Tandem inverse-electron-demand hetero-/retro-Diels–Alder reactions for aromatic nitrogen heterocycle synthesis
Radleigh A. A.
Foster
and
Michael C.
Willis
*
Department of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: michael.willis@chem.ox.ac.uk; Fax: +44 (0)1865 285002; Tel: +44 (0)1865 285126
First published on 19th October 2012
Abstract
The merged inverse-electron-demand hetero-Diels–Alder (ihDA)/retro-Diels–Alder (rDA) reaction sequence can be used to rapidly synthesise highly functionalised nitrogen heteroaromatics. The reaction offers many advantages: high atom economy, high levels of regioselectivity, it rarely requires a catalyst and, in some cases, can be performed in the absence of solvent. In this tutorial review we discuss the range of commonly used dienophiles and aza-dienes for this process whilst highlighting the reactivity trends, and illustrating their applications.
 Radleigh A. A. Foster | Radleigh Foster received his MChem in Chemistry from Jesus College, University of Oxford. During this he completed a Part II project under the supervision of Michael Willis, studing palladium-catalysed tandem processes in heterocycle synthesis. He has remained a member of the Willis group and is now in the final year of his DPhil studies. His research interests involve the novel synthesis of heterocycles capable of ihDA/rDA reactions to furnish alternative nitrogen heterocycles. |
 Michael C. Willis | Michael Willis received his undergraduate education at Imperial College London, and his PhD from the University of Cambridge working with Prof. Steven V. Ley. After a postdoctoral stay with Prof. David A. Evans at Harvard University, as a NATO/Royal Society Research Fellow, he was appointed to a lectureship at the University of Bath in November 1997. In January 2007 he moved to the University of Oxford, where he is a University Lecturer and Fellow of Lincoln College. His group's research interests are based on the development and application of new catalytic processes for organic synthesis. |
1. Introduction and overview
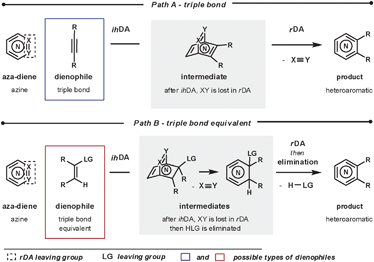
The impact that pericyclic reactions have had in the field of synthetic organic chemistry is enormous. This reaction class has been used to construct complex structures with outstanding degrees of control, and, importantly, the accompanying theory allows a high level of predictability for these processes.1 Intermolecular pericyclic reactions combine two units in a convergent synthetic sequence, they are highly atom economic, and proceed with no overall change in redox state. It is therefore unsurprising that they are frequently employed in target orientated syntheses.2The Diels–Alder (DA) reaction is arguably the most used of the pericyclic reactions. Within this class there are several subclasses: normal-, neutral- and inverse-electron-demand-, hetero-, and retro-Diels–Alder reactions. The focus of this review is the merged inverse-electron-demand hetero-/retro-Diels–Alder (ihDA/rDA) sequence for the synthesis of nitrogen heteroaromatics; an important motif in designed bioactive molecules and natural products (Scheme 1).3
 | ||
| Scheme 1 Starting material requirements for heteroaromatic synthesis using ihDA/rDA sequence. | ||
In this tutorial review we highlight the range of dienophiles employed and the most commonly used azines as aza-dienes for this process. We begin with a brief discussion about the different subclasses of DA transformations, the observed trends in reactivity for ihDA/rDA reactions, and discuss the degree of aromaticity and stability of the azines. Following this, each class of aza-diene will be reviewed individually in terms of synthesis, inter-, and intra-molecular ihDA/rDA reactions (when applicable), together with illustrative applications.
1.1 Types of Diels–Alder reactions
For all Diels–Alder reactions two new bonds are simultaneously formed between the two cycloaddends in a [π4s + π2s] fashion. Fortunately, it is this strict requirement that provides the high level of predictability and degree of stereocontrol attainable from this cycloaddition reaction.1 The reaction kinetics are governed by the energy difference between one cycloaddend's HOMO and the other cycloaddend's LUMO; the smaller the energy difference between the frontier molecular orbitals, the faster the reaction.4 To establish whether the reaction is normal-, neutral- or inverse-electron-demand, which cycloaddend contains the interacting HOMO and which contains the LUMO must be determined.If the reactive frontier orbital on the diene (4π system) is the LUMO, it is an inverse-electron demand reaction, whereas, if it is the HOMO, it is classified as a normal-electron demand reaction, as shown in Fig. 1. Thus, for iDA reactions, functional groups that lower the LUMO on the diene and raise the energy of the HOMO on the dienophile decrease the HOMO–LUMO energy gap (ΔELUMO–HOMO). Electron-withdrawing groups (EWG) lower the energy of both the HOMO and LUMO on a cycloaddend, whilst electron-donating groups (EDG) have the opposite effect. Therefore, iDA feature electron-poor dienes and electron-rich dienophiles. Neutral-electron-demand DA reactions are rarer and describe those that fall into neither of the previously mentioned subcategories. Their reactivity cannot be enhanced by electronically modifying the cycloaddends so it is unclear which orbitals on each cycloaddend are interacting.
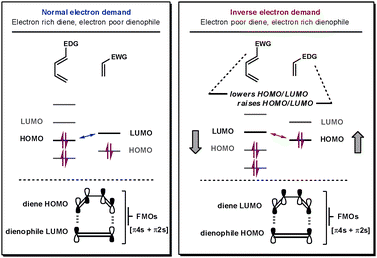 | ||
| Fig. 1 Frontier orbitals in normal- and inverse-electron-demand DA reactions. | ||
To obtain heterocyclic products, at least one of the π-components has to contain a heteroatom, and the process is classified as a hetero-Diels–Alder reaction (hDA). In terms of the content of this review, the nitrogens in the aza-dienes’ framework lower both the HOMO and LUMO energy so making them ideal substrates for ihDA reactions.
A common feature to all of the sub-classes mentioned so far is that they are in equilibrium with the reverse step, called the retro-DA reaction (rDA). Normally, the forward reaction is thermodynamically favoured as two strong σ-bonds replace two weaker π-bonds. However, the equilibrium can shift to favour the rDA when the diene and/or dienophile are particularly stable molecules, such as the formation of aromatic rings or gaseous molecules; both of which occur in the merged ihDA/rDA sequence for heteroaromatic synthesis.
1.2 Trends in reactivity for ihDA/rDA reactions
To form heteroaromatic products using a merged ihDA/rDA sequence, the starting materials require certain structural features. Firstly, the aza-diene needs to have both a diene unit for the ihDA step as well as a leaving group for the rDA step. Secondly, the dienophile needs to be a triple bond (Path A) or triple bond equivalent (Path B); this is often a double bond with a leaving group appropriately placed, facilitating a subsequent elimination reaction, as shown in Scheme 1.In this review we focus on cyclic aza-diene starting materials, where the group expelled in the rDA step is part of the ring itself. The aza-dienes discussed are azines where the number of nitrogen atoms within the ring is between two and four. The cyclic nature of the azines forces a cisoid geometry for the dienes, which enhances their reactivities.
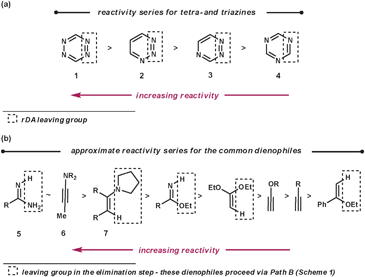
To make the aza-diene units more electron-deficient, electron-withdrawing substituents can be introduced on to the ring and/or the number of nitrogen atoms within the azine can be increased (Fig. 2a). The reactivity trend of the azines 1–4 (functioning as aza-dienes) can be rationalised by the following observations. Firstly, 1 is more reactive than 2–4 as it contains four nitrogens rather than three; secondly, 4 is less reactive than 2 and 3 because it expels the less favourable nitrile group rather than nitrogen; finally, 2 is more reactive than 3 as it breaks weaker bonds in the rDA step (N–N bonds are weaker than C–N bonds due to vicinal lone pair repulsion).
 | ||
| Fig. 2 Reactivity trends for dienes and dienophiles in ihDA/rDA reactions. | ||
The common dienophiles used in ihDA/rDA chemistry along with their approximate relative reactivities are shown in Fig. 2b.5 The less reactive dienophiles are generally able to react well in an intramolecular fashion with most azines, and react intermolecularly with reactive azines under milder conditions but need more forcing conditions with less reactive azines. However, dienophiles 5–7 are more readily reactive for intermolecular ihDA reactions as they are more electron-rich. Amidines, 5, can be thought of as being a reactive ‘nitrile’ equivalent and enamines, 7, as a reactive ‘alkyne’ equivalent, as their reaction sequences proceed via the elimination pathway (Path B, Scheme 1). Adding strain to the dienophile, which will be released upon reacting, can greatly enhance reactivity. It has been shown that highly strained alkynes, such as cyclooctyne, can be even more reactive than ynamines (6).6
Regioselectivity can be high for dienophiles that are polarised, where the difference in coefficients of atomic orbitals on the two atoms are large, such as that in 5–7.4 This will be discussed and demonstrated in further detail later in the review.
1.3 Aromaticity and azine properties
Aromatic compounds, such as benzene, typically undergo substitution reactions to retain the aromatic stabilisation. However, not all aromatic compounds behave alike. In most ‘aromatic’ compounds, especially heteroaromatics, there are degrees of aromaticity.7 It is a reduction in aromaticity that allows the di-, tri- and tetrazines, to break aromaticity and undergo ihDA reactions. In all the ihDA/rDA reactions the heteroaromatic product is more stable than the starting material, providing a significant driving force for the reaction. The thermal stability of heteraromatic compounds which contain multiple nitrogen atoms is frequently a concern, and they are often treated with caution. However, this may not be necessary, for example, 1,3,5-triazine, 4, decomposes at 600 °C and 1,2,3-triazine, 2, at 200 °C.8 Thus, under normal laboratory conditions, in most cases, only tetrazines, 1, should be treated with special care.9 Nevertheless, the more thermally labile the azine, the milder the conditions required for cycloaddition reactions.2. ihDA/rDA Reactions for nitrogen heteroaromatic synthesis using cyclic aza-dienes
There are a range of available cyclic aza-dienes that are capable of ihDA/rDA reactions.10 We shall only discuss those that are particularly reactive, have had widespread applications, and can furnish nitrogen heteroaromatic compounds; for example, 1,2-diazines are generally used to synthesise all-carbon aromatic compounds and so will not be considered.Scheme 2 shows the range of substitution patterns available in the heteroaromatic products attainable from the use of aza-dienes. The aza-dienes 9 and 10 will not be discussed in this review, but a recent report has compared the reactivities and regioselectivities of dienes 9, 10 and 3.11 We begin with the less reactive diazines (8 and 11), illustrating the techniques employed to achieve reaction. These diazines will be considered together, as the principles of their reactivities are similar and the amount of literature on these substrates is limited. Following this, we shall move to the triazines and then the tetrazines, in order of their increasing reactivity (4 to 1).
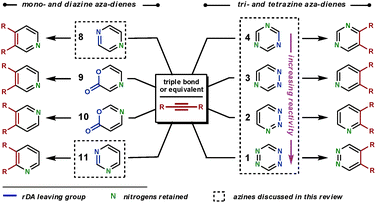 | ||
| Scheme 2 The aza-dienes discussed in this review and the substitution patterns of the heteroaromatic products after the ihDA/rDA sequence. | ||
2.1 1,3- and 1,4-Diazines
 | ||
| Scheme 3 The regioselectivity of ynamines and amidines with 1,3-diazines. | ||
Van der Plas investigated the reactivity of intramolecular reactions with the 1,3-diazine core to confirm that the reaction sequence was proceeding via the ihDA/rDA pathway. His results showed the characteristic features of DA reactions: there was a small solvent effect due to a non-ionic TS, and, in this case, the electronics of the ring had only a small effect on rate (using a Hammett plot) but it was influenced more so by the nature of the tether.17
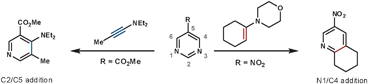
As shown in Scheme 4, encouraging the dienophile on the tether to sit above the aza-diene enhances the rate of the ihDA/rDA reaction. This was achieved by preventing molecular planarity between the azine and tether. Thus, non-conjugated tethers react faster than conjugated ones (12vs13), groups with a large steric influence that prevent planarity react rapidly (14), and the lone pair–lone pair repulsion in 15 causes it to react faster than 16.18 Similarly, placing an α phenyl group onto the tether was shown to increase the intramolecular ihDA/rDA reaction rate as a result of the cycloaddends being in closer proximity (17 reacts 3 to 4 times faster than 18 and 19).19
 | ||
| Scheme 4 Tether effects on the reaction kinetics. | ||
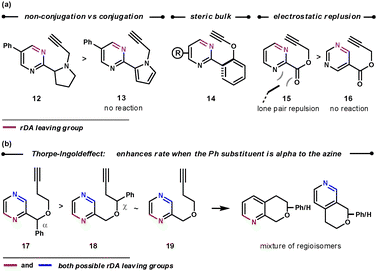
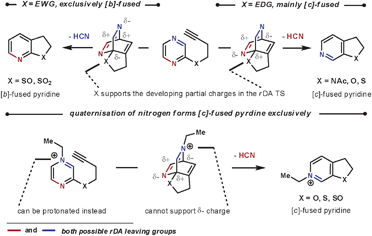
As well as the conformational properties of the tether, the electronic properties can also have a pronounced effect on the outcome of the reaction. This is especially true for 1,4-diazines, as there are two possible nitrile leaving groups in the rDA step, each of which would generate distinct products. By using a heteroatom to join the tether to the 1,4-diazine, the charge distribution in the TS can favour one nitrile expulsion over the other in the rDA step. The nitrile group that is lost more favourably is the one that can stabilise the partial negative charge on the nitrogen more easily. Therefore, for the more electron-withdrawing O-, S- and NAc groups the [c]-annulated pyridine is the dominant product, and for the more reactive electron-donating SO and SO2 groups the [b]-annulated pyridine is formed exclusively, as illustrated in Scheme 5.20 A similar outcome is seen after quaternising a nitrogen on the azine core, which can be used to both increase the rate of reaction by reducing electron density, and also increase the regioselectivity of the rDA step. In this case, HCN is expelled more favourably than HCNR+, Scheme 5.21 Quaternisation overrides the influence of the adjoining heteroatom to furnish solely the [c]-annulated pyridine. These reactions can be compared with the reactions in Scheme 4b, where without an added electronic influence a mixture of regioisomers is formed.
 | ||
| Scheme 5 Electronic effects on the eliminated group in the rDA step. | ||
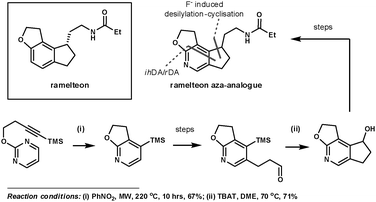
Koike et al. employed 1,3-diazines to synthesise an aza-analogue of ramelteon, which has been marketed for the treatment of insomnia.22 Its tricyclic core appears in many other biologically active compounds. This inspired the group to investigate synthetic techniques to access its structural analogues, which are potentially useful for further SAR studies. Their key ihDA/rDA step proceeded in 67% yield using microwave heating to activate the reaction, providing an improvement on the previous 14% yield when refluxed in nitrobenzene at 200 °C for 24 h. This simultaneously formed two of the three rings in the product. The third ring was formed using a fluoride-induced desilylation–cyclisation reaction, after a regioselective bromination reaction and further functionalisation (Scheme 6).
 | ||
| Scheme 6 Koike et al.'s synthesis of a ramelteon aza-analogue. | ||
As can be seen from the previous example, these unreactive diazines require forcing conditions to obtain satisfactory yields. However, the typical solvents used to achieve the high temperatures needed, such as nitrobenzene and chlorobenzene, can be toxic and difficult to remove. Recent efforts have used flow chemistry to superheat more practical solvents, such as toluene, in a controlled and safe manner for the ihDA/rDA process.23 Martin et al. used flow technology to make the intramolecular ihDA/rDA reaction between 1,3-diazines and a tethered alkyne more facile; a reaction they considered previously underutilised due to its challenging nature. A particularly impressive result was the tolerance of an NH linker atom between the tether (2-position) and the 1,3-diazine. This is normally a challenging, strongly electron-donating functionality, to employ on diazines for ihDA/rDA reactions.20,24
Hoornaert et al. have used impressive DA cascades to construct a series of novel architectures.25 They used a 5-chloropyrazine-2(1H)-one as the aza-diene where the chloro-nitrile group leaves more favourably than the isocyanic acid moiety in the rDA step. By using a tether with a sulfur group appropriately positioned, subsequent oxidation to the sulfone allowed the cheletropic loss of SO2, which leaves a heteroaromatic o-quinodimethane (o-QDM) intermediate, which is suitable for further DA reactions. A selected example is shown in Scheme 7, whereby two isomeric products are formed selectively from regioselective deprotonations then trapping with 5-bromopent-1-ene. Under thermal conditions, these compounds were able to expel sulfur dioxide by a cycloreversion process to form the reactive o-QDM intermediate, which then reacts intramolecularly with the alkene dienophile. Thus, three contiguous rings are formed with a high level of control.
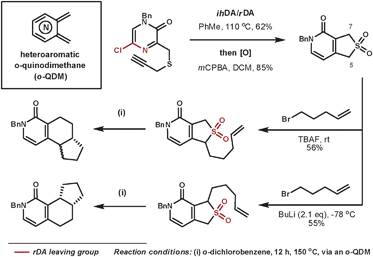 | ||
| Scheme 7 Hoornaert's cycloaddition/cycloreversion sequences to form 3 new rings systems. | ||
2.2 1,3,5-Triazines
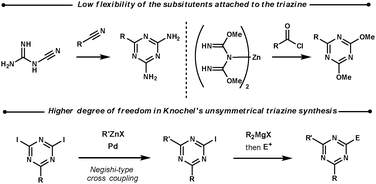 | ||
| Scheme 8 Unsymmetrical 1,3,5-triazine syntheses. | ||
Boger et al. investigated the reactivity difference between electron-donating (methyl thiol) and electron-withdrawing (ester) substituents on the triazine core.30 Ynamine dienophiles undergo ihDA reactions readily with both, however, the intermediate formed does not undergo the rDA as easily as the first step, with the ester intermediate decomposing at 40–100 °C compared with 150–230 °C for the methyl thiol triazine. Enamines are less reactive than ynamines and therefore are only able to perform the ihDA reaction with the electron-poor triazine, and require high temperatures for the rDA step. Promoting the rDA step with both dienophiles was achieved by either making the substituents more electron-withdrawing (sulfide to sulfone), by using an acid catalyst, or by using an acid/solvent mix for the one-pot procedure. Removing the remaining ester and methyl thiol functionality from the pyrimidine products was achieved by decarboxylation and RANEY® nickel conditions, respectively.
The reaction between enamines and 1,3,5-triazines is regioselective. If the enamine is cyclic, the process represents a regioselective pyrimidine annulation methodology.31 Masaguer et al. elected to use such an approach during their synthesis of an intermediate for the synthesis of quinazolinones, an aza-analogue of the antipsychotic butyrophenones (Scheme 9).32 Enaminones are not common dienophiles due to their reduced electron density compared with enamines, however, they performed well in this case. During their optimisation, the best result was achieved using a primary enaminone (others investigated include morphilino and pyrrolidino) and refluxing in acetic acid to give an 87% conversion (other screened solvents include dioxane, DMF, DME and acetonitrile). Interestingly, an unexpected reaction was discovered during the solvent screen, as when using water as the solvent – a solvent rarely used for ihDA/rDA reactions – they witnessed a double merged cycloaddition/cycloreversion process to give exclusively the double annulated pyridine product. An additional benefit was that the dimeric product, unlike the starting materials, was insoluble in water and so was purified by simple filtration in a 65% yield.
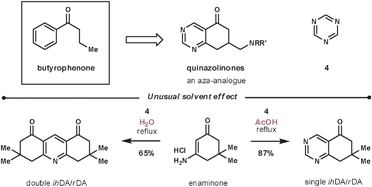 | ||
| Scheme 9 Unusual solvent effect during enaminone and 1,3,5-triazine reaction. | ||
Amidines (usually used as an acid salt) are commonly employed as nitrile equivalents as they react via Path B (Scheme 1) with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond functioning as the dienophile. Interestingly, with the less reactive 1,3,5-triazines, amidines react with the more reactive and less abundant tautomer via the CC bond (Scheme 10a), so acting as an ynamine equivalent following the elimination step. Amidines react regioselectively to deliver the amine functionality in the 6-position of the pyrimidine products. The tautomerisation of the amidine salts is promoted by DMF and high temperatures (80–110 °C). Freeing the amidine salt by base addition only hindered the reaction, probably by obstructing the rDA step. The reaction is tolerant to a range of functionalities at the β-position of the amidine which equates to the 5-position in the pyrimidine products (e.g. aryl, alkyl and SMe groups). Imidates show no reactivity, whereas thioimidates are slightly less reactive than amidines but retain the SMe group in the 6-position of the products.33 This chemistry was used by Boger during the preparation of pyrimidines 20 and 21 for their evaluation as potential antineoplastic agents and inhibitors of GAR Tfase and AICAR Tfase (Scheme 10b).34
N bond functioning as the dienophile. Interestingly, with the less reactive 1,3,5-triazines, amidines react with the more reactive and less abundant tautomer via the CC bond (Scheme 10a), so acting as an ynamine equivalent following the elimination step. Amidines react regioselectively to deliver the amine functionality in the 6-position of the pyrimidine products. The tautomerisation of the amidine salts is promoted by DMF and high temperatures (80–110 °C). Freeing the amidine salt by base addition only hindered the reaction, probably by obstructing the rDA step. The reaction is tolerant to a range of functionalities at the β-position of the amidine which equates to the 5-position in the pyrimidine products (e.g. aryl, alkyl and SMe groups). Imidates show no reactivity, whereas thioimidates are slightly less reactive than amidines but retain the SMe group in the 6-position of the products.33 This chemistry was used by Boger during the preparation of pyrimidines 20 and 21 for their evaluation as potential antineoplastic agents and inhibitors of GAR Tfase and AICAR Tfase (Scheme 10b).34
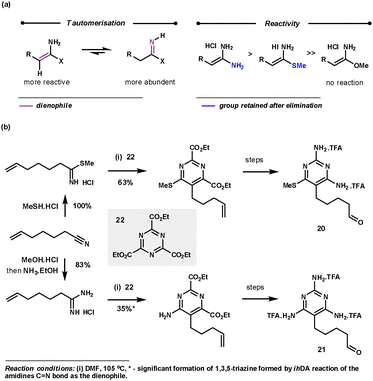 | ||
| Scheme 10 Amidine reactivity with 1,3,5-triazines and Boger's application for the synthesis of 20 and 21. | ||
A comparison of reactivity between amidines and ynamines was directly made during Boger's synthesis of (−)-pyrimidoblamic acid, the heterocyclic core of bleomycon A2, and its derivatives (Scheme 11).35 The target of the first step, 23, could be prepared by the union of the tri-ester-azine with either ethylamidine or benzyl ynamine and a subsequent deprotection. They found that both methodologies gave similar overall yields. Interestingly, in the following step the ester at C2 could be selectively reduced as it is more sterically available than the ester at C4.
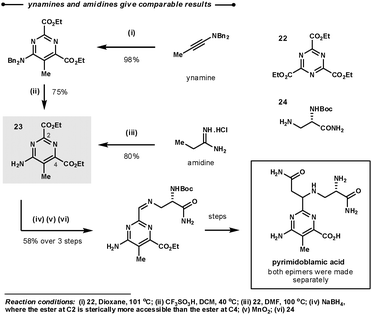 | ||
| Scheme 11 Boger's synthetic strategy for the construction of the heterocyclic core of bleomycon A2. | ||
Purines are the most widely distributed nitrogen heterocycle in nature, with their derivatives being involved in many metabolic pathways.36 A facile method to access purine cores involved union of 1,3,5-triazines with amino substituted heterocyclic dienophiles. Wu et al. performed studies which suggested that the elimination of ammonium occurs before the rDA step for these substrates.37 There have been many purine derivatives prepared in this way; a selected example by Iaroshenko, shown in Scheme 12, demonstrates the vast array of amino substituted heterocycles that can couple regioselectively under mild conditions.38 The paper focuses on fluorinated 1,3,5-triazines which are highly reactive and furnish purines and purine isosteres that exhibit a broad range of biological activities.
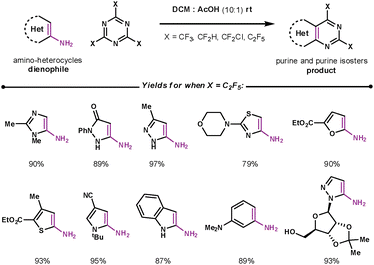 | ||
| Scheme 12 Using 1,3,5-triazines to make purines and purine isosters. | ||
2.3 1,2,4-Triazines
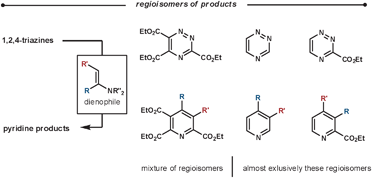 | ||
| Scheme 13 Regioselectivity of the reaction between enamines and various 1,2,4-triazine substitution patterns. | ||
Typically, the enamines are formed from pyrrolidine and the relevant carbonyl compound. The enamines are introduced into the reaction either pre-formed or by using catalytic pyrrolidine; an addition of molecular sieves can encourage both enamine formation and aromatisation. The ihDA/rDA then proceeds via Path B (Scheme 1). In some cases, it is reported that the reaction stalls before the elimination/aromatisation step has occurred. Over recent years, there have been some improvements that encourage the final step. Using a silica additive can help product formation;43 alternatively, increasing the reaction temperatures using microwave conditions has a similar promoting effect.44 Another approach involves oxidation of intermediate 26 to an N-oxide in a two-step procedure to promote a Cope elimination.45 Taylor et al. have also developed a one-pot mimic of the Cope procedure using a tethered-imine-enamine technique, illustrated in Scheme 14.44
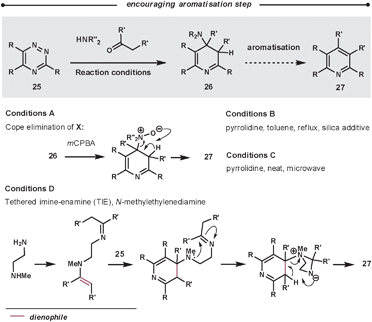 | ||
| Scheme 14 Methodologies to promote the aromatisation of 26. | ||
If the aromatisation step doesn't occur in the Path B reaction sequence, another aza-diene becomes available in the ring system, shown in Scheme 15. Taylor et al. trapped this aza-diene intermediate by using a second dienophile on the tether of either the secondary amine or carbonyl, to form the enamine. They were able to form some highly complex polycyclic architectures very rapidly.46
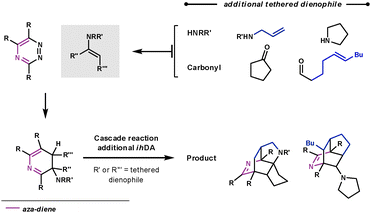 | ||
| Scheme 15 Cascade reaction for complex nitrogen-containing polycycle synthesis. | ||
Palmer et al. synthesised a library of compounds based on the structure of SCH28080 – a clinical prototype for the treatment of gastroesophageal reflux disease – that were intended to resolve the compound's metabolic and toxicity issues.47 Their synthesis of these targets demonstrates the typical convergent nature of a Diels–Alder synthetic sequence. The key ihDA/rDA sequence proceeded regioselectively in an excellent 84% yield (Scheme 16).
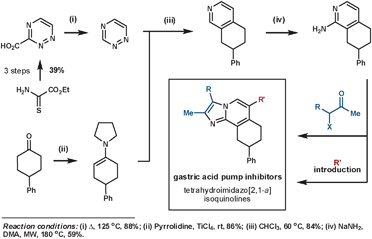 | ||
| Scheme 16 Palmer's synthesis of gastric acid pump inhibitors. | ||
Other dienophiles that have been used with 1,2,4-triazines include benzynes,48 2,3-dihydrofuran,49 and 2,5-norbornadiene (acting as an ethyne equivalent).50 When acting as a dienophile 2,3-dihydrofuran proceeds via Path B, and so eliminates a tethered alcohol group which can been used to synthesise annulated lactones if an ester group is positioned appropriately.
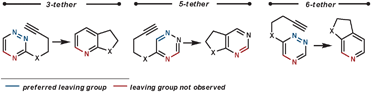 | ||
| Scheme 17 Eliminated groups in the rDA in tethered 1,2,4-triazines. | ||
The conformational properties and electronic influence of the tether has a similar effect on the reaction rates to that discussed for the diazine systems. However, the tether itself does not simply play the role of positioning the dienophile, as in subsequent steps it has been oxidised,52 and cleaved,53 as well as undergone rearrangements,54 as shown in Scheme 18. The rearrangement of the imidazole dienophile is particularly interesting when compared to that of indoles. After the tandem cycloaddition/cycloreversion reactions the imidazole decomposes to release a nitrile, thus acting as an ynamine equivalent.
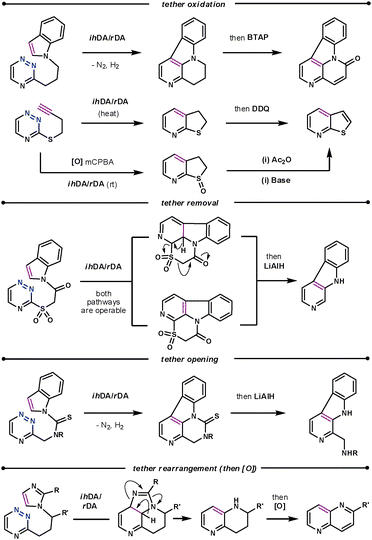 | ||
| Scheme 18 Subsequent manipulations of the tether. | ||
Nitriles are not normally used as dienophiles, however, they are able to react in an intramolecular fashion. Taylor et al. attempted to use oximes as a more reactive nitrile-equivalent, based on the rationalisation that they are more electron-rich. However, they obtained disappointing results in their intramolecular reaction. They proposed two possible explanations; the added bulk of the OMe group could disturb the formation of the TS, and the geometry of the required [π4s + π2s] orbital interaction was harder to achieve for a double bond than a triple bond (for this tether).55 Nitrile, as well as alkyne, dienophiles have been used by Akritopoulou-Zanze et al. in tandem Ugi/ihDA/rDA processes to form a range of unique scaffolds (Scheme 19).56
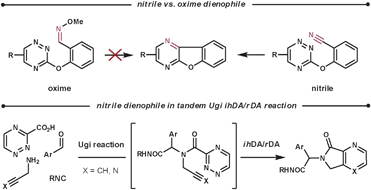 | ||
| Scheme 19 Nitrile dienophiles in ihDA/rDA cascade reactions. | ||
Guillaumet showed that terminal bromine and tin groups can be tolerated on the alkyne dienophile during the cycloaddition/cycloreversion process. This provides direct access to fused heterocycles where the para-position lends itself readily for further functionalisation using cross-coupling methodologies.57
The α-carboline core has been shown to have a range of biological properties. Synder et al. recently demonstrated a robust ihDA/rDA methodology to prepare a library (88 compounds) of lactam and lactone fused α-carbolines in which the average yield, determined by UPLC, was 94% (Scheme 20).58 They took advantage of the convergent synthetic nature of this methodology to modify the five main diversification sites on the core, while using readily available or easily prepared starting materials. As could be predicted, the pericyclic reactions were helped by reducing the electron density of the triazine core. The merged cycloaddition/cycloreversion reactions were sensitive to tether length. By increasing the tether length to form the 6-membered annulated ring the reactivity was vastly decreased. The effective molarity for 5-membered ring closures can be as high as 1000-fold greater than 6-membered closures.59 Synder found that the amide tether could H-bond to the triazine core, which promoted molecular planarity and thus reduced overall reactivity. However, switching the reaction solvent to DMF meant the solvent could compete with the azine for H-bonding to the amide, as DMF has a greater capacity to accept H-bonding than the azine.
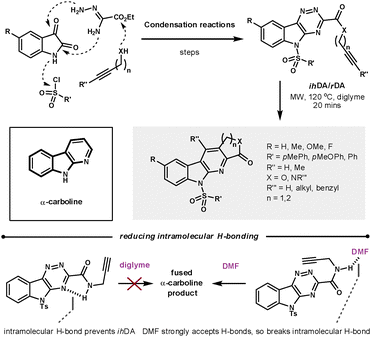 | ||
| Scheme 20 Using the ihDA/rDA sequence to synthesis an α-carboline library. | ||
2.4 1,2,3-Triazines
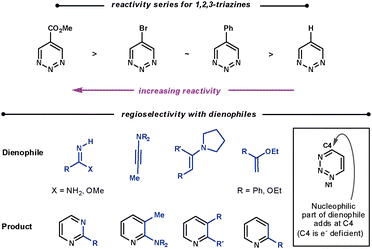 | ||
| Scheme 21 Reactivity and regioselectivity of 1,2,3-triazines. | ||
With all dienophiles a regioselective N1/C4 addition is observed, shown in Scheme 21. Boger found that the reaction with unsubstituted 1,2,3-triazines and enamines was limited compared with the other triazines. They suspected that the initial ihDA reaction was slow compared with aromatisation. The liberated pyrrolidine was found to add to the triazine, and accounts for the modest conversions. However, amidine dienophiles are particularly reactive with these triazines.5
Calculations suggest the cycloaddition should occur along N2/C5 as these sites have the largest LUMO coefficients. However, a N1/C4 cycloaddition is observed as C4 is the most electrophilic site. This suggests that the mechanism proceeds via a concerted ionic pathway,61 although no isolated reaction by-products to support this possibility have been reported.
Koyama et al. used the ihDA/rDA reaction between 4-methyl-1,2,3-triazine and aldehyde-derived enamines to synthesise fusaric acid (Scheme 22). During their initial studies, they found that a zinc bromide additive (1.5 eqv.) was crucial for the success with aldehyde enamines. They showed a good scope of aryl and alkyl β-substituted aldehydes in yields ranging from 32–82%.62
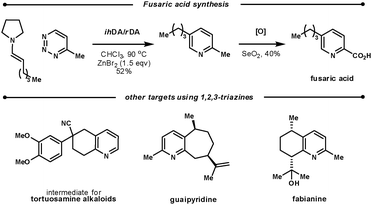 | ||
| Scheme 22 Targets using the ihDA/rDA reaction of 1,2,3-triazines. | ||
Several other synthetic efforts have used the ihDA/rDA sequence with 1,2,3-triazines as they key step: a tortuosamine alkaloid intermediate, guaipyridine, and fabianine.63
2.5 1,2,4,5-Tetrazines
Enamine dienophiles and tetrazines provide direct access to pyridazines. In an effort to find a secondary amine organocatalyst that would aid the union of ketones and 1,2,4,5-tetrazines in a universal and mild manner, Wang et al. concluded that the use of proline in DMSO at 100 °C for 1 h, was ideal.69 They were able to couple a range of cyclic and acyclic ketones. For unsymmetical ketones, however, the enamine can form on either side of the carbonyl functionality so an unavoidable mixture of regioisomers was returned.
Boger found that oxidation of one of the methane thiol groups at the 3-, or 6-position of the tetrazine to the sulfoxide increases the reaction rate, broadens the scope of the potential dienophiles, increases regioselectivity and, in some cases, can give complementary regioselectivity to the unoxidised starting material. For example, reaction of dienophile 28 with the oxidised70 and unoxidised66 benzyl 4-(methylthio)phenylcarbamate leads to regioisomeric products. It is also possible to perform a tactical reversal of regioselectivity by careful selection of the dienophiles, as demonstrated in the Scheme 23.70
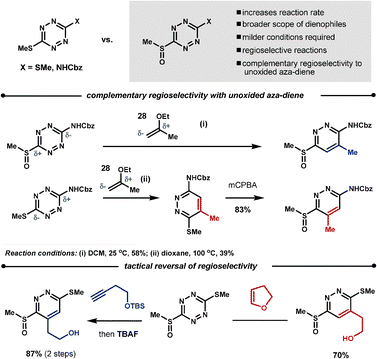 | ||
| Scheme 23 Controlling the dienophiles' regioselectivity with tetrazines. | ||
Harrity et al. made use of boron substituted alkyne dienophiles in order to exploit the functionality as a handle for further manipulation after the ihDA/rDA sequence. In their initial work they studied the use of alkynylboronates which couple with tetrazines in high yields, however, high temperatures (140 °C) were required.71 In subsequent work they studied the more electron-rich trifluorborates.72 They discovered conditions that could give outstanding results at room temperature in just 10 minutes. A mild Lewis acid additive was used to form the difluoro species that was able to coordinate to the N-directing group on the tetrazine to form pyridazines (29) with outstanding degrees of regioselectivity (Scheme 24). In a testimony to the strength of this reaction, they were also able to couple the less reactive 1,2,4-triazine in a similar manner in a 67% yield.
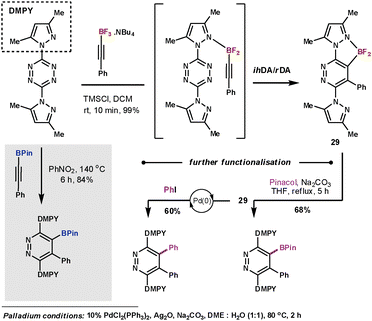 | ||
| Scheme 24 Harrity's use of trifluorborates as dienophiles | ||
Despite tetrazines being highly reactive, they still require high reaction temperatures when reacting with unreactive dienophiles such as alkynes. The products of these reactions, 1,2-diazines (pyridazines), can undergo a reductive ring contraction reaction using zinc to form highly substituted pyrroles. This has been used to synthesise a number of highly complex structures in a very efficient manner. This methodology was applied to the synthesis of ningalin D, a structurally interesting and biologically active marine natural product. Boger took advantage of the ihDA/rDA/ring contraction process to quickly and efficiently synthesise its fully substituted pyrrole core (Scheme 25). The symmetrical alkyne coupled with the electron-poor 3,5-diester substituted tetrazine in 87% yield followed by the ring contraction (Zn, TFA) in 64%. Following this a Dieckmann cyclisation, Suzuki cross coupling and oxidative decarboxylation led to a key intermediate towards the natural product. These reactions constructed the target in 9 overall steps in 19% yield.73
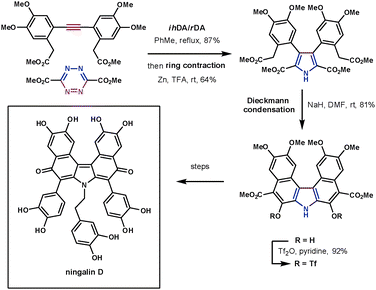 | ||
| Scheme 25 Boger's synthesis of ningalin D. | ||
Unsymmetrical dienophiles lead to unsymmeterical pyrroles. In an example reported by Boger et al. they completed the asymmetrical synthesis of the unnatural enantiomer of rosephilin, a novel antitumor antibiotic.74 Their synthesis started with an ihDA/rDA reaction at room temperature using an unsymmetrical chiral dienophile prepared using Wittig chemistry, which furnished the intermediate in 91% over two-steps. After the ring contraction had provided the pyrrole, and further elaboration of its side chains, the next key step was the closure of the 13-membered macrocycle using Grubbs catalyst. The remaining ester was converted to a selenoester for a 5-exo-trig acyl radical-alkene cyclisation to form the fused cyclopentanone. This furnished the desired intermediate as a single diastereoisomer. The final heterocyclic side chain was condensed onto the cyclopentanone (Scheme 26). A second example of this methodology was used to prepare a range of α-helix mimetics. The ester containing tetrazines could be coupled with alkynes, enamines, and enol ethers under simple heating conditions in good yields (65–87%), followed by ring-contraction in poor to good yields (27–70%).75
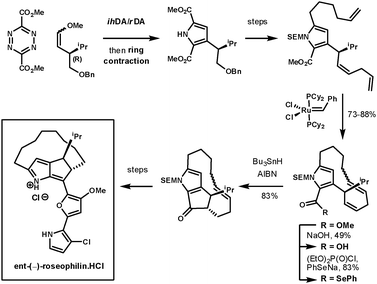 | ||
| Scheme 26 Boger's synthesis of ent-(−)-roseophilin.HCl. | ||
Further synthetic utility following the reductive ring-contraction is available by oxidative decarbonylation of the 2,5-ester substituted pyrrole products.76
A bioorthogonal reaction is one that can proceed inside a living organism without disturbing the normal biochemical processes. This branch of chemistry is used for the labelling of proteins or DNA, for example, to grasp a greater understanding of important biological processes. Additional requirements for in vivo labelling reagents include being unreactive to cell culturing conditions (stable to nucleophiles such as water and thiols), be non-toxic, be quantifiable even at low concentrations, be able to diffuse through cell membranes and form stable products that can facilitate labelling for a diverse set of probes. Ideally, the labelling reaction would also not require an additional catalyst. Recent work in this field has used the reaction between tetrazines and highly reactive strained unsaturated dienophiles. This method has some advantages over other existing methodologies such as rapid coupling rates, no catalyst needed, and non-bulky components.77 Crucially, the rate of reaction can be tailored and fine tuned on both components of the reaction. This is important as the reactions inside cells occur at a variety of rates and ideally there should be a labelling reaction that suits each biological reaction that is to be monitored. The fine-tuning of the rate is achieved by manipulating the LUMOdiene–HOMOdienophile energy gap. Lowering the LUMO can be easily achieved by increasing the electron-withdrawing nature of the groups on the tetrazine, while the HOMO energy can be increased by adding strain and/or electron density on the dienophile. Wang et al. investigated this and reported the following reaction rates for strained dienophiles (Scheme 27), the most reactive dienophiles in ihDA reactions.78
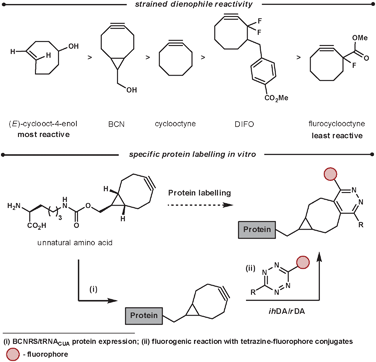 | ||
| Scheme 27 Reactivity and application of strained dienophiles. | ||
A recent application of this is the work was reported by Chin et al.79 They were able to use a range of tetrazines in aqueous conditions at room temperature for the fluorogenic reaction of BCN (Scheme 27). The ihDA/rDA reaction with BCN forms a single product, unlike strained alkenes which form diastereoisomers and regiosiomers. This is advantageous when homogeneity in the orientation of the probe is important. They were able to incorporate the strained dienophiles site specifically into proteins of E. coli and mammalian cells by the discovery of aminoacyl-tRNA synthease/tRNA pairs. From this they were able to react, and thus fluorogenically label, the proteins in vivo (and in vitro) specifically and efficiently with cell-permeable tetrazine fluorophore conjugates.
Psoralens are used for phototherapy for a range of skin diseases. In an attempt to avoid their unwanted side-effects, caused by the furan ring, Uriarte et al. have had positive results by fusing an aromatic ring to it. Their initial attempted synthesis employed benzofuran as the dienophile, which released phenol in the elimination step.80 By using a benzofuran-3(2H)-one type dienophile (via its enol form) instead, water is eliminated in place of phenol which furnishes the desired heteroaromatic fused psoralen (Scheme 28).81
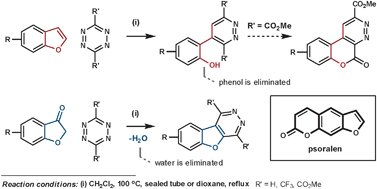 | ||
| Scheme 28 Uriarte's synthesis of psoralens. | ||
Another remarkable display of the application of ihDA/rDA chemistry was shown by Boger et al. during their formal synthesis of streptonigrin in 1985 (Scheme 29).82 If the final steps had followed Kende's methodology then the synthesis would have been completed in 17 overall steps. The strength of Boger's synthetic strategy lies in the convergent nature in which he assembled the central pyridine core using two ihDA/rDA reactions. The A and B rings on one side of the core were introduced by an S-methyl thioimidate and a tetrazine. The resulting 1,2,4-triazine then lends itself nicely to construct the C and D rings with an enamine dienophile.
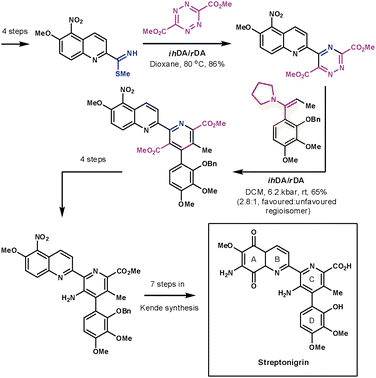 | ||
| Scheme 29 Boger's formal synthesis of steptronigrin. | ||
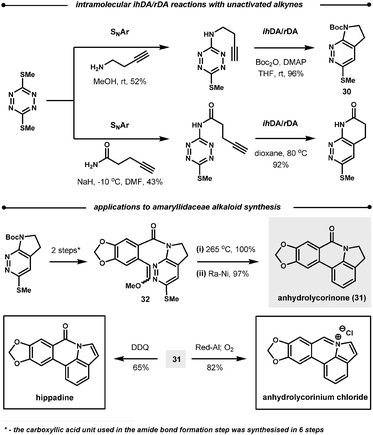 | ||
| Scheme 30 Intramolecular reactions of 1,2,4,5-tetrazines. | ||
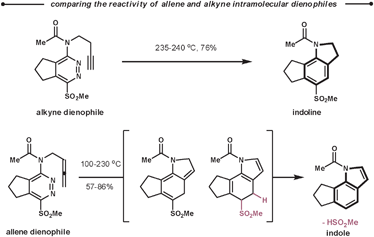
Following the reaction with tetrazines, a contrasting feature of the resultant 1,2-diazines is observed in their intramolecular ihDA/rDA reactions with tethered alkynes and allenes. Firstly, the allenes are more reactive than the alkynes but secondly, allenes generate indole products after elimination of HSO2Me, as opposed to the indoline products observed with alkynes (Scheme 31).84 This approach was employed in the total synthesis of (±)-cis and (±)-trans-trikentrin A.85
3. Conclusions
In this tutorial review we have highlighted the key features and applications of the merged inverse-electron-demand hetero-Diels–Alder (ihDA)/retro-Diels–Alder (rDA) reactions. These reaction cascades bring many advantages: convergent synthetic sequences, little waste, cheap/no catalysts required, high regioselectivities, and clean reactions with few side reactions observed. Importantly, these transformations often result in a significant increase in molecular complexity. Given these features it is perhaps surprising that there are not more examples of these cascades being employed in syntheses. One possible explanation is that the ihDA/rDA reaction sequence is not always straightforward to visualise during the retrosynthetic analysis of a target. We hope that this review raises awareness of this powerful class of transformations, and inspires chemists to further develop and utilise this subcategory of pericyclic chemistry.Notes and references
- J. Poulin, C. M. Grisé-Bard and L. Barriault, Chem. Soc. Rev., 2009, 38, 3092 RSC.
- T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010 RSC.
- A. F. Pozharskii, A. T. Soldatenkov and A. R. Katritzky, Heterocycles in Life and Society, John Wiley & Sons, Ltd, West Sussex, 1997 Search PubMed.
- I. Fleming, Frontier Orbitals and Organic Chemical Reactions, John Wiley & Sons, Ltd, West Sussex, 1976 Search PubMed.
- E. D. Anderson and D. L. Boger, J. Am. Chem. Soc., 2011, 133, 12285 CrossRef CAS.
- J. Sauer, D. K. Heldmann, J. Hetzenegger, J. Krauthan, H. Sichert and J. Schuster, Eur. J. Org. Chem., 1998, 2885 CrossRef CAS.
- A. T. Balaban, D. C. Oniciu and A. R. Katritzky, Chem. Rev., 2004, 104, 2777 CrossRef CAS.
- J. A. Joule and K. Mills, Heterocyclic Chemistry, 5th edn, Wiley-Blackwell, West Sussex, 2010 Search PubMed.
- D. E. Chavez, M. A. Hiskey and D. L. Naud, Propellants, Explos., Pyrotech., 2004, 29, 209 CrossRef CAS.
- D. L. Boger, Chem. Rev., 1986, 86, 781 CrossRef CAS.
- P. Rooshenas, K. Hof, P. R. Schreiner and C. M. Williams, Eur. J. Org. Chem., 2011, 983 CrossRef CAS.
- Selected examples to synthesise pyrizines using: (a) 1,2-diamines: I. Flament and M. Stoll, Helv. Chim. Acta, 1967, 50, 1754 CrossRef CAS; E. B. van der Tol, H. J. van Ramesdonk, J. W. Verhoeven, F. J. Steemers, E. G. Kerver, W. Verboom and D. N. Reinhoudt, Chem.–Eur. J., 1998, 4, 2315 Search PubMed; (b) L. Birkofer, Chem. Ber., 1947, 80, 83 CrossRef CAS; F. Y. Miyake, K. Yakushijin and D. A. Horne, Org. Lett., 2000, 2, 3185 Search PubMed.
- M. D. Hill and M. Movassaghi, Chem.–Eur. J., 2008, 14, 6836 CrossRef CAS.
- A. T. M. Marcelis and H. C. van der Plas, J. Org. Chem., 1986, 51, 67 CrossRef CAS.
- H. C. van der Plas and A. T. M. Marcelis, J. Org. Chem., 1986, 51, 4070 CrossRef CAS.
- H. Neunhoeffer and G. Werner, Liebigs Ann. Chem., 1974, 1190 CrossRef.
- A. E. Frissen, A. T. M. Marcelis, W. C. Melger and H. C. van der Plas, Tetrahedron, 1989, 45, 6891 CrossRef CAS.
- W. A. W. Stolle, A. E. Frissen, A. T. M. Marcelis and H. C. van der Plas, J. Org. Chem., 1992, 57, 3000 CrossRef CAS.
- M. Biedrzycki, D. A. de Bie and H. C. van der Plas, Tetrahedron, 1989, 45, 6211 CrossRef CAS.
- D. A. de Bie, A. Ostrowicz, G. Geurtsen and H. C. van der Plas, Tetrahedron, 1988, 44, 2977 CAS.
- A. E. Frissen, G. Geurtsen, A. T. M. Marcelis and H. C. van der Plas, Tetrahedron, 1990, 46, 595 CrossRef CAS.
- T. Koike, Y. Hoashi, T. Takai and O. Uchikawa, Tetrahedron Lett., 2011, 52, 3009 CrossRef CAS.
- R. E. Martin, F. Morawitz, C. Kuratlim, A. M. Alker and A. I. Alanine, Eur. J. Org. Chem., 2012, 47 CrossRef CAS.
- W. A. W. Stolle, A. T. M. Marcelis, A. Koetsier and H. C. van der Plas, Tetrahedron, 1989, 45, 6511 CrossRef CAS.
- T. C. Govaerts, I. A. Vogels, F. Compernolle and G. J. Hoornaert, Tetrahedron, 2003, 59, 5481 CrossRef CAS.
- 1,3,5-triazine synthesis using: (a) acidic conditions: 2,4,6-tris(ethoxycarbonyl)[1,3,5]triazine; E. Ott, Chem. Ber., 1919, 52, 656 Search PubMed; (b) triflic anhydride conditions: amine, alkyl, aryl and thiol substituted 1,3,5-triazines; A. Herrera, R. Martínez-Alvarez, P. Ramiro, M. Chioua and R. Chioua, Synthesis, 2004, 503 CAS.
- J.-J. Shie and J.-M. Fang, J. Org. Chem., 2007, 72, 3141 CrossRef CAS.
- S. Oudir, B. Rigo, J.-P. Hénichart and P. Gautret, Synthesis, 2006, 2845 CAS.
- Z. Peng, B. A. Haag and P. Knochel, Org. Lett., 2010, 12, 5398 CrossRef CAS.
- D. L. Boger and Q. Dang, Tetrahedron, 1988, 44, 3379 CrossRef CAS.
- D. L. Boger, J. Schumacher, M. D. Mullican, M. Patel and J. S. Panek, J. Org. Chem., 1982, 47, 2673 CrossRef CAS.
- E. R. Bilbao, M. Alvarado, C. F. Masaguer and E. Raviña, Tetrahedron Lett., 2002, 43, 3551 CrossRef CAS.
- D. L. Boger and M. J. Kochanny, J. Org. Chem., 1994, 59, 4950 CrossRef CAS.
- D. L. Boger, M. J. Kochanny, H. Cai, D. Wyatt, P. A. Kitos, M. S. Warren, J. Ramcharan, L. T. Gooljarsingh and S. J. Benkovic, Bioorg. Med. Chem., 1998, 6, 643 CrossRef CAS.
- D. L. Boger, T. Honda and Q. Dang, J. Am. Chem. Soc., 1994, 116, 5619 CrossRef CAS.
- H. Rosemeyer, Chem. Biodiversity, 2004, 1, 361 CAS.
- Z.-X. Yu, Q. Dang and Y.-D. Wu, J. Org. Chem., 2005, 70, 998 CrossRef CAS.
- V. O. Iaroshenko, Synthesis, 2009, 3967 CrossRef CAS.
- D. L. Boger, J. S. Panek and M. Yasuda, Org. Synth., 1988, 66, 142 CAS.
- N. Catozzi, M. G. Edwards, S. A. Raw, P. Wasnaire and R. J. K. Taylor, J. Org. Chem., 2009, 74, 8343 CrossRef CAS.
- E. Badarau, F. Suzenet, A.-L. Fînaru and G. Guillaumet, Eur. J. Org. Chem., 2009, 3619 CrossRef CAS.
- B. Shi, W. Lewis, I. B. Campbell and C. J. Moody, Org. Lett., 2009, 11, 3686 CrossRef CAS.
- N. Catozzi, W. J. Bromley, P. Wasnaire, M. Gibson and R. J. K. Taylor, Synthesis, 2007, 2217 CAS.
- Y. F. Sainz, S. A. Raw and R. J. K. Taylor, J. Org. Chem., 2005, 70, 10086 CrossRef CAS.
- B. L. Chenard and R. T. Ronau, J. Org. Chem., 1988, 53, 5175 CrossRef CAS.
- S. A. Raw and R. J. K. Taylor, J. Am. Chem. Soc., 2004, 126, 12260 CrossRef CAS.
- A. M. Palmer, B. Grobbel, C. Brehm, P. J. Zimmermann, W. Buhr, M. P. Feth, H. C. Holst and W. A. Simon, Bioorg. Med. Chem., 2007, 15, 7647 CrossRef CAS.
- S. Diring, P. Retailleau and R. Ziessel, Tetrahedron Lett., 2007, 48, 8069 CrossRef CAS.
- S. P. Stanforth, B. Tarbit and M. D. Watson, Tetrahedron Lett., 2002, 43, 6015 CrossRef CAS.
- V. N. Kozhevnikov, D. N. Kozhevnikov, O. V. Shabunina, V. L. Rusinov and O. N. Chupakhin, Tetrahedron Lett., 2005, 46, 1791 CrossRef CAS.
- E. C. Taylor and J. L. Pont, J. Org. Chem., 1987, 52, 4287 CrossRef CAS.
- Oxidation with (a) BTAP (benzyltriethylammonium permanganate): J.-H. Li and J. K. Snyder, Tetrahedron Lett., 1994, 35, 1485 CrossRef CAS; (b) DDQ: E. C. Taylor and J. E. Macor, J. Org. Chem., 1987, 52, 4280 CrossRef CAS.
- (a) Tether removal: Z.-K. Wan and J. K. Snyder, Tetrahedron Lett., 1998, 39, 2487 CrossRef CAS; (b) Tether ring opening: W.-H. Fan, M. Parikh and J. K. Snyder, Tetrahedron Lett., 1995, 36, 6591 CrossRef CAS.
- B. R. Lahue, S.-M. Lo, Z.-K. Wan, G. H. C. Woo and J. K. Snyder, J. Org. Chem., 2004, 69, 7171 CrossRef CAS.
- E. C. Taylor, J. L. Pont and J. C. Warner, Tetrahedron, 1987, 43, 5159 CrossRef CAS.
- I. Akritopoulou-Zanze, Y. Wang, H. Zhao and S. W. Djuric, Tetrahedron Lett., 2009, 50, 5773 CrossRef CAS.
- Y. Hajbi, F. Suzenet, M. Khouili, S. Lazar and G. Guillaumet, Synlett, 2009, 92 CAS.
- Z. Ma, F. Ni, G. H. C. Woo, S.-M. Lo, P. M. Roveto, S. F. Schaus and J. K. Snyder, Beilstein J. Org. Chem., 2012, 8, 829 CrossRef CAS.
- M. A. Casadei, C. Galli and L. Mandolini, J. Am. Chem. Soc., 1984, 106, 1051 CrossRef CAS.
- A. Ohsawa, H. Arai, H. Ohnishi, T. Itoh, T. Kaihoh, M. Okada and H. Igeta, J. Org. Chem., 1985, 50, 5520 CrossRef CAS.
- T. Itoh, M. Okada, K. Nagata, K. Yamagucki and A. Ohsawa, Chem. Pharm. Bull., 1990, 38, 2108 CrossRef CAS.
- J. Koyama, T. Ogura and K. Tagahara, Heterocycles, 1994, 38, 1595 CrossRef CAS.
- T. Okatani, J. Koyama and K. Tagahara, Heterocycles, 1989, 29, 1809 CrossRef CAS , and references therein.
- D. L. Boger, R. S. Coleman and J. S. Panek, J. Org. Chem., 1985, 50, 5377 CrossRef CAS; D. L. Boger, J. S. Panek and M. Patel, Org. Synth., 1992, 70, 79 Search PubMed.
- J. Sandström, Acta Chem. Scand., 1961, 15, 1575 CrossRef.
- D. L. Boger, R. P. Schaum and R. M. Garbaccio, J. Org. Chem., 1998, 63, 6329 CrossRef CAS.
- S. M. Sakya, K. K. Groskopf and D. L. Boger, Tetrahedron Lett., 1997, 38, 3805 CrossRef CAS.
- J. Sołoducho, J. Doskocz, K. Cabaj and S. Roszak, Tetrahedron, 2003, 59, 4761 CrossRef.
- H. Xie, L. Zu, H. R. Oueis, H. Li, J. Wang and W. Wang, Org. Lett., 2008, 10, 1923 CrossRef CAS.
- A. Hamaski, R. Ducray and D. L. Boger, J. Org. Chem., 2006, 71, 185 CrossRef.
- M. D. Helm, J. E. Moore, A. Plant and J. P. A. Harrity, Angew. Chem., Int. Ed., 2005, 44, 3889 CrossRef CAS.
- J. F. Vivat, H. Adams and J. P. A. Harrity, Org. Lett., 2010, 12, 160 CrossRef CAS.
- A. Hamasaki, J. M. Zimpleman, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2005, 127, 10767 CrossRef CAS.
- D. L. Boger and J. Hong, J. Am. Chem. Soc., 2001, 123, 8515 CrossRef CAS.
- L. Moisan, S. Odermatt, N. Gombosuren, A. Carella and J. Rebek Jr, Eur. J. Org. Chem., 2008, 1673 CrossRef CAS.
- D. L. Boger and C. M. Baldino, J. Am. Chem. Soc., 1993, 115, 11418 CrossRef CAS.
- J. L. Seitchik, J. C. Peeler, M. T. Taylor, M. L. Blackman, T. W. Rhoads, R. B. Cooley, C. Refakis, J. M. Fox and R. A. Mehl, J. Am. Chem. Soc., 2012, 134, 2898 CrossRef CAS.
- W. Chen, D. Wang, C. Dai, D. Hamelberg and B. Wang, Chem. Commun., 2012, 48, 1736 RSC.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317 CrossRef CAS.
- J. C. González, T. Dedola, L. Santana, E. Uriarte, M. Begala, D. Copez and G. Podda, J. Heterocycl. Chem., 2000, 37, 907 CrossRef.
- J. C. González-Gómez, L. Santana and E. Uriarte, Synthesis, 2002, 43 CrossRef.
- D. L. Boger and J. S. Panek, J. Am. Chem. Soc., 1985, 107, 5745 CrossRef CAS.
- D. L. Boger and S. E. Wolkenberg, J. Org. Chem., 2000, 65, 9120 CrossRef CAS.
- D. L. Boger and S. M. Sakya, J. Org. Chem., 1988, 53, 1415 CrossRef CAS.
- D. L. Boger and M. Zhang, J. Am. Chem. Soc., 1991, 113, 4230 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |