Solid state structures of these plerotopic molecules confirmed that at higher aqueous concentrations the hydrophobic tert-butyl group was located inside the hydrophobic cavity of another β-cyclodextrin and superstructures like dimers, trimers and also polymers were present (Fig. 6). In solution it was shown that a dimer was present up to a concentration of 2 × 10−4M, which already confirms the strong hydrophobic effect. This study was further corroborated by crystal structures of other, for the use in modeling enzymes synthesized, modified cyclodextrins.56 In further studies it was demonstrated by 1H-NOESY that also in solution hydrophobic linkers attached to β-cyclodextrins are incorporated into the cavity.57 The strong hydrophobic binding of aromatic guests to cyclodextrins was used to assemble a variety of daisy chains based on this binding motif.57,58 Furthermore Liu et al. could show that orientation, alignment and helicity of the self-assembly can be controlled by tuning or exchanging the pivot heteroatom and the tether length.58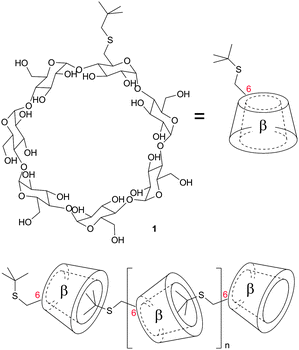 | ||
| Fig. 6 Linear acyclic daisy chain array of in 6 position tert-butyl sulfane functionalized β-cyclodextrins (n = 2). The tBuS groups are intermolecularly included in the hydrophobic cavity of the macrocycle.55 | ||
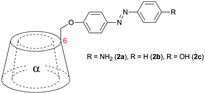
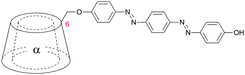
Kaneda et al. coupled 4′-hydroxyazobenzenes to permethylated, in 6 position tosylated α-cyclodextrins (2a–c, Table 1).45,49,50 The self-aggregation of the resulting plerotopic monomer was studied by 1H-NMR-titration experiments and an aggregation number of n = 2 was found in water–methanol mixtures. 2D-NOESY-NMR confirmed the formation of a cyclic dimeric daisy chain ([c2]daisy chain) in a mechanically interlocked structure. Fixation of the dimer was achieved by attaching 2-naphthol-3,6-disulfonic acid to the remaining free amines (3) (Fig. 7).
| Compound | Binding position | Aggregation number n | |
|---|---|---|---|
|
|||
| 2 |
|
6 | trans n = 2 |
| cis n = 1 | |||
| 4 |
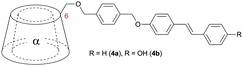
|
6 | 2 |
| 5 |
|
6 | 2, 4 |
| 6 |
|
3 | 2 |
| 7 |
|
6 | 2 |
| 8 |
|
8a,b 6 | 3 |
| 8c 6 | 2 | ||
| 8c 3 | 12 | ||
| 8d 3 | >15 | ||
| 9 |
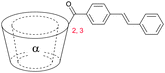
|
2 | trans n = 2 |
| 2 | cis oligomer | ||
| 3 | trans oligomer | ||
| 3 | cis n = 2 | ||
| 10 |
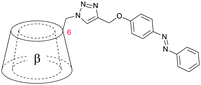
|
6 | 3 |
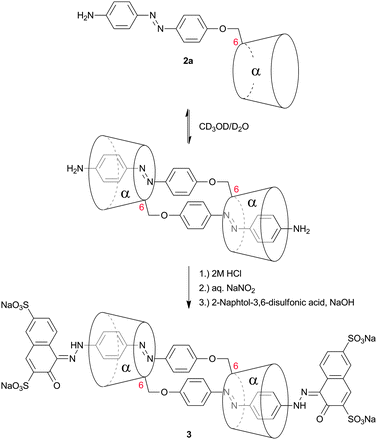 | ||
| Fig. 7 Self-aggregation of azobenzene substituted α-cyclodextrin 2a. Attachment of 2-naphthol-3,6-disulfonic acid to the remaining free amines prevented deaggregation.45 | ||
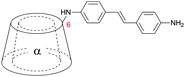
The effect of E/Z isomerization in compounds 2b and 2c on the aggregation behavior was studied revealing that photoisomerization of the central azo-bond from the thermodynamically stable E-isomer to the Z-isomer caused a dissociation of the pseudorotaxane.59 After thermal back-reaction the [c2]daisy chain was restored. The self-aggregation behavior was therefore dynamically controlled by an external physical input which resulted in a dramatic change in aggregate formation. Elongation of the rod (compounds 4 and 5, Table 1) provided mainly [c2]daisy chains, whereas for compound 5 traces of a tetrameric complex were found by NMR.49,50 Similarly it was demonstrated for stilbene substituted α-cyclodextrin (compound 6, Table 1) that the photoisomerization from trans to cis caused a dissociation of the [c2]daisy chain and the formation of nonthreaded supramolecular self-assemblies.60
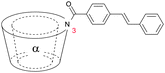
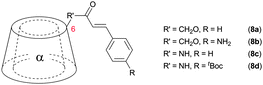
Also 4,4′-diaminostilbene was coupled to an α-cyclodextrin (7) which formed [c2]daisy chains in aqueous solution in a monomer:dimer ratio of 1:50 and a 65% yield after capping with 2,4,6-trinitrobenzenesulfonate.61 By several analytic methods only the presence of dimer was observed providing further evidence that the cyclic dimer is the thermodynamically most stable oligomer. Additionally cinnamoyl amines and alcohols were used as tails for plerotopic cyclodextrin based monomers (8a–d).51,62–64 Interestingly the different substitution pattern used where the tail was linked to the 3 position of α-cyclodextrin caused formation of supramolecular oligomers with up to 15 repeat units in aqueous solution (compound 8c and 8d) (Fig. 8).51,64 The mean averaged molecular weight and the hydrodynamic radius of self-aggregated higher oligomers were determined by vapor pressure osmometry (VPO), mass spectrometry and by determination of the self-diffusion coefficient with PFG-NMR. Additional studies on cinnamoyl substituted α-cyclodextrins (CiO-α-CD) where the guest was coupled in 2 or 3 position demonstrated that 2-CiO-α-CD formed double-threaded dimers in aqueous solution, whereas supramolecular oligomers were obtained from 3-CiO-α-CD.65 Interestingly, by mixing 2-CiO-α-CD and 3-CiO-α-CD only hetero-correlations were observed by 1H-ROESY-NMR instead of homo-correlations. This indicates the formation of supermolecules (n = 8–11) with alternating 2-CiO-α and 3-CiO-α-CDs, which was confirmed by a larger association constant of the mixture compared to the individuals alone.65
![Formation of (A) [c2]daisy chains by 6-cinnamoyl α-cyclodextrins and (B) oligomers with n = 15 by 3-cinnamoyl α-cyclodextrins.51](/image/article/2013/CS/c2cs35217f/c2cs35217f-f8.gif) | ||
| Fig. 8 Formation of (A) [c2]daisy chains by 6-cinnamoyl α-cyclodextrins and (B) oligomers with n = 15 by 3-cinnamoyl α-cyclodextrins.51 | ||
The importance of the linking position on the cyclodextrin was demonstrated by studies on photoisomerizable stilbenes (9).54 Linking of trans-stilbene to the hydroxyl group in 2 position (9a) afforded double threaded dimers at high concentrations, whereas cis-stilbene obtained after photoisomerization gave acyclic oligomers. In contrast, linkage of trans-stilbene to the hydroxyl group in 3 position (9b) led to formation of acyclic oligomers at high aqueous concentrations and cis-stilbene to [c2]daisy chains (Fig. 9).
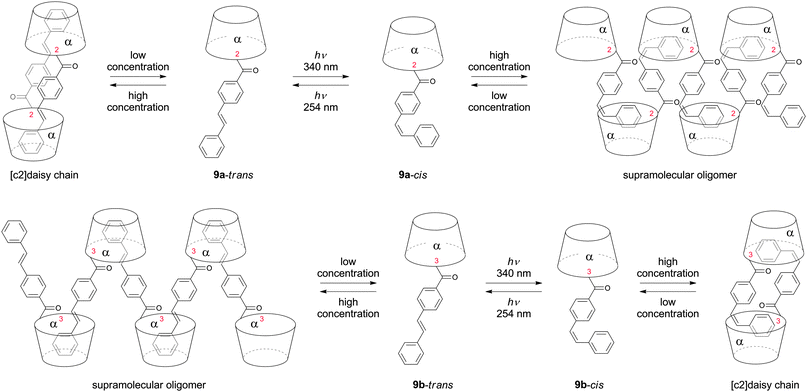 | ||
| Fig. 9 Aggregation behavior of in 2 or 3 position photoisomerizable stilbene functionalized α-cyclodextrins. Depending of the substitution position either the trans- or cis-isomers form supramolecular oligomers.54 | ||
Studies of Liu et al. have shown that not only the linking position but also the spatial arrangement of the tail can be of great importance for the self-assembling behaviour of daisy chain monomers. Azobenzenes substituted with propargyl alcohols were coupled to azide functionalized β-cyclodextrin hydrothermally and copper catalysed. Hydrothermal linkage led to self-threading (10a), whereas Cu-catalyzed click chemistry gave the unthreaded daisy chain monomer (10b). Surprisingly, the [1]rotaxane assembled in aqueous solution into [c2]daisy chains as demonstrated by ROESY-NMR and solid state structure. The β-cyclodextrin with a free cavity gave linear oligomeric self-assemblies in water–DMSO mixtures (Fig. 10).66
![Aggregation behavior a self-threaded [1]-rotaxane 10a and azobenzene substituted β-cyclodextrin 10b.66](/image/article/2013/CS/c2cs35217f/c2cs35217f-f10.gif) | ||
| Fig. 10 Aggregation behavior a self-threaded [1]-rotaxane 10a and azobenzene substituted β-cyclodextrin 10b.66 | ||
Another interesting and impressive method using cyclodextrin based head–tail monomers to construct oligomeric daisy chain-like supramolecular assemblies was reported by Miyauchi et al.67 In this study an α-cyclodextrin was equipped with an admantyl guest in 3 position and a β-cyclodextrin with a tBoc-cinnamoyl group. The guests are in this case not perfectly suited in size for inclusion in the attached cavity. In contrast, the admantyl group binds strongly to β-cyclodextrin and the tBoc-cinnamoyl to α-cyclodextrin and not to β-cyclodextrin. By mixing the two daisy chain monomers in a 1:1 ratio intermolecular complexes were observed by NMR leading to alternating α, β-cyclodextrin oligomeric daisy chains. By vapor pressure osmometry the molecular weight of these cyclodextrin based supramolecular polymers was estimated to be about 10000.67
Calixarene/pillar[5]arene based daisy chains
Floriani et al. modified calix[4]arenes with tungsten which was bound by the oxomatrix of these bowl-shaped molecular hosts (11).68 This functionalization allowed not only the alteration of the calixarene shape but also further complexation of phenoxy groups which act as aromatic guest molecules (Fig. 11). By X-ray analysis the self-assembly in columnar structures of these compounds was shown. Since the tungsten complex blocked the oxo-side of the calixarene cavity no formation of cyclic dimeric species was possible.Engbersen and Reinhoudt demonstrated by merging the cyclodextrin binding motif with calix[4]arenes (12) – functionalized with polyethylene glycols – to which a chromophore was attached in the opposite side to the oxomatrix that two possible superstructures are formed (Fig. 11).69 One where the chromophore is incorporated into the cavity of the cyclodextrin intramolecularly which forms vesicles (12a) and a second one where poly[n]daisy chains are formed by intermolecular recognition leading to fibers (12b).
![(A) Tungsten functionalized calix[4]arene (11) which forms columnar stacks in the solid state; (B) merged cyclodextrin and calixarene motifs. Substituent X can either be included in the cyclodextrin intra- or intermolecularly.](/image/article/2013/CS/c2cs35217f/c2cs35217f-f11.gif) | ||
| Fig. 11 (A) Tungsten functionalized calix[4]arene (11) which forms columnar stacks in the solid state; (B) merged cyclodextrin and calixarene motifs. Substituent X can either be included in the cyclodextrin intra- or intermolecularly. | ||
Recently the formation of long polymeric fibers using pillar[5]arenes, a calixarene analogue, as host molecules was reported. In pillararenes the aromatic walls are interconnected in 1,4 instead of 2,6 position like in calixarenes.70
In contrast to the “traditional” host–guest motifs where hydrogen bonding, π–π interactions, hydrophobic interactions or metal–ligand complexation are used for supramolecular complexation, pillararenes complex electron poor guests by C–H⋯π interactions, the weakest hydrogen bonds. The rigidity of pillararenes compared to calixarenes, crown ethers or cyclodextrins allows for selective binding to specially designed guests and to low entropic costs upon oligomerization. Huang and co-workers were able to synthesize an octyl monofunctionalized dimethoxybenzene copillar[5]arene 13 (Fig. 12).461H-NMR-titrations in chloroform displayed a chemical upfield shift of the octyl-protons, indicating the inclusion of the alkane moiety into the electron rich cavity. The formation of the inclusion complex was further confirmed by 2D-NOESY-NMR. By using the Carother's equation aggregation numbers of n = 40 were calculated from these 1H-NMR-titrations. DOSY-NMR and viscosity measurements corroborated the high degree of polymerization at higher concentrations. Furthermore even long fibers were detected by scanning electron microscopy. The tendency of such systems to form polymers instead of small cyclic aggregates was explained by a positive change in entropy due to exclusion of the solvent from the cavity and a strong enthalpic driving force for the inclusion of the octyl chain. Bromide substitution of the octyl chain at the terminus resulted in the formation of smaller cyclic aggregates like [c2]daisy chains composed of mirror images of the pillararene.71 Similar results were obtained by Stoddart et al. for pillar[5]arenes substituted with viologen rods 14.33 UV/Vis spectra showed charge-transfer bands which were assigned to the penetration of the electron poor thread into the electron rich cavity. At lower temperatures the intensity of this charge-transfer band increased which is indicative for the shift of the equilibrium to the associated form. An association constant of 1.3 × 105M−1 was calculated from fluorescence titrations depicting a strong inclusion complex. By 1H-NMR-titrations, diffusion-ordered 1H-NMR-titrations, viscosimetry and dynamic light-scattering the aggregation of the daisy chain monomer to long polymeric structures was confirmed.
![Monofunctionalized pillar[5]arenes 13 and 14 reported by Huang et al. and Stoddart et al. respectively forming long acyclic polymeric aggregates at high concentrations in chloroform.33,46](/image/article/2013/CS/c2cs35217f/c2cs35217f-f12.gif) | ||
| Fig. 12 Monofunctionalized pillar[5]arenes 13 and 14 reported by Huang et al. and Stoddart et al. respectively forming long acyclic polymeric aggregates at high concentrations in chloroform.33,46 | ||
The availability of rather few studies about the self-aggregation behavior of pillar[5]arene based daisy chain monomers is directly linked to the low synthetic yields using a statistical pillararene assembly. Therefore a synthetic procedure developed by Ogoshi et al. towards monofunctionalized pillar[5]arenes where permethylated precursors are selectively mono de-O-methylated has great potential to lead towards an increased number of studies based on pillar[5]arene daisy chains.72
Crown ether based daisy chains
The crown ether/dialkylammonium recognition motif was already used successfully in molecular shuttles and was first investigated for daisy chain formation by Stoddart and Williams in 1998.23 A 2-formyl monofunctionalized dibenzo[24]crown-8 ether (DB24C8) was synthesized which was coupled with benzyl amine by reductive amination (15, Table 2). The heteroditopicity was established by final protonation of the free amine. In this study the methodology of daisy chains was introduced and the question came up whether such self-complementary molecules will form cyclic or acyclic chains. By solid state structures and mass spectrometry the formation of cyclic [c2]daisy chains was confirmed in the solid state as well as in the gas phase (Fig. 13). The recognition in such binding motifs is based on cooperative stabilizing π–π interactions of the catechol units and by hydrogen bonds between R2NH2+ and the polyether oxygens.| Compound | Aggregation number n | |
|---|---|---|
| 15 |
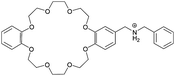
|
2 |
| 16 |
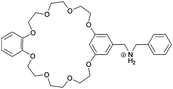
|
2, 3 |
| 17 |
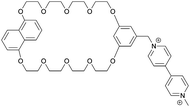
|
1−5 |
| 18 |
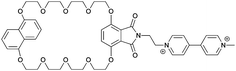
|
1−5 |
| 19 |
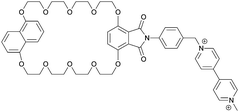
|
1−4 |
| 20 |
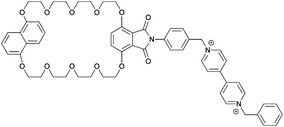
|
1−4 |
| 21 |
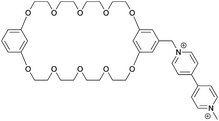
|
50 |
| 23 |
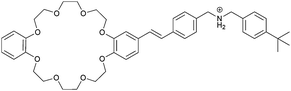
|
5 |
| 24 |
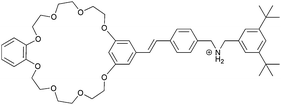
|
2, 3 |
| 25 |
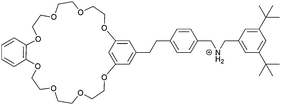
|
2 |
| 26 |
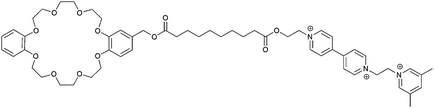
|
45 |
| 27 |
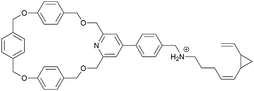
|
2 |
| 29 |
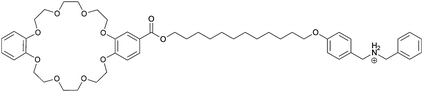
|
Oligomers |
| 30 |
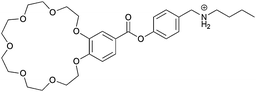
|
2 |
| 31 |
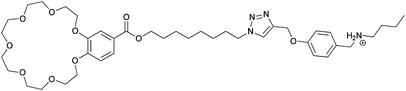
|
Oligomers |
![Crystal structure of a [c2]daisy chain formed by a plerotopic crown ether/dialkylammonium monomer (15). Reprinted with permission of Stoddart et al.23](/image/article/2013/CS/c2cs35217f/c2cs35217f-f13.gif) | ||
| Fig. 13 Crystal structure of a [c2]daisy chain formed by a plerotopic crown ether/dialkylammonium monomer (15). Reprinted with permission of Stoddart et al.23 | ||
The formation of [c2]daisy chains demonstrated the difficulty in generating infinite supramolecular arrays in non-covalent synthesis under thermodynamic control and therefore posed the question how to overcome enthalpic and entropic costs. Based on this study demonstrating the possibility of daisy chain formation using the mentioned recognition motif, several chemical modifications on the dibenzo[24]crown ether moiety as well as on the guest molecule were made to overcome the aggregation to the thermodynamically more stable [c2]daisy chain (also called Janus[2]rotaxane) (Table 2; compound 16 and 17).29,30,44 Since a π–π stacking between the host catechol units was observed in solid state structures of the parent test molecule (Fig. 13) which potentially favored dimer formation, these units were replaced by phthalimides and the “tail” was exchanged to viologens (paraquat) (18–20).30 By preventing the stabilizing interaction between two parts of the hosts, acyclic pentamers to dimers were detected by mass spectrometry in solution. This finding was further confirmed by concentration dependent 1H-NMR shifts where a peak broadening was observed at high concentrations and UV/Vis spectroscopy which showed a nonlinear correlation of absorbance with changing concentration and therefore gives evidence for aggregation. In contrast [c2]daisy chains were observed in the solid state structures. Additionally it was shown by Gibson et al. that expanding the crown ethers cavity to dibenzo[32]crown-10 and using a rigid viologen-tail (compound 21) to avoid self-complexation cause the formation of oligomers with up to 50 repeat units in a 2 M acetone solution (Fig. 14).44 The observed signal broadening in 1H-NMR at high concentration was related to the increased viscosity of the solution when polymers are present.
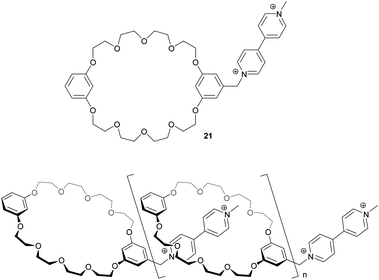 | ||
| Fig. 14 Formation of linear acyclic daisy chains with up to 50 repeat units by use of a crown ether/viologen binding motif.44 | ||
The inclusion of the viologen in the crown ether cavity was demonstrated by monitoring nuclear-overhauser-effects (NOE) of the viologen protons with protons inside the cavity. The presence of small cyclic aggregates was excluded by viscosity measurements and the higher viscosity compared to the unsubstituted crown ether. Because of their smaller hydrodynamic radius small aggregates should have a decreased viscosity compared to the linear aggregates.
By differential scanning calorimetry (DSC) two glass transitions were observed which were higher than the reference system further supporting the formation of daisy chains with a higher extend of self-organization. Another important finding by using such binding motifs was the observation of diastereoselective dimer formation in the parent catechol-based crown ether host (Fig. 15) (compound 15).29 The stereoisomers arise from the prochirality of the interacting cations. The absence of meso-forms in the solid state arising from maintenance of opposed prochiralities was explained by the diastereoselective crystallization process.
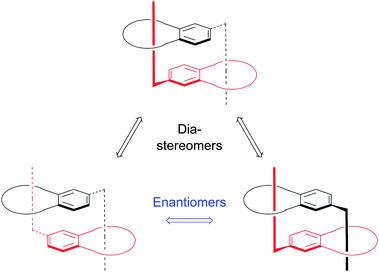 | ||
| Fig. 15 Schematic representation of the three stereoisomers which can form upon dimerization of compound 15.29 | ||
To overcome the aggregation of self-complementary head–tail monomers to the thermodynamically favored cyclic dimer several attempts were reported. Rowan and Stoddart used so-called surrogate-stoppered rotaxanes to construct linear polymeric acyclic daisy chains.24,73,74 This attempt makes use of the conventional covalent polymerization. Based on the dibenzo[24]crown-8 ether/dialkylammonium motif a crown ether monofunctionalized with an aldehyde on the catechol was threaded with a dibenzylammonium ion which was stoppered at one end with tert-butyl groups. After threading, the pseudorotaxane was capped with triphenylphosphine (22).
By treatment with sodium hydride the phosphonium was converted into an ylid which was able to undergo a Wittig reaction with the aldehyde moiety attached to the host molecule (compounds 23, 24, 25).
After regenerating the binding site by addition of acid, aggregates from [c2]daisy chains to acyclic pentamers were obtained in dichloromethane depending on the concentration. At lower concentrations mainly [c2]daisy chains were observed because the macrocyclization was favored over the chain propagation (Fig. 16). At higher concentrations oligomers were obtained including 5 monomers (compound 23).
![[c2]Daisy chain synthesized with the “surrogate-stoppered” approach. The preformed crown ether/dialkylammonium complex was capped with triphenylphospine and then cyclized by an intermolecular Wittig-reaction. Hydrogenation followed by restoring the binding site yielded [c2]daisy chains. Using similar conditions also yielded longer acyclic daisy chains with up to five repeat units.24,74](/image/article/2013/CS/c2cs35217f/c2cs35217f-f16.gif) | ||
| Fig. 16 [c2]Daisy chain synthesized with the “surrogate-stoppered” approach. The preformed crown ether/dialkylammonium complex was capped with triphenylphospine and then cyclized by an intermolecular Wittig-reaction. Hydrogenation followed by restoring the binding site yielded [c2]daisy chains. Using similar conditions also yielded longer acyclic daisy chains with up to five repeat units.24,74 | ||
Interestingly only cyclic species were detected when the reaction was performed in dimethylformamide, maybe because of the insolubility of the deprotonated intermediate. The preformation of rotaxanes prior to interlinking them was also performed by using cyclobis(paraquat-p-phenylene) cyclophanes encircling a polyethylene glycol based thread containing two hydroquinone recognition sites.53 Coupling of the functionalized cyclophane to one of the rotaxane stoppers afforded dimers. Repetition of this protocol has a high potential towards longer main-chain polymers. This assembling methodology was also used in a method called threading-followed-by-polymerization. Dibenzo[24]crown-8 ether monosubstituted with an acid chloride was threaded with a 1,2-bis(pyridinium)ethane unit substituted with a hydroxyl group, followed by esterification (compound 26).35 The temperature in the reaction was kept low to slow down the esterification and maximize the threading. Afterwards the temperature was raised to obtain full conversion (Fig. 17). By this acyclic oligomers including 45 monomer units were synthesized which was observed in UV/Vis, NMR and viscosity measurements.
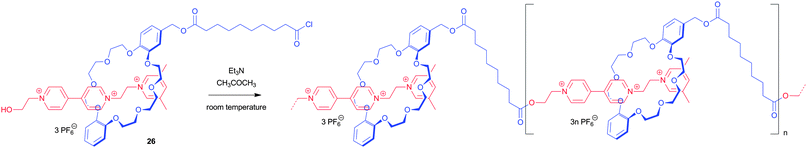 | ||
| Fig. 17 Schematic representation of the preparation of a long acyclic mechanically interlocked daisy chain with up to 45 repeat units. This threading-followed-by-polymerization approach is based on the polycondensation of a dynamic bifunctional pseudorotaxane.35 | ||
In other studies it was proposed that making the host and the guest more rigid can avoid homo dimer formation. Even though this approach was unsuccessful and only [c2]daisy chains were obtained an interesting stoppering method was used, the so-called threading-followed-by-swelling. For this the rigid tails were functionalized with dialkenylcyclopropanes (compound 27) and after aggregation heated to form a cycloheptadiene by a rearrangement (28) (Fig. 18).75 The clipping methodology used for synthesis of poly[n]rotaxanes was also applied to assemble [c2]daisy chains.27 To an isophthalamide based half cycle functionalized at the two ends with alkenes which can undergo ring-closing metathesis a hydrocarbon rod containing a 3,5-dicarbamoyl-1-methylpyridinium chloride was attached (29). The isophthalamide is then able to complex the chloride in apolar solvents. By ring-closing metathesis the macrocycle was “clipped” around the complexed thread which resulted in a product mixture of [c2]daisy chains and “self-biting” monomers (Fig. 19). Even though the yields of dimer formation were low, this approach represents a novel potential route towards mechanically interlocked molecules.
![Fixing a preformed [c2]daisy chain by swelling stoppers.75](/image/article/2013/CS/c2cs35217f/c2cs35217f-f18.gif) | ||
| Fig. 18 Fixing a preformed [c2]daisy chain by swelling stoppers.75 | ||
![Schematic illustration of the synthesis of a pyridinium/isophthalamide [c2]daisy chain using an anion-templating approach.27](/image/article/2013/CS/c2cs35217f/c2cs35217f-f19.gif) | ||
| Fig. 19 Schematic illustration of the synthesis of a pyridinium/isophthalamide [c2]daisy chain using an anion-templating approach.27 | ||
In a similar attempt Grubbs et al. used ring-closing metathesis to form well defined [c2]daisy chains and then to further incorporate these mechanically interlocked units into a polymeric scaffold by conventional polymerization. In this case an open polyethylene glycol to which the latter molecular rod equipped with a suitable stopper already attached was used. Precomplexation followed by ring-closing metathesis yielded the desired [c2]daisy chains.76
The widely used DB24C8/dibenzylammonium (DBA) motif was also part of a hermaphroditic monomer investigated by Huang et al.77 In contrast to the previous discussed motifs where a rigid tail was used to overcome the entropic costs of polymerization, the molecular tail was made more flexible to allow the system to find its perfect alignment. Therefore the host (DB24C8) and the guest (DBA) were covalently linked by a flexible alkyl chain (29). 1H-NMR titrations showed the formation of small cyclic oligomers at low monomer concentrations. However at high concentrations (>60 mM in MeCN) polymeric species were observed, which was further confirmed by DOSY-NMR and viscosity measurements. It was thus demonstrated that indeed a more flexible tail lowers the critical polymerization concentration (CPC) dramatically. By cooling of the solution long fibers and even 3D networks were observed by SEM.77 This daisy chain monomer showed gelation to super gelation (gelation below 0.1 wt%) properties in various organic solvents, where the gel-sol transition could be triggered by addition of base, K+, Fe3+ or heating. The process could be reversed by addition of acid, B18C6, oxidation or cooling respectively, leading to a multi stimulus responsive gelator.78 Despite the DB24C8/DBA motif also a benzo-21-crown-7 (B21C7)/secondary ammonium salt was used to construct crown ether based daisy chains (30).79 The advantage of B21C7 compared to DB24C8 is that the smaller crown ether (B21C7) binds the dialkylammonium guest more efficient. The host was merged covalently with a benzyl-butyl-ammonium guest by esterification. Solid state structures obtained from a CH2Cl2/MeOH mixture confirmed the formation of [c2]daisy chains. The intermolecular inclusion of the tail into the macrocycle was demonstrated by 1H-NMR COSY and NOESY-NMR. Furthermore it was shown that this binding motif is multi-responsive because addition of a base or K+ caused dethreading of the dimer.79 Similar to the larger DB24C8 crown ether motif introduction of a long alkyl spacer between the host and the guest (31) allowed for the formation of acyclic polymeric aggregates in chloroform above a concentration of 55 mM. The electrospinning technique was used to obtain nanofibers as observed by SEM.80 Furthermore it was even possible to orthogonally self-assemble these long polymeric daisy chains to an elastic polymer network by addition of a palladium salt. Thereby the 1,2,3-triazoles of 31, introduced by click chemistry, were used as ligands for the transition metal.81 These gels could be molded into shape-persistent, free-standing objects which exhibit pH-, thermo-, cation- and metal induced gel-sol transitions.82 These studies are remarkable examples where it was possible to obtain not only acyclic, long polymeric daisy chains, but also to include these pseudorotaxanes into 3D polymer networks.
Molecular muscles
A further dimension of material manipulation can be obtained by the inclusion of two binding motifs on the molecular thread. It is then not only possible to generate macromolecules where the mechanical bond is an integral part of the main-chain polymer but also to alter the properties by an external stimulus. In analogy to molecular shuttles, where two binding sites for a macrocycle are present in the thread of a rotaxane, the external stimulus can disturb the thermodynamical equilibrium leading to the movement of the macrocycle to the new global energy minimum. Therefore chemical, electrochemical or photochemical inputs can be converted to mechanical linear motion in daisy chains what leads to artificial muscular behavior due to the reversible contraction and extension (Fig. 20). [c2]Daisy chains are ideal candidates to investigate such processes because of their compactness, their kinetic and thermodynamic stability and the linear arrangement of the two threads with respect to each other. The dibenzo[24]crown-8 ether/dibenzylammonium binding motif was used to generate pseudo[c2]daisy chain rotaxanes.31 The end standing benzyl was further functionalized with a benzylic bromide which enabled to cap the supermolecule with a 3,5-di-tert-butylbenzyl substituted paraquat to generate a kinetically stable [c2]daisy chain. In the favored co-conformation the two hosts complexed the ammonium centers by [NH+–O] hydrogen bonds. By deprotonation of the recognition site a migration to the paraquat station was obtained which caused a contraction of the [c2]daisy chain. The process was monitored by 1H-NMR where a downfield shift due to association of the two electron poor paraquat units was observed. Reprotonation of the amine reversed the whole process.![Acid/base switchable [c2]daisy chain as contracting and extending subunit in an oligomeric chain.87](/image/article/2013/CS/c2cs35217f/c2cs35217f-f20.gif) | ||
| Fig. 20 Acid/base switchable [c2]daisy chain as contracting and extending subunit in an oligomeric chain.87 | ||
Oligomers consisting of up to 11 such daisy chain units in series were obtained by an [AA + BB]-type conventional polymerization when the stoppers in this system were further functionalized with acetylenes (32).32 For polymerization a copper mediated 1,3-dipolar cycloaddition between attached alkenyls and diazides was used (33). The averaged molecular weight and the polydispersity of the polymers obtained were estimated by Zimm-plot analysis from size exclusion chromatography. The switching mechanism was monitored by 1H-NMR, UV/Vis and cyclic voltammetry. Quantitative, efficient and reversible switching in solution was observed of the oligomer where the contraction/extension was even faster compared to the “monomeric” [c2]daisy chain (Fig. 20). The contraction and extension were identified as the rate limiting steps which are slower than the acid/base reaction.83 This approach is a perfect example that correlated molecular motion in oligomers is possible and therefore can be regarded as a first step towards conversion of molecular properties to the macroscopic scale.
As further examples of acid/base switchable [c2]daisy chain artificial molecular muscles the work of Coutrot on dimannosyl capped DB24C8/dialkylammonium salts have to be mentioned.84 There also more than 2 binding sites where introduced into the molecular tail leading to molecular muscles with different contraction modes.85
The use of a clipping methodology by Grubbs et al. to encircle a molecular rod by an half-cycle followed by ring-closing metathesis allowed for the synthesis of a [c2]daisy chain where the ammonium center was placed near the stopper, giving an extension and contraction of amazing 48%.86
Based on the observation that heteroditopic cyclodextrins can form [c2]daisy chains with azobenzenes and that photochemical E/Z isomerization leads to dissociation of the pseudorotaxanes, the described systems were capped with bulky anilines and then polymerized with para-bis(bromomethyl)benzene to obtain oligomers including up to five [c2]daisy chains (34).25 The stoppers and the N,N-linkage prevented dethreading of the Z-isomer when photochemically switched. The Z-form (34b) is significantly shorter than the E-form (34a) which results in contraction of the oligomer by irradiation and can therefore be regarded as the first light-driven molecular muscle (Fig. 21). The same was possible by photoisomerization of stilbene based cyclodextrin [c2]daisy chains.52 By photoisomerization the trans-stilbene was converted into the cis-form which caused a slipping of the cyclodextrin towards the alkyl chains attached to the stilbenes. Harada and co-workers elongated their cinnamoyl substituted α-cyclodextrins with long aliphatic chains bearing bulky stoppers at each side. This mechanically interlocked artificial molecular muscle showed an elongation upon increasing the solvent polarity by addition of water. By PFGSE-NMR the variation in size was monitored as a function of the solvent polarity.88
![Light-driven molecular muscle based on an azobenzene functionalized permethylated α-cyclodextrin [c2]daisy chain motif.25](/image/article/2013/CS/c2cs35217f/c2cs35217f-f21.gif) | ||
| Fig. 21 Light-driven molecular muscle based on an azobenzene functionalized permethylated α-cyclodextrin [c2]daisy chain motif.25 | ||
The very first example of artificial molecular muscles was based on a metal-templating approach and represents until today the [c2]daisy chain system with the highest contraction efficiency.26 Sauvage and co-workers coupled a linear, bidentate phenanthroline moiety to a macrocycle containing a second phenanthroline binding site. The monomers were templated by addition of CuI to form a [c2]daisy chain where the bidentate ligands form a tetrahedral metal complex. The linear rods were then capped by a terpyridine substituted 4-(tris(4-(tert-butyl)-phenyl)methyl)phenol to install the second – in this case tridentate – binding site (35a). Metal exchange promoted by potassium cyanide, and subsequent addition of ZnII resulted in a penta-coordinated metal complex between the bidentate ligands of the macrocycles and the terpyridine stations (35b) (Fig. 22). By this the whole system was contracted by an amazing 27% which is in the dimension of natural muscle contraction.
![The first [c2]daisy chain motif acting as unimolecular muscle triggered by the exchange of the coordinating ion.26](/image/article/2013/CS/c2cs35217f/c2cs35217f-f22.gif) | ||
| Fig. 22 The first [c2]daisy chain motif acting as unimolecular muscle triggered by the exchange of the coordinating ion.26 | ||
Molecular design analysis
By careful analysis of the systems used to prepare poly[n]daisy chains it is possible to gain evidence which structural features are necessary to obtain oligomerization (Tables 1 and 2). Functionalization of the cylindrical cyclodextrin structures in 6 position – the smaller side of the cavity – results almost exclusively in formation of [c2]daisy chains (Table 1). In contrast to this, functionalization with a rigid rod in 3 position on the secondary hydroxyl side leads to oligomer formation as found by Harada et al.51 Furthermore in all modified cyclodextrins the molecular tail enters the hydrophobic cavity from the primary hydroxyl side which can be explained by the formation of a stronger supramolecular inclusion complex by minimizing unfavorable dispersion forces. As a result the rods coupled in 6 position enter the cyclodextrin cavity from the primary hydroxyl side which places the second rod in ideal position to form stable [c2]daisy chains and therefore to find its thermodynamic global minimum. If the rod is included from the secondary hydroxyl side the reversibility of the system prevents formation of long acyclic species (Fig. 23). The same situation occurs if the molecular tail is placed in 3 position, but in contradiction the formation of cyclic dimers is now thermodynamically not the most favored situation which causes again deaggregation because of the reversibility of the system (Fig. 23). Inclusion from the primary hydroxyl side of the cyclodextrin can be therefore seen as the first propagation step towards oligomers. The dynamic behavior thus leads to self-sorting oligomers. Furthermore evidence for the importance of the pivot atom to the formation of oligomers can be obtained by comparing compounds 6 and 9. Even though both stilbene rods are attached to the cyclodextrin in 3 position, compound 6 with an amide linkage forms dimers and compound 9 with an ester linkage oligomers. Also the rigidity of the rod seems to play an important role since the attachment of stilbenes, azobenzenes and cinnamoyls on the primary alcohol in 6 position leads to cyclic dimer formation. Unfortunately no example is known were a rigid molecular rod is attached on the secondary alcohol in a benzylic position. Therefore no conclusion can be drawn about the influence of such a flexible part in the molecular rod.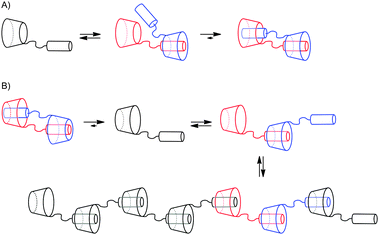 | ||
| Fig. 23 Schematic illustration of the aggregation behavior of hermaphroditic cyclodextrins with: (A) thread attached in 6 position; (B) thread attached in 3 position. | ||
The situation in the case of the crown ether based hermaphroditic molecules is somehow more complex since no discrimination of one side of the cavity can be expected. Comparison of the systems used as heteroditopic monomers (Table 2) provides some indications about the structure-property relation of such compounds. In solid state structures of [c2]daisy chains formed by dibenzo24-crown-8 ethers and short, more or less rigid molecular tails (compounds 15, 23, 24) several stabilizing interactions between the host and the guest were observed (Fig. 24A, B and D). Additionally host–host (Fig. 24D) and guest–guest interactions (Fig. 24A and B) were found, demonstrating the stability of the smallest possible cyclic daisy chain. The exchange of catechols (compound 15) by 1,5-dihydroxynaphthyl units (compounds 17–20) results in formation of pentamers. Solid state structures showed that the naphthyl moiety is able to complex the electron poor paraquat tails by π–π stacking, and the resorcinol units stabilizes the tail by edge-to-face interaction (Fig. 24C).30 No interaction in the solid state structure of compound 17 between the two resorcinol moieties could be observed in contrast to compound 15 where the two functionalized catechols stack with each other (Fig. 24C and D).23 Furthermore no interactions between the two viologen tails were present like in compound 23 (Fig. 24B).
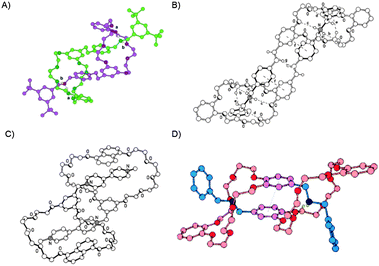 | ||
| Fig. 24 Solid state structures of compound 24 (A),7423 (B),8917 (C)30 and 15 (D)23 displaying the different stabilizing interactions between the host and the guest. Reprinted with permission of the corresponding publishers. | ||
Thus prevention of the host–host and the guest–guest interaction seems to enhance the probability for formation of acyclic aggregates. The same holds true for compounds 18–20 where the phthalimide units prevent host–host stacking. The reason why hermaphroditic compound 21 forms daisy chains with aggregation numbers up to 50 remains unclear. Unfortunately only in a few cases the association constants were determined and therefore no conclusions about the stability of these complexes can be drawn. The inclusion complex in 21 seems to be stable enough to overcome the entropic costs of oligomer formation. In the beginnings of daisy chain research the hypothesis came up that a rigid molecular rod is crucial for the formation of acyclic oligomeric daisy chains by heteroditopic monomer.5,8,75 This hypothesis was based on the idea that a high degree of preorganization can overcome the entropic costs of polymerization. Indeed it was shown by crown ether based cryptands to which a viologen was coupled that a well defined guest leads to stronger binding of the molecular rod. By introduction of a flexible alkyl linker, at high concentrations or low temperature linear oligomeric daisy chains were observed in acetonitrile.81 Recently Huang and co-workers could demonstrate that introduction of a long flexible alkyl linker between the host and the guest of a hermaphroditic monomer (compounds 29 and 31) results in the formation of supramolecularly bonded oligomeric daisy chains in contrast to the parent systems (compound 15, 16, 23–25 and 30 respectively).77,78,80 Therefore it seems like that by introduction of flexible spacers the supramolecular complex has more degrees of freedom to find its perfect alignment. Furthermore host–host and guest–guest interactions are prevented. Nevertheless a rigid part which enters the cavity seems to be necessary in order to overcome the entropic costs of polymerization. Compound 26, which also contains a long aliphatic part and hence not a rigid rod, assembled to long oligomers, but in this case a different synthetic route was chosen making it impossible to compare the system with daisy chains assembled from heteroditopic monomers (Fig. 17). Nevertheless, formation of the supramolecular assembly and introduction of the stopper prior to attaching the molecular rod seems to be a reasonable pathway towards mechanically interlocked polymeric daisy chains since the competition between small cyclic and long acyclic aggregates can be controlled by the reaction conditions used for interlinking the two counterparts.
Conclusions
In conclusion, for the daisy chain systems reported so far, namely: cyclodextrins/aromatic guests and crown ethers/cations mainly the dimeric cyclic aggregate was formed. Nevertheless, indications were gained that preventing host–host and guest–guest interactions, as well as introduction of flexible aliphatic linkers might favor the ability to form higher daisy chain aggregates. Furthermore the size of the cavity and the stability of the inclusion complexes seem to play an important role in the formation of oligomeric species. In the pillararene based systems the entropic costs of polymerization seems to be compensated by a large gain in enthalpic energy hence leading to long polymeric daisy chains. Analysis of the relation between molecular structure and aggregation behavior allows us to propose a heteroditopic monomer which should in principle consist of a well defined, rigid cavity to minimize the entropic costs of polymerization. Furthermore strong host–guest interactions are crucial to obtain a strong inclusion complex, which is preferentially formed by an induced complexation like change of solvent, structural change of the cavity by an external trigger or external stimulation of the thread. Furthermore the molecular rod should be chosen such that host–host, as well as guest–guest interactions are prevented and formation of strong inclusion complexes is guaranteed. As in the case of cyclodextrin systems discrimination of complexation from the side of the cavity where the tail is attached should also lead to formation of higher oligomers. Additionally binding motifs where the solvent induces complexation should be considered, like for example making use of a non-classical hydrophobic effect. The strong cohesive forces of for example water then lead to a large gain in enthalpy upon formation of a host–guest complex which can overcome the entropic costs. Nevertheless, the assembly to [c2]daisy chains is thermodynamically favored and therefore more precise. Polymerization of such dimers using conventional covalent coupling allows the preparation of well defined oligomers and is thus maybe the most promising route towards novel functional materials.25,32Despite the fact that only a few examples are known where longer oligomeric species could be observed, these novel types of polymers are expected to have unique properties. Improved stability, exterior functionalization, better processability and especially the internal motion can potentially lead to novel materials with applications in all areas of our daily life were high-performing plastics are of great interest.
Generous support by the University of Basel, the Swiss National Science Foundation (SNF), the Swiss Nano Institute, the NCCR “Nanoscale Sciences” of the SNF and the Karlsruhe Institute of Technology is greatfully acknowledged. The authors thank Michel Rickhaus for beautiful artwork.
Notes and references
- J. Kaur, J. H. Lee and M. L. Shofner, Polymer, 2011, 52, 4337–4344 CrossRef CAS.
- H. W. Gibson, M. C. Bheda and P. T. Engen, Prog. Polym. Sci., 1994, 19, 843–945 CrossRef CAS.
- H.-H. Kausch-Blecken von Schmeling, Colloid Polm. Sci., 2011, 289, 1407–1427 CrossRef CAS.
- J. M. Cowie, Polymers: Chemistry and Physics of Modern Materials, 1974 Search PubMed.
- L. Fang, M. A. Olson, D. Benítez, E. Tkatchouk, W. A. Goddard III and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17 RSC.
- T. Takata, N. Kihara and Y. Furusho, in Polymer Synthesis, Springer Berlin/Heidelberg, 2004, vol. 171, pp. 1–75 Search PubMed.
- A. Harada, Y. Takashima and H. Yamaguchi, Chem. Soc. Rev., 2009, 38, 875 RSC.
- B. Zheng, F. Wang, S. Dong and F. Huang, Chem. Soc. Rev., 2012, 41, 1621 RSC.
- J. Beck B and S. J. Rowan, Supramolecular Polymers, Taylor & Francis Group, 2nd edn, 2005 Search PubMed.
- F. Huang and H. W. Gibson, Prog. Polym. Sci., 2005, 30, 982–1018 CrossRef CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- G. Wenz, B. H. Han and A. Müller, Chem. Rev., 2006, 106, 782–817 CrossRef CAS.
- F. M. Raymo and J. F. Stoddart, Chem. Rev., 1999, 99, 1643–1664 CrossRef CAS.
- J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802 RSC.
- J. Watanabe, Chem. Lett., 1998, 1031 CrossRef CAS.
- H. Gibson, Macromolecules, 1997, 30, 3711 CrossRef CAS.
- C. Gong, Macromolecules, 1998, 31, 5278 CrossRef CAS.
- I. Yamaguchi, Polym. Bull. (Berlin), 2000, 44, 247 CrossRef CAS.
- M. J. Frampton and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 1028–1064 CrossRef CAS.
- J. Li and X. J. Loh, Adv. Drug Deliv. Rev., 2008, 60, 1000–1017 CrossRef CAS.
- S. S. Zhu, P. J. Carroll and T. M. Swager, J. Am. Chem. Soc., 1996, 118, 8713–8714 CrossRef CAS.
- J. J. Li, F. Zhao and J. Li, Appl. Microbiol. Biotechnol., 2011, 90, 427–443 CrossRef CAS.
- P. R. Ashton, I. Baxter, S. J. Cantrill, M. C. T. Fyfe, P. T. Glink, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 1294–1297 CrossRef CAS.
- S. J. Rowan and J. F. Stoddart, Polym. Adv. Technol., 2002, 13, 777–787 CrossRef CAS.
- S. Tsuda, Y. Aso and T. Kaneda, Chem. Commun., 2006, 3072 RSC.
- M. C. Jiménez, C. Dietrich-Buchecker and J. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284–3287 CrossRef.
- N. H. Evans and P. D. Beer, Chem.–Eur. J, 2011, 17, 10542–10546 CrossRef CAS.
- J. Voignier, J. Frey, T. Kraus, M. Buděšínský, J. Cvačka, V. Heitz and J.-P. Sauvage, Chem.–Eur. J, 2011, 17, 5404–5414 CrossRef CAS.
- S. J. Cantrill, G. J. Youn, J. F. Stoddart and D. J. Williams, J. Org. Chem., 2001, 66, 6857–6872 CrossRef CAS.
- P. R. Ashton, I. W. Parsons, F. M. Raymo, J. F. Stoddart, A. J. P. White, D. J. Williams and R. Wolf, Angew. Chem., Int. Ed., 1998, 37, 1913–1916 CrossRef CAS.
- J. Wu, K. C.-F. Leung, D. Benítez, J.-Y. Han, S. J. Cantrill, L. Fang and J. F. Stoddart, Angew. Chem., Int. Ed., 2008, 47, 7470–7474 CrossRef CAS.
- L. Fang, M. Hmadeh, J. Wu, M. A. Olson, J. M. Spruell, A. Trabolsi, Y.-W. Yang, M. Elhabiri, A.-M. Albrecht-Gary and J. F. Stoddart, J. Am. Chem. Soc., 2009, 131, 7126–7134 CrossRef CAS.
- N. L. Strutt, H. Zhang, M. A. Giesener, J. Lei and J. F. Stoddart, Chem. Commun., 2012, 48, 1647 RSC.
- F. Diederich, Angew. Chem., Int. Ed. Engl., 1988, 27, 362–386 CrossRef.
- M. Zhang, S. Li, S. Dong, J. Chen, B. Zheng and F. Huang, Macromolecules, 2011, 44, 9629–9634 CrossRef CAS.
- R. Margalit, N. Shaklai and S. Cohen, Biochem. J., 1983, 209, 547 CAS.
- F. M. Menger and L. G. Whitesell, J. Org. Chem., 1987, 52, 3793–3798 CrossRef CAS.
- J. W. Park, H. E. Song and S. Y. Lee, J. Phys. Chem. B, 2002, 106, 5177–5183 CrossRef CAS.
- S. R. McAlpine and M. A. Garcia-Garibay, J. Am. Chem. Soc., 1998, 120, 4269–4275 CrossRef CAS.
- D. Delmarre, N. Hioka, R. Boch, E. Sternberg and D. Dolphin, Can. J. Chem., 2001, 79(5), 1068–1074 CrossRef CAS.
- T. W. Davey, W. A. Ducker and A. R. Hayman, Langmuir, 2000, 16, 2430–2435 CrossRef CAS.
- S. Anderson and H. L. Anderson, Angew. Chem., Int. Ed. Engl., 1996, 35, 1956–1959 CrossRef CAS.
- S. Anderson, R. T. Aplin, T. D. W. Claridge, T. Goodson III, A. C. Maciel, G. Rumbles, J. F. Ryan and H. L. Anderson, J. Chem. Soc., Perkin Trans. 1, 1998, 2383–2398 RSC.
- N. Yamaguchi, D. S. Nagvekar and H. W. Gibson, Angew. Chem., Int. Ed., 1998, 37, 2361–2364 CrossRef CAS.
- T. Fujimoto, Y. Sakata and T. Kaneda, Chem. Commun., 2000, 2143–2144 RSC.
- Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1397–1401 CrossRef CAS.
- B. O. Persson, T. Drakenberg and B. Lindman, J. Phys. Chem., 1976, 80, 2124–2125 CrossRef CAS.
- E. M. Seward, R. B. Hopkins, W. Sauerer, S. W. Tam and F. Diederich, J. Am. Chem. Soc., 1990, 112, 1783–1790 CrossRef CAS.
- T. Fujimoto, Y. Uejima, H. Imaki, N. Kawarabayashi, J. H. Jung, Y. Sakata and T. Kaneda, Chem. Lett., 2000, 29, 564–565 CrossRef.
- T. Fujimoto, Y. Sakata and T. Kaneda, Chem. Lett., 2000, 29, 764–765 CrossRef.
- M. Miyauchi, Y. Kawaguchi and A. Harada, J. Inclusion Phenom. Macrocyclic Chem., 2004, 50, 57–62 CrossRef CAS.
- R. E. Dawson, S. F. Lincoln and C. J. Easton, Chem. Commun., 2008, 3980 RSC.
- M. P. L. Werts, M. van den Boogaard, G. M. Tsivgoulis and G. Hadziioannou, Macromolecules, 2003, 36, 7004–7013 CrossRef CAS.
- A. Kanaya, Y. Takashima and A. Harada, J. Org. Chem., 2010, 76, 492–499 CrossRef.
- K. Hirotsu, T. Higuchi, K. Fujita, T. Ueda, A. Shinoda, T. Imoto and I. Tabushi, J. Org. Chem., 1982, 47, 1143–1144 CrossRef CAS.
- D. Mentzafos, A. Terzis, A. W. Coleman and C. de Rango, Carbohydr. Res., 1996, 282, 125–135 CrossRef CAS.
- Y. Liu, Z. Fan, H.-Y. Zhang and C.-H. Diao, Org. Lett., 2003, 5, 251–254 CrossRef CAS.
- Y. Liu, Z. Fan, H.-Y. Zhang, Y.-W. Yang, F. Ding, S.-X. Liu, X. Wu, T. Wada and Y. Inoue, J. Org. Chem., 2003, 68, 8345–8352 CrossRef CAS.
- T. Fujimoto, A. Nakamura, Y. Inoue, Y. Sakata and T. Kaneda, Tetrahedron Lett., 2001, 42, 7987–7989 CrossRef CAS.
- K. Yamauchi, Y. Takashima, A. Hashidzume, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2008, 130, 5024–5025 CrossRef CAS.
- H. Onagi, C. J. Easton and S. F. Lincoln, Org. Lett., 2001, 3, 1041–1044 CrossRef CAS.
- L.-H. Tong, Z.-J. Hou, Y. Inoue and A. Tai, J. Chem. Soc., Perkin Trans. 2, 1992, 1253 RSC.
- T. Hoshino, M. Miyauchi, Y. Kawaguchi, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2000, 122, 9876–9877 CrossRef CAS.
- M. Miyauchi, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2005, 127, 2984–2989 CrossRef CAS.
- N. Tomimasu, A. Kanaya, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2009, 131, 12339–12343 CrossRef CAS.
- Y. Liu, Z.-X. Yang and Y. Chen, J. Org. Chem., 2008, 73, 5298–5304 CrossRef CAS.
- M. Miyauchi and A. Harada, J. Am. Chem. Soc., 2004, 126, 11418–11419 CrossRef CAS.
- A. Zanotti-Gerosa, E. Solari, L. Giannini, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Chem. Commun., 1996, 119 RSC.
- J. Bügler, N. A. J. M. Sommerdijk, A. J. W. G. Visser, A. van Hoek, R. J. M. Nolte, J. F. J. Engbersen and D. N. Reinhoudt, J. Am. Chem. Soc., 1999, 121, 28–33 CrossRef.
- T. Ogoshi, S. Kanai, S. Fujinami, T. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS.
- Z. Zhang, G. Yu, C. Han, J. Liu, X. Ding, Y. Yu and F. Huang, Org. Lett., 2011, 13, 4818–4821 CrossRef CAS.
- T. Ogoshi, K. Demachi, K. Kitajima and T. Yamagishi, Chem. Commun., 2011, 47, 7164 RSC.
- S. J. Rowan, S. J. Cantrill, J. F. Stoddart, A. J. P. White and D. J. Williams, Org. Lett., 2000, 2, 759–762 CrossRef CAS.
- S.-H. Chiu, S. J. Rowan, S. J. Cantrill, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem. Commun., 2002, 2948–2949 RSC.
- S.-H. Ueng, S.-Y. Hsueh, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Chem. Commun., 2008, 817 RSC.
- E. N. Guidry, J. Li, J. F. Stoddart and R. H. Grubbs, J. Am. Chem. Soc., 2007, 129, 8944–8945 CrossRef CAS.
- S. Dong, Y. Luo, X. Yan, B. Zheng, X. Ding, Y. Yu, Z. Ma, Q. Zhao and F. Huang, Angew. Chem., Int. Ed., 2011, 50, 1905–1909 CrossRef CAS.
- S. Dong, B. Zheng, D. Xu, X. Yan, M. Zhang and F. Huang, Adv. Mater., 2012, 24, 3191–3195 CrossRef CAS.
- B. Zheng, M. Zhang, S. Dong, J. Liu and F. Huang, Org. Lett., 2011, 14, 306–309 CrossRef.
- X. Yan, M. Zhou, J. Chen, X. Chi, S. Dong, M. Zhang, X. Ding, Y. Yu, S. Shao and F. Huang, Chem. Commun., 2011, 47, 7086 RSC.
- F. Wang, J. Zhang, X. Ding, S. Dong, M. Liu, B. Zheng, S. Li, L. Wu, Y. Yu, H. W. Gibson and F. Huang, Angew. Chem., Int. Ed., 2010, 49, 1090–1094 CrossRef CAS.
- X. Yan, D. Xu, X. Chi, J. Chen, S. Dong, X. Ding, Y. Yu and F. Huang, Adv. Mater., 2012, 24, 362–369 CrossRef CAS.
- M. Hmadeh, L. Fang, A. Trabolsi, M. Elhabiri, A.-M. Albrecht-Gary and J. F. Stoddart, J. Mater. Chem., 2010, 20, 3422 RSC.
- F. Coutrot, C. Romuald and E. Busseron, Org. Lett., 2008, 10, 3741–3744 CrossRef CAS.
- C. Romuald, E. Busseron and F. Coutrot, J. Org. Chem., 2010, 75, 6516–6531 CrossRef CAS.
- P. G. Clark, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 13631–13633 CrossRef CAS.
- L. Fang, M. A. Olson, D. Benítez, E. Tkatchouk, W. A. Goddard III and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17 RSC.
- W. Deng, H. Yamaguchi, Y. Takashima and A. Harada, Angew. Chem., Int. Ed., 2007, 46, 5144–5147 CrossRef CAS.
- D. G. Amirsakis, A. M. Elizarov, M. A. Garcia-Garibay, P. T. Glink, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2003, 42, 1126–1132 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |