DOI:
10.1039/C2CS35206K
(Review Article)
Chem. Soc. Rev., 2013,
42, 305-341
Nucleotide, c-di-GMP, c-di-AMP, cGMP, cAMP, (p)ppGpp signaling in bacteria and implications in pathogenesis
Received 8th June 2012
First published on 28th September 2012
Abstract
For an organism to survive, it must be able to sense its environment and regulate physiological processes accordingly. Understanding how bacteria integrate signals from various environmental factors and quorum sensing autoinducers to regulate the metabolism of various nucleotide second messengers c-di-GMP, c-di-AMP, cGMP, cAMP and ppGpp, which control several key processes required for adaptation is key for efforts to develop agents to curb bacterial infections. In this review, we provide an update of nucleotide signaling in bacteria and show how these signals intersect or integrate to regulate the bacterial phenotype. The intracellular concentrations of nucleotide second messengers in bacteria are regulated by synthases and phosphodiesterases and a significant number of these metabolism enzymes had been biochemically characterized but it is only in the last few years that the effector proteins and RNA riboswitches, which regulate bacterial physiology upon binding to nucleotides, have been identified and characterized by biochemical and structural methods. C-di-GMP, in particular, has attracted immense interest because it is found in many bacteria and regulate both biofilm formation and virulence factors production. In this review, we discuss how the activities of various c-di-GMP effector proteins and riboswitches are modulated upon c-di-GMP binding. Using V. cholerae, E. coli and B. subtilis as models, we discuss how both environmental factors and quorum sensing autoinducers regulate the metabolism and/or processing of nucleotide second messengers. The chemical syntheses of the various nucleotide second messengers and the use of analogs thereof as antibiofilm or immune modulators are also discussed.
 Dimpy Kalia | Dimpy Kalia (née Sikriwal) obtained her MSc (with honors) in organic chemistry from Panjab University, and PhD in organic chemistry from the Central Drug Research Institute (CDRI), Lucknow under Dr Dinesh K. Dikshit in 2011. Subsequently, she worked in a biotechnology industry in Bangalore for a period of 1 year and 2 months before joining the Sintim research group in 2012. Her current research interests focus on the chemical biology of bacterial signaling, specifically on the development of novel antibacterial compounds that function by modulating cyclic dinucleotide signaling. |
 Gökçe Merey | Gökçe Merey received both her BS and PhD (advisor Prof. Olcay Anaç) from Istanbul Technical University. She also worked in the laboratory of Prof. Albert Padwa at Emory University as an exchange student between August 2006 and August 2007. She joined the Sintim laboratory as a postdoctoral research associate in 2011. Currently, she is working as an instructor and researcher at Istanbul Technical University. Her primary interests are syntheses of biologically active compounds. |
 Shizuka Nakayama | Shizuka Nakayama received her BS and MS from Kobe Gakuin University. In 2007, she obtained her PhD from the University of Kyushu, under Prof. Shigeki Sasaki, where she studied nucleic acid chemistry. After a brief stint at Tohoku University, in the laboratory of Prof. Fumi Nagatsugi, she joined the Sintim laboratory as a postdoctoral research associate in 2007. Her current research interests are focused on small molecules that are involved in bacterial communication and the development of isothermal methods to detect disease biomarkers. |
 Benjamin Roembke (top left), Min Guo (top right), Yiling Luo (bottom left), Yue Zheng (bottom, middle), and Jie Zhou (bottom, right) | Benjamin Roembke (top left), Min Guo (top right), Yiling Luo (bottom left), Yue Zheng (bottom, middle), and Jie Zhou (bottom, right) are graduate students in Dr Sintim's laboratory. Benjamin obtained his BS in Chemistry from Towson University, MD, in 2010. Min Guo received his BS (2006) and MS (2009) in Chemistry from Nanjing University. Yiling (a joint student in the Sintim and Dayie laboratories) obtained her BS in Biomedical Chemistry at the University of Warwick. Yue received her BS degree in pharmacy from Fudan University in China. Jie received her BS degree from Nanyang Technological University, Singapore. |
 Herman O. Sintim | Herman O. Sintim received his BS in medicinal chemistry from University College London and DPhil in organic chemistry from University of Oxford under Prof. David Hodgson. He held postdoctoral positions at Oxford and Stanford Universities (with Profs Tim Donohoe and Eric Kool respectively) before joining the University of Maryland at College Park (UMD) in 2006 as an assistant professor. In 2012, Herman received tenure and was promoted to associate professor. Herman's research interests include the chemical biology of bacterial virulence and biofilm formation, isothermal detection of disease biomarkers and synthesis of drug-like molecules. |
1. Introduction
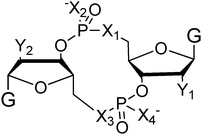
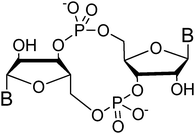
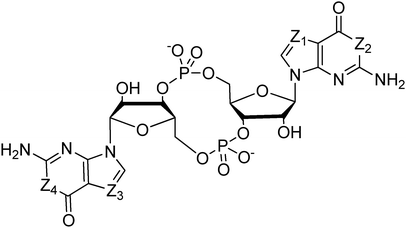
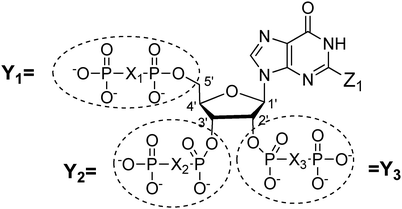
Both prokaryotes and eukaryotes utilize linear and cyclic nucleotides to regulate diverse cellular processes in response to extracellular cues. In bacteria, cyclic nucleotides such as cyclic dimeric guanosine 3′,5′-monophosphate (c-di-GMP),1 cyclic dimeric adenosine 3′,5′-monophosphate (c-di-AMP),2 cyclic guanosine 3′,5′-monophosphate (cGMP),3 cyclic adenosine 3′,5′-monophosphate (cAMP)4 as well as linear nucleotides such as guanosine 3′,5′-bispyrophosphate (ppGpp) and guanosine 3′-diphosphate, 5′-triphosphate (pppGpp)5,6 (see Fig. 1 for structures) have emerged as important second messengers involved in the regulation of processes that regulate virulence factor production or biofilm formation. Population-dependent bacterial cell-to-cell communication, also called quorum sensing (QS), regulate these same processes and accumulating evidence now points to an interaction between signaling by nucleotides and QS.7 Several excellent reviews on QS8 and second messenger nucleotides9–13 have been published but there is a paucity of reviews that have addressed the cross-talks between the majority of the small molecules that bacteria use to make “decisions”. This review aims to provide an update of nucleotide signaling in bacteria14 and also provides an overview of how the different environmental cues, QS autoinducers and nucleotide second messengers interlink to regulate physiological processes in bacteria (Fig. 2 and 3).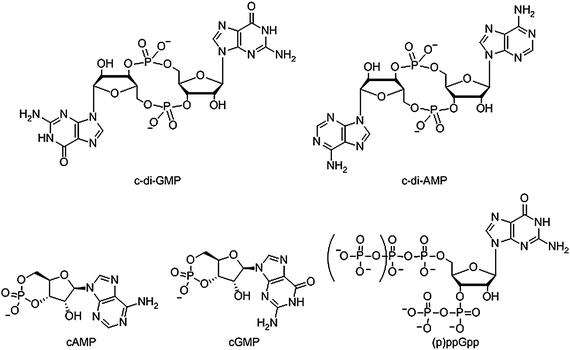 |
| | Fig. 1 Structure of nucleotide second messengers found in bacteria. | |
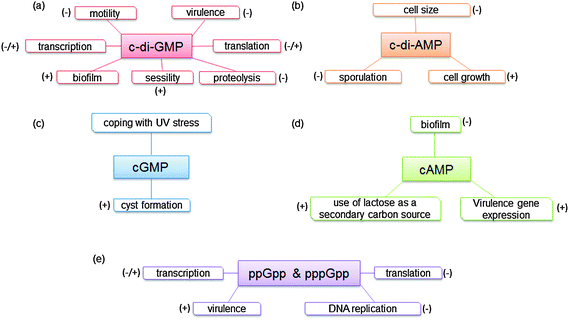 |
| | Fig. 2 Phenotypes or processes that are controlled by (a) c-di-GMP. (b) c-di-AMP. (c) cGMP. (d) cAMP. (e) ppGpp and pppGpp. | |
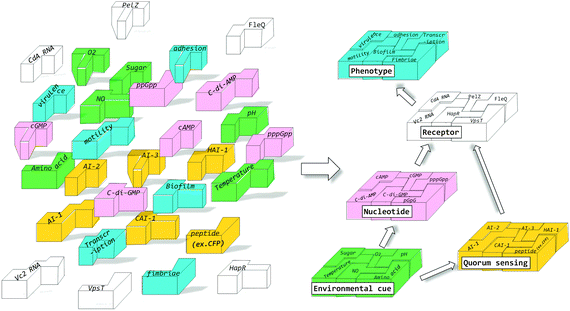 |
| | Fig. 3 Different environmental cues and signaling molecules interact to produce specific bacterial phenotypes. | |
In 1957, Earl Sutherland, who would later receive the Nobel prize in 1971, discovered that cAMP mediated the hyperglycemic effects of epinephrine and glucagon.15 A few years later, in 1963, cGMP was isolated from rat urine16 and it took a decade before it was demonstrated in 1974 that cGMP was not unique to eukaryotes but also existed in prokaryotes.3 In 1970, Cashel and co-workers first reported the existence of ppGpp and pppGpp in E. coli5 and it was later shown that the concentrations of ppGpp and pppGpp increased during stress.6 It would take another decade before the next nucleotide second messenger would be discovered in bacteria. In 1987 Benziman discovered that c-di-GMP acted as an allosteric modulator of cellulose synthase in G. xylinus (formerly called A. xylinum).1 C-di-GMP is associated with biofilm formation as well as virulence factor production in a good number of clinically important bacteria and hence there is immense interest in c-di-GMP signaling.17 Almost two decades after Benziman's seminal discovery of cyclic dinucleotide signaling in bacteria, Hopfner reported that bacteria also use c-di-AMP for signaling.2Fig. 2 summarizes the general description of phenotypes expressed through the various signal transduction pathways.
2. C-di-GMP
Transition between two mutually exclusive lifestyles, motile single cells and sedentary multicellular communities (biofilms) is regulated by secondary messenger c-di-GMP (Fig. 4). C-di-GMP is a ubiquitous signaling molecule and many bacteria contain genes that code for c-di-GMP associated proteins.10 Although Benziman discovered c-di-GMP more than two decades ago, it is only in the last few years that the molecular details of c-di-GMP signaling, as well as the phenotypes associated with increased intracellular c-di-GMP concentrations,18–27 have begun to emerge (see Fig. 5).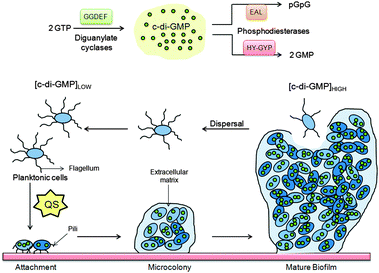 |
| | Fig. 4 C-di-GMP regulate several processes that control biofilm maturation pathways. | |
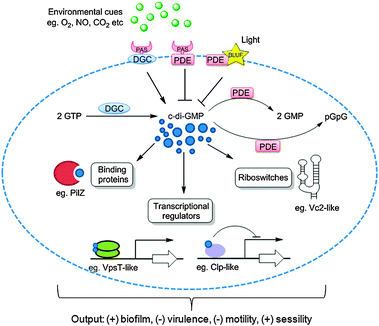 |
| | Fig. 5 C-di-GMP synthesis, degradation and signaling. Two molecules of GTP are converted to c-di-GMP by DGC proteins containing the GGDEF domain and c-di-GMP is degraded by PDE proteins with EAL or HD-GYP domains. Multiple numbers of DGCs and PDEs are often found associated with sensor domains such as PAS for sensing gaseous ligands such as O2, CO2, NO etc. and BLUF for sensing light. C-di-GMP binds to receptor proteins such as PilZ or riboswitches or transcriptional regulators to regulate bacterial “lifestyle”. | |
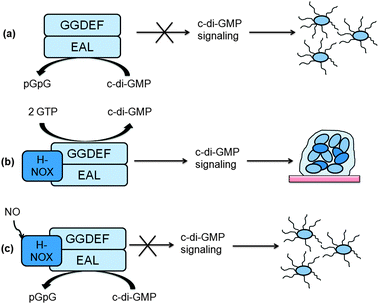
The intracellular concentration of c-di-GMP is controlled by synthases (called diguanylate cyclases (DGC), which contain a GGDEF11 or GGEEF28 domain) and hydrolytic enzymes (phosphodiesterases (PDE), which contain either EAL29 or HD-GYP30 domains). These proteins are sometimes fused to well-recognized regulatory motifs, such as a phosphorylation receiver domain (REC) or oxygen/redox potential/light sensor domain (PAS), which regulate the activities of the PDEs and DGCs (Table 1).
Table 1 Domain organizations and cellular locations of GGDEF and EAL containing proteins
| Protein | Domain organization | Organism | Cellular location |
|---|
| Predicted based on highly similar gene that is experimentally verified. Computationally predicted. Experimental verified. Note that not all EAL or GGDEF domain proteins are enzymatically active. For example TipF is an EAL domain protein but not a phosphodiesterase. Similarly, FimX is neither a DGC nor a PDE. |
|---|
| PleD31 | 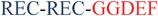 | C. crescentus | Cytoplasma |
| WspR32 |  | P. aeruginosa | Cytoplasmicb |
| PvrR33 | 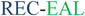 | P. aeruginosac | Cytoplasmic membraneb |
| TipF34 | 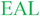 | C. crescentus | Cytoplasmic membranec |
| FimX35 |  | P. aeruginosa | Cytoplasmic membraneb |
| MbaA36 |  | V. cholerae | Cell inner membraneb |
2.1 Diguanylate cyclases
As already mentioned, c-di-GMP is synthesized from two equivalents of GTP by diguanylate cyclases (DGC), which are characterized by active site (A-site) amino acid motifs GGDEF (Gly-Gly-Asp-Glu-Phe) or GGEEF (Gly-Gly-Glu-Glu-Phe).37–39 Any point mutation of the GGDEF motif eliminates enzymatic activity.40 DGC proteins often contain an inhibitory site (I-site) which is characterized by a RxxD motif (x: any amino acid). Binding of c-di-GMP to the I-site of DGC allosterically inhibits c-di-GMP synthesis (product inhibition, see Fig. 6).41 This system of allosteric modulation helps to avoid excessive GTP consumption and ultimately limits production of c-di-GMP.42 The first example of a DGC containing both an A- and I-site was PleD (pleiotropic) protein in C. crescentus.41,43 PleD has REC1–REC2–GGDEF domains; REC1 and REC2 are phosphorylation receiver domains, which modulate the activity of PleD. Binding of c-di-GMP dimer immobilizes the domains and prevents rotation of the two active sites towards one another, which is necessary for the chemical condensation to occur.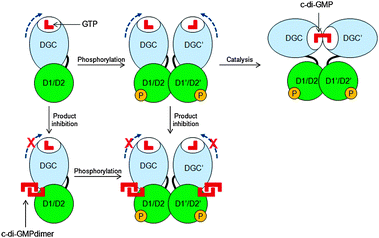 |
| | Fig. 6 Inhibition of c-di-GMP synthesis via product inhibition. Blue broken arrows indicate free rotation of the DGC domain when no c-di-GMP dimer is bound to the I-site. D1 and D2 refer to REC1 and REC2 domains.43 (Copied from ref. 43 with permission. Copyright 2004, National Academy of Sciences, U.S.A.) | |
Several other functional DGCs that are allosterically modulated by an I-site have also been described. For example WspR from P. aeruginosa,32 PleD from C. crescentus43 and DgcA from R. sphaeroides44 are other well-characterized DGCs that are inhibited by higher concentrations of c-di-GMP. Not all active DGCs have I-sites and some, such as XCC4471GGDEF from X. campestris can synthesize c-di-GMP without any product inhibition.45 Computational analysis with over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 GGDEF sequences revealed that about half of GGDEF sequences (potentially with DGC activity) lack the canonical allosteric inhibition sites.46 It is probable that these DGCs without an I-site are regulated via alternative pathways.
000 GGDEF sequences revealed that about half of GGDEF sequences (potentially with DGC activity) lack the canonical allosteric inhibition sites.46 It is probable that these DGCs without an I-site are regulated via alternative pathways.
2.2 Phosphodiesterases
HD-GYP domain PDEs hydrolyze c-di-GMP directly into GMP30,47 whereas EAL domain PDEs cleave c-di-GMP into pGpG and further into two GMP molecules.1,29 Both classes of PDEs require Mg2+ ion for the phosphodiester hydrolysis, and Mn2+ can also replace Mg2+.48 Conversely, cations such as Ca2+, Ni2+, Fe2+ and Zn2+ inhibit the activity of PDE.29,49,50 A detailed mechanism for the cleavage of c-di-GMP by PDEs has not been worked out yet but analyses of crystal structures of PDEs, with or without bound c-di-GMP, have provided some clues regarding how c-di-GMP is cleaved by phosphodiesterases. For example, the crystal structures of BlrP1 (K. pneumonia) and TBD1265 (T. denitrificans) reveal the presence of two metal ions that are ligated by the exocyclic phosphate oxygens of c-di-GMP, hydroxide anion and conserved residues of EAL-domain proteins (Asp, Glu and Asn), (see Fig. 7 and alignment Fig. 8).50,51 Yakunin and co-workers have proposed that the metal ions in the active site of PDE are involved in Lewis acid activation of the phosphate center, as well as activating the bound water molecule (with the assistance of a neighboring lysine to generate a catalytic hydroxide), which then attacks the phosphate center of c-di-GMP to lead to cleavage. The 3′-alkoxide leaving group could then be protonated by a bound water molecule (which is coordinated by a conserved aspartic acid see Fig. 7). Sequence alignments of EAL domains (Fig. 8) reveal that the majority of the residues, which are postulated to be involved in the ligation of the catalytic cations or generation/stabilization of the catalytic hydroxide or bound water, which would then protonate the 3′-alkoxide leaving group are conserved (see Fig. 8) and therefore provides a strong indirect evidence for the mechanism proposed by Yakunin.51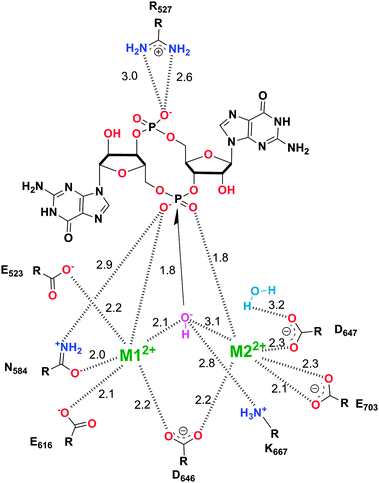 |
| | Fig. 7 Mechanism of c-di-GMP hydrolysis. C-di-GMP in the active site of TBD1265.51 M1 and M2 represent divalent metals. The numbers represent interatomic distances (in Å). (Adapted from ref. 51 with permission. Copyright 2010, Elsevier.) | |
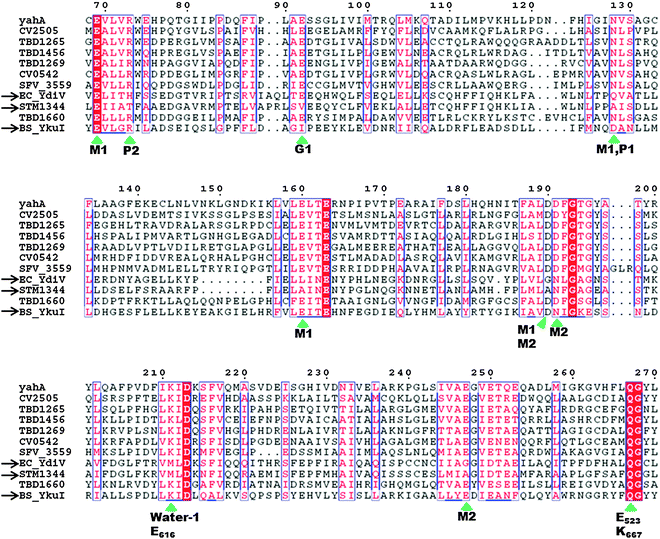 |
| | Fig. 8 Sequence alignment of EAL domains TBD1265 (T. denitrificans), YahA and YdiV (E. coli), CV0542 and CV2505 (C. violaceum), TBD1456, TBD1269 and TBD1660 (T. denitrificans), YkuI (B. subtilis), STM1344 (S. typhimurium). Absolutely conserved residues are boxed in white font on a red background, and partially conserved residues are boxed in red font on a white background. Conserved residues, which are predicted (based on the crystal structure of TBD1265) to contact the “catalytic” metals, phosphate moiety of c-di-GMP, “catalytic” water, and the nucleobase of c-di-GMP are marked by the green arrows. Inactive EAL domains are marked with black arrows. Because some of the inactive EAL-domain proteins contain the conserved active site residues, it implies that other factors (residues), apart from these conserved residues, affect catalytic proficiencies of EAL-containing proteins. M1: metal 1; M2: metal 2; water-1: catalytic water molecule; G1: guanine 1; P1: phosphorus atom attacked by water-1; P2: the other phosphorus atom in c-di-GMP that is not cleaved by EAL-domain proteins.51 Alignment was done using ClustalW252 and ESPript.53 (Adapted from ref. 51 with permission. Copyright 2010, Elsevier.) | |
In contrast to EAL-domain PDEs, for which there are a few crystal structures, HD-GYP has so far resisted high-resolution crystallography and Bd1817 from B. bacteriovorus is currently the only HD-GYP protein for which a good resolution crystal structure data is available.54 Bd1817 however lacks the active-site tyrosine present in most HD-GYP proteins but shows some sequence similarity to other HD-GYP proteins (see alignment Fig. 9) and so is the best model currently available to deduce the catalytic mechanism of c-di-GMP cleavage by HD-GYP-containing proteins. The crystal structure of Bd1817 reveals the presence of a binuclear iron center (see Fig. 10). Sequence alignment of Bd1817 with other HD-GYP-containing proteins (Fig. 9) reveals that the majority of residues that contact the iron ions or phosphate moiety of c-di-GMP are conserved. As more high-resolution crystal data for catalytically active HD-GYP become available, the strategy that this class of enzyme uses to cleave both phosphate moieties of c-di-GMP would emerge.
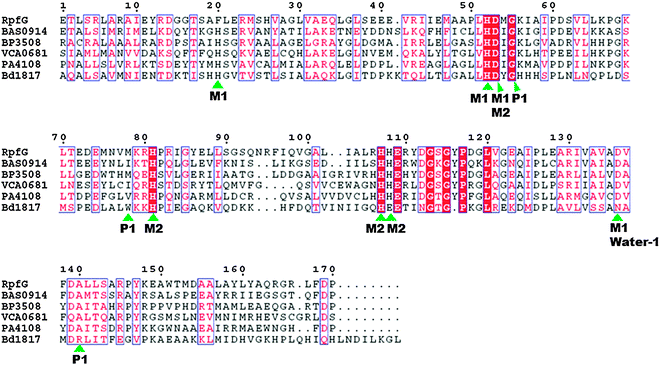 |
| | Fig. 9 Sequence alignment of the HD-GYP domain proteins Bd1817 (B. bacteriovorus), RpfG (X. campestris), BAS0914 (B. anthracis), BP3508 (B. pertussis), VCA0681 (V. cholerae) and PA4108 (P. aeruginosa). Absolutely conserved residues are boxed in white font on a red background, and partially conserved residues are boxed in red font on a white background. The conserved motif residues that contact the metal, phosphate moiety of c-di-GMP and water in the crystal structure of Bd1817 are indicated by the green arrows. M1: metal 1; M2: metal 2; water-1: putative catalytic water molecule; P1: phosphorus atom attacked by water-1.54 Alignment was done using ClustalW252 and EsPript.53 (Adapted from ref. 54 with permission. Copyright 2011, American Society for Microbiology.) | |
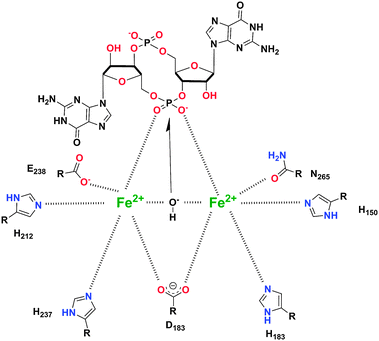 |
| | Fig. 10 Proposed binding model for c-di-GMP substrate in HD-GYP protein (PDB code (Bd1817): 3TM8).54 (Adapted from ref. 54 with permission. Copyright 2011, American Society for Microbiology.) | |
2.3 Hybrid proteins containing both DGC and PDE domains
Hybrid proteins that contain both GGDEF and EAL or HD-GYP domains also exist in several bacteria (see Table 2). Some of the hybrid proteins have dual activities (i.e. act as both DGC and PDE, depending on cellular state) or only one activity whereby the other domain is inactive.46,55,56 In some instances the inactive domain acts as regulatory domain by binding to a guanine nucleotide to modulate the activity of the other domain. A typical example is hybrid protein CC3396, from C. crescentus, which contains both GGDEF and EAL domains but the GGDEF domain is catalytically inactive. When GTP binds to the inactive GGDEF domain of CC3396, the adjacent EAL domain is allosterically activated and PDE activity is enhanced.48 Overall, a higher proportion of GGDEF domains (2235 proteins; 23.7%) have lost their catalytic motif as compared to EAL domains (935 proteins; 16.8%).57
Table 2 Number of GGDEF, EAL, HD-GYP and hybrid domains in different bacteria
| Bacteria | GGDEF | EAL | HD-GYP | GGDEF-EAL (hybrid) |
|---|
| May contain hybrid domains. n/a: Exact number not known, to the best of our knowledge. |
|---|
| E. coli K1258 | 12 | 17 | 0 | 7 |
| B. subtilis59 | 3 | 3 | 0 | 1 |
| R. prowazekii59 | 1 | 1 | n/a | 0 |
| V. vulnificus YJ01660 | 46 | 14 | n/a | 18 |
| V. cholerae61 | 26 | 22 | 9 | 15 |
| S. sp. PCC680359 | 23a | 13 | 2 | n/a |
| P. aeruginosa61 | 19 | 21 | 3 | 14 |
| B. halodurans61 | 2 | 2 | 2 | 2 |
| C. crescentus61 | 11a | 10 | 0 | n/a |
| S. oneidensis62 | 51 | 27 | 1 | 20 |
| S. flexneri63 | 3 | 6 | n/a | 4 |
| S. Typhimurium61 | 12 | 14 | 0 | n/a |
| V. fischeri60 | 28 | 9 | n/a | 10 |
| D. radiodurans61 | 16a | 5 | 4 | n/a |
| A. aeolicus61 | 11a | 8 | 1 | n/a |
| S. aureus64 | 1 | 0 | n/a | n/a |
| S. typhimurium65 | 12a | 14 | n/a | n/a |
2.4 Effector proteins
C-di-GMP must regulate the biofilm phenotype by binding to receptors.66 So far five types of c-di-GMP receptors have been found: (a) effector proteins, such as PilZ domain proteins that do not have any enzymatic activities of their own but bind to c-di-GMP to regulate other proteins or enzymes via domain–domain (if the PilZ domain is covalently linked to the enzymatic domain) or protein–protein interactions;66–68 (b) degenerate GGDEF proteins that are no longer catalytically proficient;23,69–71 (c) proteins with enzymatic activity that bind to c-di-GMP and upon binding, enzymatic proficiency is enhanced;72 (d) transcription factors or repressors that bind to c-di-GMP to regulate gene expression73–75 and (e) riboswitches that bind to c-di-GMP to regulate transcription76 or translation.77,78PilZ is a protein found in P. aeruginosa and regulates pili formation and twitching motility.79 Amikam and Galperin upon realizing that domains, which were similar to the PilZ protein were found to be associated with c-di-GMP metabolism domains (GGDEF, EAL and HD-GYP domains) proposed that these domains (named PilZ due to the semblance to the PilZ protein) were the long-sought after c-di-GMP binding proteins.68 Subsequent experimental work by Gomelsky and co-workers verified that proteins containing the PilZ domain could indeed bind to c-di-GMP.66 PilZ proteins bind to c-di-GMP and undergo either structural or aggregation change, which presumably facilitates the activation of other proteins, which directly or indirectly modulates the biofilm phenotype.80 So these proteins are in essence “adaptor” proteins, in that they do not have enzymatic activities on their own but bind to c-di-GMP to modulate other proteins via protein–protein interactions. The crystal structures of a few PilZ domain-containing proteins (VCA0042,80 PP439781 and PA460882) have been solved (see Fig. 11) and like the I-site of DGCs, the PilZ proteins contain arginine fingers (RxxxR) that bind to c-di-GMP. VCA0042, from V. cholerae, contains a C-terminal PilZ domain and N-terminal YcgR-N domain. This protein exists as a dimer and binds monomeric c-di-GMP. The binding event causes a conformational change in the loop between the PilZ and YcgR-N domains, leading to c-di-GMP being sandwiched between the two domains.80 PP4397 (from P. putida) is another protein containing YcgR-N and PilZ domains. This protein, like VCA0042, exists as a dimer and binds to c-di-GMP in the region between the YcgR-N and PilZ domains. Upon binding to dimeric c-di-GMP, the protein transitions from being a dimer into a monomer.81 PA4608 (from P. aeruginosa) is a single domain PilZ protein and binds to dimeric c-di-GMP. Upon ligand binding, significant structural rearrangements in both termini (C- and N-regions) occur. This leads to the clustering of surface charges on PA4608 (where one face of the protein is strongly negatively charged).82 It has been proposed that this c-di-GMP-facilitated surface charge clustering is a readout of c-di-GMP signaling and provides charged surface for tight binding to regulatory downstream target proteins.82 Other PilZ containing proteins are, Alg44 (found in P. aeruginosa and controls alginate synthesis),57 DgrA (found in C. crescentus and controls flagellar motor function,83 BcsA (found in G. xylinus and regulates the synthesis of cellulose)66 YcgR (found in E. coli and regulates flagellum-based motility).66 The molecular details of how YcgR integrates the c-di-GMP signal into biofilm-related processes have now been worked out. This protein inhibits flagellar motility via modulation of the motor–stator complex of the flagellum. The flagellum (a large and complex structure) is made up of a propeller and a rotary motor. Bacteria swim or tumble using this complex macromolecular apparatus. The rotating rotary motor of the bacterial flagellum consists of rotor and stator proteins. YcgR-c-di-GMP complex inhibits the motor–stator complex of the flagellum and reduces torque generation19 by binding to MotA (a stator protein), which presumably interacts with FliG (a rotor protein).18 Therefore when c-di-GMP concentration increases, the occupancy of the YcgR protein by c-di-GMP increases and swimming motility is inhibited.20
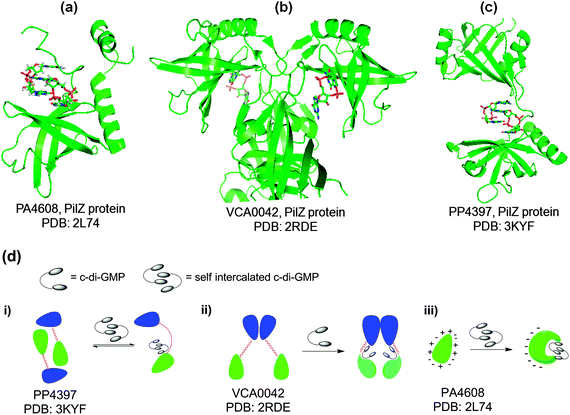 |
| | Fig. 11 (a) Crystal structure of c-di-GMP bound to PA4608, PilZ protein (PDB code: 2L7482); (b) crystal structure of c-di-GMP bound to VCA0042, PilZ protein (PDB code: 2RDE80); (c) crystal structure of c-di-GMP bound to PP4397, PilZ protein (PDB code: 3KYF81); (d) c-di-GMP modulation of the conformation or aggregation of PilZ-containing proteins; blue and green represent YcgR-N domain and PilZ domain, respectively. | |
Transcription factors are known to respond to environmental cues by binding to second messengers that are synthesized in response to changing environment. Hiekman and Harwood were the first to discover a c-di-GMP-dependent transcriptional regulator, FleQ.73 Interestingly, FleQ represses the pel biosynthetic genes in the absence of c-di-GMP and activate these same genes in the presence of c-di-GMP, using the same promoter sites.84 Pel is a polysaccharide that is a component of the biofilm matrix of P. aeruginosa. Harwood has proposed an interesting model to explain how FleQ, working with FleN, can both repress (in the absence of c-di-GMP) and activate (in the presence of c-di-GMP) genes, whiles still bound to the same promoter sites (see Fig. 12 for the model).84
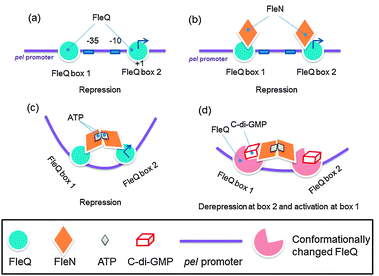 |
| | Fig. 12 FleQ binds to two pel promoter sites and each FleQ associates with FleN monomer. ATP binds to FleQ and causes association of the FleN monomers, causing structural changes that distort the pel DNA to repress pel. Upon c-di-GMP binding to FleQ, the FleQ-c-di-GMP/FleN-ATP complex undergoes a conformational change to reverse the pel DNA distortion, leading to pel expression. Key points: FleQ can still interact with FleN, even when ATP is absent and the binding of FleQ to box2 is essential for repression whereas binding of FleQ to box1 is essential for activation of pel.84 (Adapted from ref. 84 with permission. Copyright 2012, Oxford University Press.) | |
Other transcriptional regulators that bind to c-di-GMP have also been found. For example Clp, which is homologous with E. coli cAMP receptor protein, CRP, (45% sequence identity), binds c-di-GMP and the protein–ligand complex no longer has an affinity for DNA.75 This is different from other CRP-like proteins, which typically bind to DNA more tightly when bound to cAMP/cGMP.
VpsT is a transcriptional regulator in V. cholerae that inversely regulates motility and biofilm formation.85 C-di-GMP, which binds to VpsT in the dimeric form, drives the dimerization of VpsT to promote DNA recognition and transcriptional modulation. Interestingly, c-di-GMP also affects the subcellular localization of VpsT.86 Recently, Johnson and co-workers, reported that the transcriptional regulator MrkI from K. pneumoniae, which co-transcribes with MrKH (predicted to contain a PilZ domain), binds to c-di-GMP to regulate fimbriae and biofilm formation.87 The molecular details of how MrkHI regulate transcription is currently unknown.
Proteins that also do not have enzymatic activities but modulate other enzymes or proteins (similar to PilZ proteins) are known. These are usually degenerate EAL or GGDEF domain proteins that do not have enzymatic activity. A typical example is LapD, which is an inner membrane protein.21,22 LapD is from P. fluorescens and contains degenerate and enzymatically inactive DGC and PDE domains, but it is capable of c-di-GMP binding via its phosphodiesterase EAL domain.22 When bound to c-di-GMP (in the cytoplasm), LapD interacts with LapG in the periplasm. LapG is a protease that cleaves the N-terminal of LapA (an outer membrane adhesion protein that is important for surface attachment, and hence biofilm formation). LapG, when sequestered by LapD is not functional; therefore increased intracellular c-di-GMP concentration (which is responsible for LapG sequestration) would lead to the accumulation and maintenance of LapA and hence the persistence of the biofilm structure. However, when inorganic phosphate (Pi) becomes limiting, then the expression of RapA (a c-di-GMP phosphodiesterase) is increased and this depletes cellular c-di-GMP. The end result is that LapD/LapG complex dissociates and the “free” LapG, which is now active, cleaves the N-terminal of LapA. Cleaved LapA is then released from the cell surface, leading to biofilm dispersal (Fig. 13).
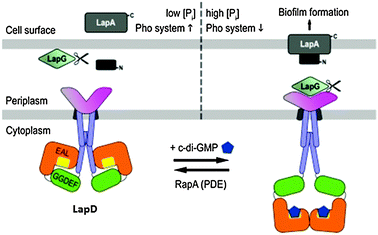 |
| | Fig. 13 Model for LapD inhibition of the protease LapG via sequestration.88 Copied from ref. 88 (open access). | |
Other degenerate GGDEF and EAL domain-containing effectors are PopA (from C. crescentus)69,89 and PelD (from P. aeruginosa).90 PopA contains a GGDEF domain, a conserved I-site and two receiver domains. During asymmetric division in C. crescentus, at the G1-to-S phase, PopA becomes concentrated at the old pole (after c-di-GMP binds to the I-site) and recruits proteins that are required for the degradation of CtrA (a master regulator of the cell cycle).69 C-di-GMP also binds to the I-site of PelD, which increases Pel exopolysaccharide and biofilm formation. FimX (from P. aeruginosa) is a degenerate GGDEF-EAL domain-containing effector and c-di-GMP binds to the EAL site.91,92 Upon c-di-GMP binding to FimX, the protein undergoes long-range conformational changes to regulate type IV pili assembly and twitching motility.35
C-di-GMP is also known to directly bind to and modulate proteins with enzymatic activities. In E. coli, c-di-GMP has been shown to bind to polynucleotide phosphorylase (PNPase), which is an important enzyme in RNA metabolism.72 PNPase can serve as a 3′-polyribonucleotide polymerase or a 3′ to 5′ exoribonuclease. When c-di-GMP binds to PNPase, RNA processing and degradation in bacteria are affected.72
Table 3 lists a few c-di-GMP related proteins the biological processes that are regulated by these proteins.
Table 3 Proteins involved in c-di-GMP signaling
| Activity | Protein | Domain | Organism | Biological output |
|---|
| PAS domain: Per-Arnt-Sim. BpeGReg: B. pertussis globin-coupled regulator. Light activates cleavage of protein into a PDE fragment and a DGC fragment. The activities for both PDE and DGC are controlled by ScrA and ScrB. PDE activity regulates swarming. Associate with H-NOX. HAMP domain: HAMP domains are found in proteins that are related to signaling transduction, for example Histidine kinases, Adenyl cyclases, Methyl accepting chemotaxis proteins and Phosphatases. |
|---|
| DGC (c-di-GMP synthase) | AxDGC2102 | PASa-GGDEF-EAL | A. xylinus | Cellulose synthesis |
| BpeGReg103b | Globin-GGDEF | B. pertussis | Biofilm formation |
| DgcA41 | GGDEF | C. crescentus | Motility inhibition |
| PleD7 | REC1–REC2–GGDEF | C. crescentus | Stalk formation, flagellum ejection |
| PelD104 | GGDEF | P. aeruginosa | Polysaccharide synthesis |
| WspR32 | REC-GGDEF | P. aeruginosa | Polysaccharide synthesis |
| DgcK105 | PAS-GGEEF | V. cholerae | Motility inhibition |
| DgcL105 | REC-GGEEF | V. cholerae | Motility inhibition |
| YdaM58 | GGDEF | E. coli | Curli fimbriae, cellulose synthesis |
| MucR106 | MHYT-GGDEF-EAL | P. aeruginosa | Alginate synthesis |
| PDE (c-di-GMP hydrolase) | AxPDEA1107 | PAS-EAL | A. xylinus | Cellulose synthesis inhibition |
| RocR108 | EAL | P. aeruginosa | Fimbrial assembly |
| YahA29 | EAL | E. coli | C-di-GMP hydrolysis |
| BlrP1(also known as KPN_01598)50,109 | BLUF-EAL | K. pneumoniae | C-di-GMP hydrolysis, upon irradiation |
| PvrR110 | CheY-EAL | P. aeruginosa PA14 | Biofilm formation inhibition |
| YciR58 | GGDEF-EAL | E. coli | Expression of curli fimbriae |
| Hybrid DGC/PDE (i.e. can act as either a DGC or PDE) | BphG156 | GGDEF-EAL | R. sphaeroides | Synthesis and degradation of c-di-GMPc |
| ScrC55 | GGDEF-EAL | V. parahaemolyticus | Regulate swarmingd |
| SwDGC111 | GGDEF-EAL | S. woodyi | Biofilm formatione |
| Effector protein (PilZ-containing) | MrKI87 | PilZ | K. pneumoniae | Type 3 fimbriae and biofilm formation |
| BcsA66 | PilZ | G. xylinus | Cellulose synthesis |
| DgrA83 | PilZ | C. crescentus | Motor function, cell mobility |
| YcgR66 | PilZ | E. coli | Flagella activity inhibition |
| Alg4480 | PilZ | P. aeruginosa | Alginate synthesis |
| PilZ79 | PilZ | P. aeruginosa | Pili formation, twitching motility |
| PlzB67 | PilZ | V. cholarea | Motility, biofilm formation |
| Effector protein (degenerate EAL or GGDEF; enzymatically inactive) | LapD21 | HAMPf-GGDEF-EAL | P. fluorescens Pf0-1 | Biofilm formation |
| FimX35 | GGDEF-EAL | P. aeruginosa | Pilusbiogenesis, twitching motility |
| Transcription regulators | FleQ84 | FLEQ/AAA/HTH | P. aeruginosa | Flagellar gene and pel expression |
| VpsT85 | REC-HTH | V. cholerea | vps gene expression, then biofilm formation |
| Clp75,112 | cNMP-HTH | X. campestris111 | Bacteria virulence |
| | | X. axopodis74 | |
| Bcam1349113 | c-NMP binding domain | B. cenocepacia | Biofilm formation |
2.5 C-di-GMP riboswitches
The first riboswitch that senses c-di-GMP, with a picomolar dissociation constant (called c-di-GMP class I riboswitch), was reported by Breaker.77 Soon thereafter, a second class of c-di-GMP riboswitch (called class II) was discovered in bacteria by the same group.78 (see Fig. 14 for the general architecture of class I and II riboswitches). These riboswitches are located upstream of the open reading frame (ORF) of DGC and PDE proteins in some organisms or upstream of some genes controlled by c-di-GMP. Binding of c-di-GMP to the riboswitch can either affect transcription or translation. For translational control, this is achieved via either c-di-GMP-mediated RNA structural changes that affect the accessibility of the ribosomal binding site or c-di-GMP-mediated self-splicing to generate a ribosomal binding site (Fig. 15).93 The crystal structures of both c-di-GMP class I and II riboswitches have been solved.94–96 In both the c-di-GMP class I and II riboswitches, a key adenosine is found in the binding pocket and this stacks between the two guanine bases of the bound c-di-GMP (Fig. 16). Mutation of this key adenine in both of the riboswitches would dramatically decrease the affinity of the riboswitch for c-di-GMP.95–97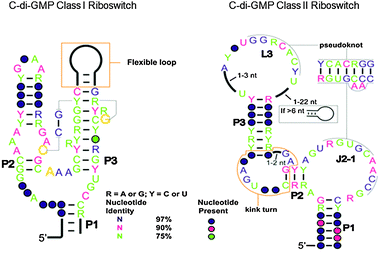 |
| | Fig. 14 Comparison of c-di-GMP class I and class II riboswitches.78 (Adapted from ref. 78 with permission. Copyright 2010, American Association for the Advancement of Science.) | |
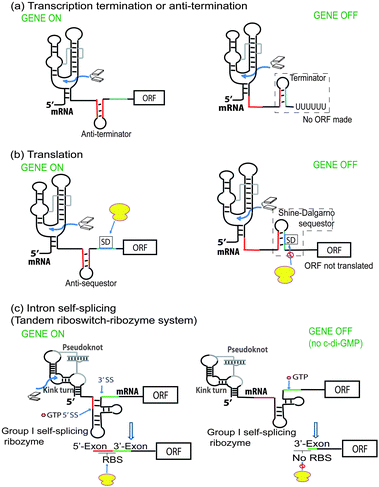 |
| | Fig. 15 Mechanism of c-di-GMP riboswitch. (Adapted from ref. 93 with permission. Copyright 2012, Cold Spring Harbor Laboratory Press.) | |
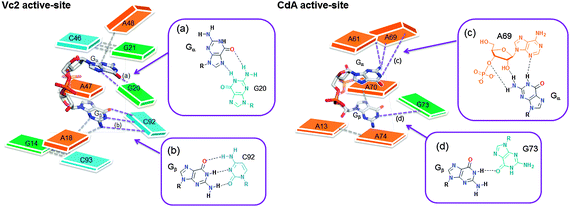 |
| | Fig. 16 Active site architectures of c-di-GMP class I (Vc2) and c-di-GMP class II (CdA) riboswitches. Guanine is shown as green color, adenine is orange and cytosine is blue. (a)–(d) displays the interaction between c-di-GMP (Gα or Gβ) and RNA bases. C-di-GMP (red represent oxygen and blue represent nitrogen). Different coloring are used just for clarity. | |
Because riboswitches play important roles in regulating mRNAs and hence the levels of proteins that are involved in c-di-GMP signaling in bacteria, there have been interests in discovering ligands that could compete with c-di-GMP binding with riboswitches. Both the groups of Strobel96,98,99 and Sintim100 have shown that it is possible to differentiate class I and class II c-di-GMP riboswitches using c-di-GMP analogs (Fig. 17). 2′-Modified c-di-GMP, such as bulky 2′-O-methyl analog96 and 2′-biotinylated analog,100 were found to be able to discriminate between c-di-GMP-I class and II class riboswitches; 2′-modified c-di-GMP bind to class II but not class I c-di-GMP riboswitch.96,100
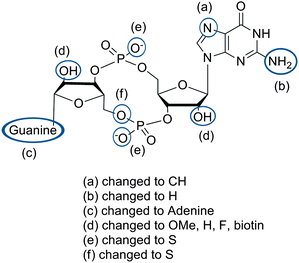 |
| | Fig. 17 Structures of c-di-GMP analogs studied by Strobel and Sintim groups. | |
In general, most modifications to c-di-GMP decreased the binding affinity to both class I and II riboswitches. Class I riboswitch was more discriminating than class II. Binding of c-di-GMP riboswitch to pGpG, the breakdown product of c-di-GMP, has also been studied.99 Both X-ray crystal analysis and biochemical studies have revealed that class I c-di-GMP riboswitch significantly favors binding of c-di-GMP over pGpG, although it was noted that the removal of the 5′-phosphate group of pGpG to give GpG, increased binding to riboswitches.99
2.6 Sigma (σ) factors
A sigma (σ) factor is a subunit of RNA-polymerase (RNAP) and enables RNAP to bind to gene promoters and initiate transcription. Environmental conditions affect the activation of different σ-factors. The “housekeeping” σ-factor (RpoD) is primarily responsible for the transcription of genes in growing cells and keeping the cell alive by making necessary proteins. Under starvation conditions σ-factor RpoS (σS) promotes the expression of genes to cope with the environmental stress. RpoS (σS) is also implicated in the role of biofilm formation. Therefore RpoS (σS) is called master regulator for stress resistance. C-di-GMP, also dubbed “a master regulator of bacterial lifestyle” has some associations with RpoS, albeit indirectly. For example, some GGDEF/EAL gene expressions are controlled by RpoS (σS) in E. coli101 and in the absence of RpoS (σS), these c-di-GMP metabolism genes (examples include genes for the GGDEF protein YdaM and the GGDEF/EAL protein YciR) are not expressed.2.7 Sensor domains of c-di-GMP
In order for bacteria to survive, they need to efficiently adapt to their surroundings. To do this they employ sensor (signal input) domains, which respond to various environmental cues. PAS (oxygen sensing), BLUF (blue light sensing), GAF-PHY (red/far light sensing), haemerythrin (gas sensing), REC (phosphorylation receiver), and GAF (cyclic nucleotide, e.g. cAMP and cGMP, and phytochrome binding) domains are examples of sensor domains. These sensor domains are linked to GGDEF or EAL (or HD-GYP) domains which are switched on or off according to the signaling input. As a result, environmental factors may affect the intracellular concentration of c-di-GMP (Table 4).59,65,114,115
Table 4 Environmental cues
| Environmental cue | Related protein | Organism | Effects | C-di-GMP level |
|---|
| O2 | DosP116 | E. coli | Hydrolysis of c-di-GMP | (−) |
| DosC116 | E. coli | Synthesis of c-di-GMP | (+) |
| BpeGReg103 | B. pertussis | Synthesis of c-di-GMP and enhanced biofilm formation | (+) |
| NO | Hnox1117 | L. pneumophila | Biofilm inhibition | (−) |
| BdlA118 | P. aeruginosa | Biofilm dispersal | (−) |
| Amino acid | TasA119 | B. subtilis | Biofilm inhibition | (−) |
| Light | PapB50 | R. palustris | PDE activation via BLUF domain | (−) |
| BlrB120 | K. pneumonia | Modulation of PDE activity | (−) |
| Carbon starvation | LapA121 | P. putida | Biofilm inhibition | (−) |
| Oxygen starvation | RbdA122 | P. aeruginosa | Biofilm inhibition | (−) |
| Phosphate starvation | RapA123 | P. fluorescens | C-di-GMP degradation | (−) |
| Temperature | HmsT124,125 | Y. pestis | Biofilm formation | (+) |
2.7.1 Oxygen sensing. GGDEF and EAL domain proteins that are also associated with heme-containing domains (such as PAS) can respond to oxygen levels to regulate the synthesis and removal of c-di-GMP.107,116,126 A well-studied example is the Dos system in E. coli. DosC (a DGC) binds to oxygen (Kd of 21 μM) via a GCS (globin-coupled sensor) domain, whereas DosP (a PDE) binds oxygen (Kd of 74 μM) via a PAS domain. Interestingly these enzymes are co-transcribed and are physically associated with each other (Fig. 18). Even more interestingly, DosC and DosP are associated with a mRNA and PNPase (which binds to c-di-GMP to modify RNA).72 It therefore appears that this system senses the concentration of oxygen (utilizing differences in binding constants for the c-di-GMP metabolism proteins, DosC and DosP) and translates this information into mRNA processing. Another example of a c-di-GMP metabolism protein that is modulated by oxygen is the DGC BpeGReg, from the whooping cough pathogen B. pertussis.103 O2 binding to the heme-containing sensor domain switches on diguanylate cyclase activity, increasing c-di-GMP level and enhancing biofilm formation.103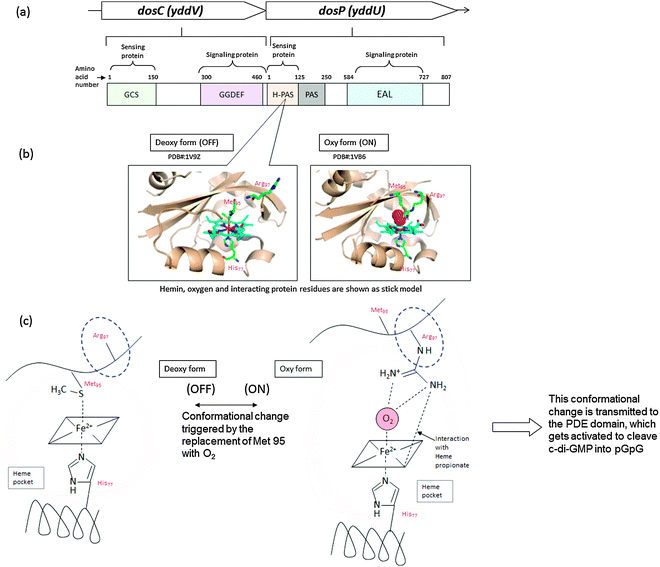 |
| | Fig. 18 Example of oxygen sensing protein, DosCP116 (a) Diguanylate cyclase (DosC) and phosphodiesterase (DosP) are co-transcribed.116 This dosCP operon is controlled by σS (RpoS). EcDosC is composed of two domains, GCS (globin coupled sensor, which contains heme domain) and GGDEF domain. EcDosP is composed of two domains, PAS (N terminal Heme binding domain) EAL domain.126 Binding of oxygen to the heme moiety leads to structural rearrangement of the oxygen-binding domain, which is transmitted to the enzymatic (DGC or PDE) domains to regulate enzymatic activities. | |
2.7.2 Nitric oxide sensing. It is now well established that low, non-toxic concentrations of nitric oxide, NO, can disperse bacterial biofilms.127 Several bacterial biofilms, such as those of P. aeruginosa,127,128E. coli,129V. cholerae,129S. woodyi,111B. licheniformis,129S. marcescens,129L. pneumophila,117N. europaea130 or N. gonorrhoeae131 have been shown to be dispersed with NO.NO regulates bacterial phenotype via the regulation of proteins involved in both c-di-GMP111,117 and quorum sensing signalings.132 NO is sensed by H-NOX (heme-nitric oxide/oxygen-binding) domain proteins. H-NOX can directly interact with DGC to regulate c-di-GMP synthesis and degradation (Fig. 19).
 |
| | Fig. 19 Boon's model for NO regulation of c-di-GMP synthesis in S. woodyi.111 (a) In the absence of H-NOX, SwDGC works primarily as a phosphodiesterase; (b) after H-NOX binds to DGC, it enhances the diguanylate cyclase activity and maintains only basal phosphodiesterase activity; (c) upon NO binding, H-NOX inhibits diguanylate cyclase activity and activates phosphodiesterase activity.111 (Adapted from ref. 111 with permission. Copyright 2012, American Chemical Society.) | |
2.7.3 Light sensing. Blue light using flavin (BLUF) photoreceptors are capable of capturing and utilizing blue light in order to induce physiological processes in bacteria. Made obvious by their name, these receptors use a flavin chromophore, which in turn interacts with the protein to carry out enzymatic processes.133,134 One such example of a BLUF protein is PapB from R. palustris.120 Irradiated PapB affects the PDE activity of the EAL domain protein, PapA, which hydrolyzes c-di-GMP.120 In the absence of blue light the EAL domain of PapA and the BLUF domain of PapB interact directly. However, upon light illumination, PapB undergoes a conformational change, which is transmitted to the interacting domain of PapA to increase the PDE activity of PapA.120 BlrP1, an EAL domain protein found in K. pneumonia, also responds to light. The kcat of BlrP1 increases four fold upon irradiation of blue light, suggesting a conformational change in the active sight of the EAL domain.50 The mechanism by which this conformational change affects enzymatic activity remains largely unknown however. Presumably, upon photoactivation, a highly conserved glutamine residue undergoes a 180° rotation, which rearranges the protein/flavin hydrogen bonding structure (see Fig. 20).109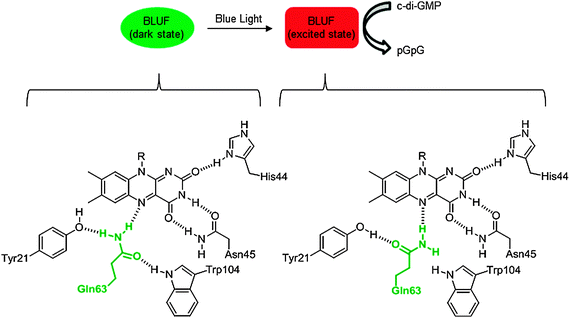 |
| | Fig. 20 Photoactivation of BLUF proteins. Bottom portion of the figure shows the dark and light state conformations of flavin/protein hydrogen-bonding network in AppA of R. sphaeroides. Emboldened, green glutamine is highly conserved in BLUF proteins and undergoes a 180° bond rotation upon irradiation with blue light. | |
2.7.4 Nutrient sensing. Nutrients such as organic carbon, glucose, nitrogen and phosphate all have an effect on c-di-GMP regulation, thus biofilm formation. The effect can be felt as an upregulation or downregulation of biofilm forming genes. For example, P. putida biofilm is dispersed during carbon starvation via decrease in intracellular c-di-GMP121 whereas the biofilm of Acinetobacter sp. GJ12 condenses during organic carbon starvation.135 Carbon starvation of P. putida decreases the level of LapA, a large adhesin that is used for the formation of biofilm.121In addition to the availability of carbon, bacteria respond through c-di-GMP mediated signaling processes, to the availability of inorganic phosphate (Pi) via the Pho regulon.136 The Pho regulon encodes the protein RapA which is has an active EAL domain.136 Elimination of the Pho regulon eliminates the ability of the bacteria to break down c-di-GMP and thus affects biofilm dispersal.
2.8 Polymorphism of c-di-GMP and binding to receptors
Jones revealed that c-di-GMP could aggregate to form different polymorphs, including dimer,137 tetramer138,139 and even octamer138,139 forms in the presence of cations (see Fig. 21–23). Alkali metals such as potassium promote G-quadruplex formation in c-di-GMP whereas divalent metals such as magnesium favor the intercalated dimer.138,139 At physiological concentrations of c-di-GMP (up to 10 μM), c-di-GMP mainly exists as monomer140 and only a small fraction is in the dimeric form141 (Fig. 23). Nonetheless the ability of c-di-GMP to form G-quadruplex tetrameric and octameric forms at sub-millimolar concentrations is interesting because simple nucleotides such as GMP, pGpG, GTP or even the analogous linear dimer pGpG do not form G-quadruplexes at micromolar concentrations.140,142,143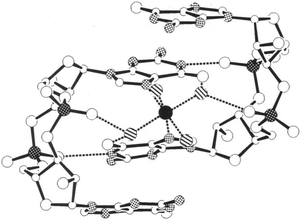 |
| | Fig. 21 Crystal structure of self-associated c-di-GMP dimer.137 Black-on-white stippled atoms: nitrogen atoms; white-on-black stippled atoms: phosphorus atoms; black: Mg2+; Hatched: waters coordinated to Mg2+; dashed lines: hydrogen bonds. (Copied from ref. 137 with permission. Copyright 1990, National Academy of Sciences, U.S.A.) | |
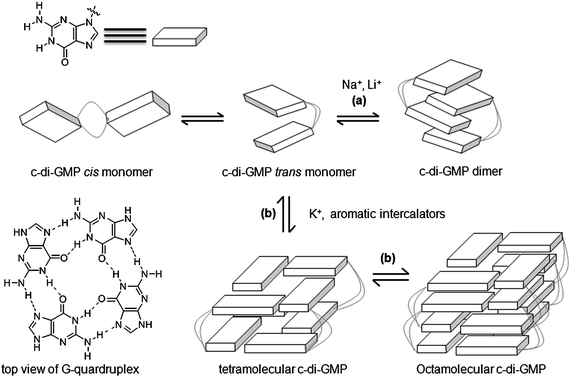 |
| | Fig. 22 Proposed model of polymorphism of c-di-GMP in presence of cations139 or intercalators.140 (Adapted from ref. 139 with permission. Copyright 2011, American Chemical Society.) | |
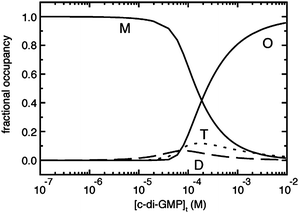 |
| | Fig. 23 Percentage of different c-di-GMP aggregation states as a function of total c-di-GMP concentration in buffer containing potassium and sodium.141 (Copied from ref. 141 with permission. Copyright 2012, American Chemical Society.) | |
Sintim and co-workers have shown that c-di-GMP is finely poised to form aggregates because a conservative replacement of one of the phosphodiester bridging oxygen in c-di-GMP with sulfur afforded an analog (called endo-S-c-di-GMP) that was not as aggregate-prone as c-di-GMP.140
Some aromatic intercalators, such as acridines, amidines and cyanines have been shown to promote c-di-GMP aggregation.143–145 Although attempts to use NMR or X-ray crystallography to structurally define intercalator/c-di-GMP complex have failed so far,146 several pieces of evidence suggest that intercalators promote G-quadruplex formation by c-di-GMP at physiological concentrations of c-di-GMP. Sintim has demonstrated that the CD spectrum of c-di-GMP changes upon the addition of some intercalators, showing positive CD peaks around 290 nm (which is usually indicative of G-quadruplex formation).140,144,145 Job plot analysis also revealed that c-di-GMP forms a 4:1 or higher aggregate with proflavine,144 thiazole orange143 and diminazene (berenil).145 The enhancement of the CD signal around 290 nm by intercalators occurs in the presence of G-quadruplex promoting cations such as potassium and sodium but not lithium.145 Recently, it was also demonstrated that in the presence of intercalators, c-di-GMP but not pGpG, has a peroxidase activity142 (a property that has been shown to be displayed by DNA/RNA G-quadruplex but not duplex and single stranded DNA/RNA).147,148 All of these evidences hints at a facilitation of G-quadruplex formation of c-di-GMP by intercalators at physiological concentrations. But without a definitive structural characterization using NMR or X-ray crystallography,146 other forms of association between intercalators and c-di-GMP can not be excluded as being responsible for the spectroscopic observations.
So far, only monomeric and dimeric, but not tetrameric or octameric, forms of c-di-GMP have been found bound to c-di-GMP receptor proteins (see Fig. 24). Analyses of the co-crystal structures of c-di-GMP and several proteins (DGC, PDE and PilZ proteins) show that c-di-GMP engages with several proteins via multiple interactions (Fig. 24). Arginine is common in the binding pocket of the majority of these proteins and forms hydrogen-bonding interactions with either the Hoogsteen face of the guanine nucleobase or the phosphate groups (see Fig. 24). Arginine can also form cation–π interactions with the guanine nucleobases of c-di-GMP.149 Other common residues that are also seen in c-di-GMP binding pockets of proteins are lysine (which forms hydrogen bonding interactions with the phosphate moiety of c-di-GMP), and aspartic and glutamic acids (which form hydrogen bonding interactions with the Watson–Crick face or phosphate moiety of c-di-GMP). Other aromatic residues, such as tryptophan, have also been seen forming π–π-stacking interactions with the guanine nucleobase of c-di-GMP.82
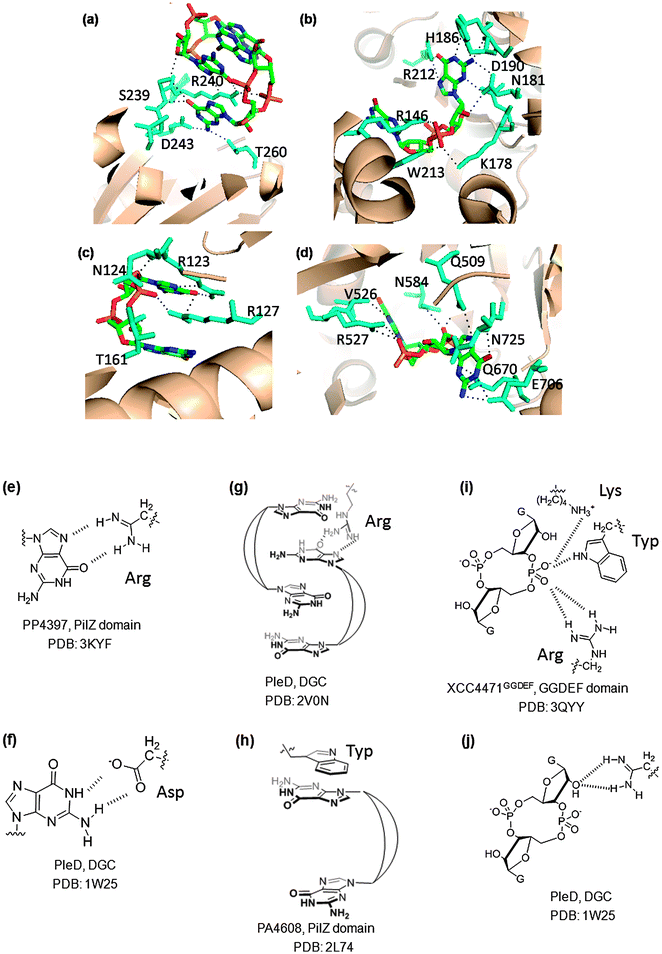 |
| | Fig. 24 (a) Dimeric c-di-GMP, bound to WspR (DGC domain I-site; PDB code: 3I5A32); (b) monomeric c-di-GMP bound to XCC4471GGDEF (GGDEF domain A-site; PDB code: 3QYY45); (c) dimeric c-di-GMP, bound to PP4397 (PilZ domain; PDB code: 3KYF81); (d) monomeric c-di-GMP bound to TBD1265 (EAL domain; PDB code: 3N3T); (e) arginine–guanine Hoogsteen H-bonding interactions in PP4397 (PilZ domain, PDB code: 3KYF81); (f) carboxylate–guanine Watson–Crick H-bonding interaction in PleD (PDB code: 1W2543); (g) cation–π interactions with arginine in PleD (PDB code: 2V0N149); (h) π–π-stacking interactions with tryptophan in PA4608 (PilZ domain, PDB code: 2L7482); (i) salt bridge between protonated lysine and phosphate group in XCC4471GGDEF (GGDEF domain A-site; PDB code: 3QYY45); (j) interaction between arginine and 2′-OH of c-di-GMP in PleD (PDB: 1W2543). | |
2.9 C-di-GMP and quorum sensing (QS)
Bacterial population-dependent cell-to-cell communication, known as quorum sensing (QS),151 is mediated by small signaling molecules, called autoinducers (AIs) to regulate gene expression in response to population density.152 There are three distinct classes of autoinducers (Fig. 25), named AI-1, oligopeptides and AI-2, and also some unique small molecules, such as 2-heptyl-3,4-dihydroxyquinoline (PQS, Pseudomonas quinolone signal), butyrolactone, CAI-1, cis-decenoic acid (also called diffusible signal factor, DSF) and DSF-like molecules (see Fig. 25 for structures).153 AI-1 or N-acylated-L-homoserine lactones (AHLs) based QS is found in Gram-negative bacteria154 while oligopeptides are produced by Gram-positive bacteria.155 QS mediated by AI-2, is found in more than 70 species of both Gram-negative and Gram-positive bacteria and hence AI-2 has been referred to as a “universal” autoinducer.156,157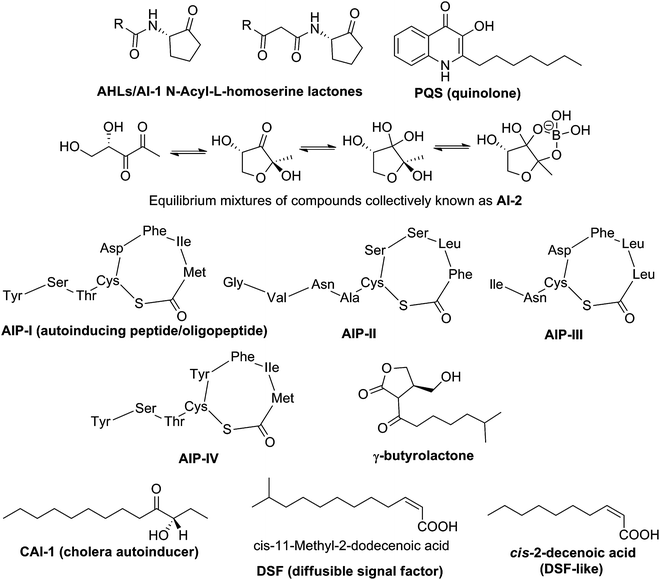 |
| | Fig. 25 Structures of representative autoinducers. | |
Since QS and c-di-GMP both involve the regulation of virulence and biofilm formation,157 it is presumed that the two signaling pathways may be linked and/or intersect. In V. cholerae, as bacterial cell density increases, so does the concentration of autoinducers AI-2 and CAI-1. These QS autoinducers in turn bind to their receptors, LuxP and CqsS, leading to increased expression of hapR (see Fig. 26). HapR (LuxR homolog, a master transcriptional regulator) represses the expression of vpsT and aphA. VpsT is a transcription factor that together with VpsR, binds to c-di-GMP to increase expression of several genes needed for biofilm development.85 Another transcription factor AphA not only induces virulence gene expression158 but also represses the expression of the acgA and acgB genes, which produce EAL and GGDEF containing proteins respectively.159 Overexpression of AcgA (a PDE) decreases biofilm formation while overproduction of AcgB (a DGC) does the opposite.159 It therefore appears that V. cholerae autoinducers integrates with c-di-GMP to modulate biofilm formation and virulence as bacterial population changes.
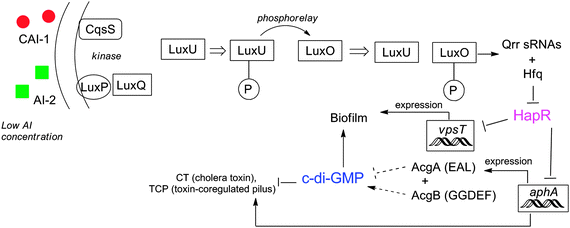 |
| | Fig. 26 Interaction between c-di-GMP and QS in V. cholerae. At low concentration of AIs, LuxU and LuxO are phosphorylated and activate expression of Qrr small RNAs, which along with Hfq represses HapR. HapR represses both biofilm formation and virulence directly and indirectly. At high concentration of AIs, dephosphorylation of LuxO results in termination of expression of sRNAs and then HapR is accumulated. | |
In P. aeruginosa, it has been demonstrated by Wood that TpbA, which is positively regulated by 3-oxo-C12-HSL and LasR complex, dephosphorylates the PDE, PA1120.160
Chopra and co-workers demonstrated that AI-1 (ahyRI dependent) positively regulates biofilm formation of A. hydrophila whereas AI-2 (luxS dependent) negatively regulates biofilm formation.161 They proposed that c-di-GMP mediates the AI-1- and AI-2-mediated biofilm formation and reduction processes, but the exact mechanism by which this is achieved remains unclear.161
The linkage between QS and c-di-GMP in X. campestris, a plant pathogen has also been investigated.47X. campestris uses diffusible signaling factor (DSF) to modulate QS system. Upon the binding of DSF to its receptor RpfC, the response regulator RpfG is activated and functions as PDE to degrade c-di-GMP.47
2.10 Detection of c-di-GMP
Earlier detection of c-di-GMP utilized HPLC-MS methods for sensitive detection (5 nM c-di-GMP could be detected using this method).162 This satisfies the need to detect intracellular concentrations of c-di-GMP, which range from 0–10 μM.11,162 However, tandem HPLC-MS of crude c-di-GMP is time consuming and expensive, requiring high resolving HPLC columns and high resolution mass spectrometers. It also requires specialist laboratories for the assay to always work. The use of fluorescent proteins that report the presence of c-di-GMP has also been reported.27The Sintim group recently described new assays to detect c-di-GMP that utilized shelf-stable reagents (intercalators) that are commercially available.144 As already discussed in Section 2.8 of this review, c-di-GMP associates with various intercalators and although structural data is not available, circumstantial evidences point to the facilitation of G-quadruplex formation by certain aromatic intercalators. Based on this working hypothesis that intercalators promote c-di-GMP G-quadruplexes, Sintim and co-workers demonstrated that thiazole orange (TO), which is weakly fluorescent in solution, becomes highly fluorescent when complexed with c-di-GMP.143 The current hypothesis to account for this observation is that thiazole orange becomes trapped between the G-tetrad planes of c-di-GMP G-quadruplex and hence rotation within the molecule, which is responsible for fluorescence deactivation, is limited in the c-di-GMP/TO complex and therefore fluorescence of TO is enhanced. In a very interesting recent report, the Sintim group also demonstrated that a mixture of hemin and proflavine aggregate c-di-GMP into a peroxidase complex and hence provides a simple colorimetric detection of c-di-GMP (see Fig. 27).142 So far, it has been shown that intercalator-based detection of c-di-GMP can detect low micromolar concentrations of c-di-GMP (∼5 μM for the TO detection and ∼1 μM for the peroxidase detection). These values are higher than the lower limit of detection obtained with HPLC-MS methods but the simplicity of the intercalator-based c-di-GMP detection is noteworthy and future developments to make the methods more sensitive are certainly warranted.
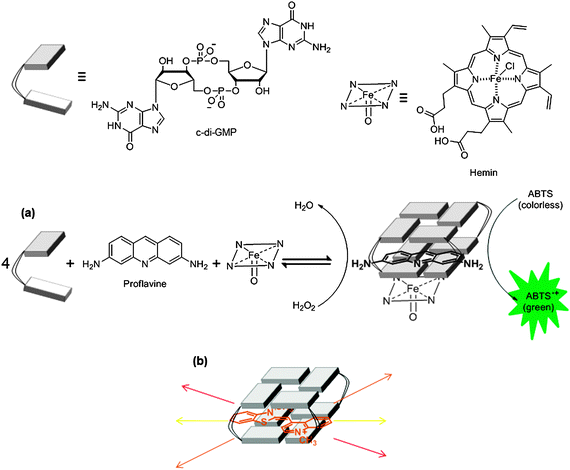 |
| | Fig. 27 (a) Colorimetric and (b) fluorescence detection of c-di-GMP utilizing aggregation strategy. ABTS: 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid). | |
Recently both the Breaker and Sintim groups independently reported the use of aptamers to detect nanomolar concentrations of c-di-GMP.163,164 Breaker's method utilizes a radiolabeled hammerhead self-cleaving ribozyme that is tethered to a c-di-GMP-binding aptamer (see Fig. 28a). Upon c-di-GMP binding, the ribozyme self cleaves and the cleaved product could be detected by PAGE. The obvious drawback of this method is the use of radiolabeling. Sintim's method utilizes a fluorescent aptamer strategy.164 For this safer alternative, a c-di-GMP aptamer is tethered to a spinach RNA. Spinach RNA binds 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI), which is a weakly-fluorescent molecule but upon binding to RNA fluorescence is enhanced appreciably (see Fig. 28b).
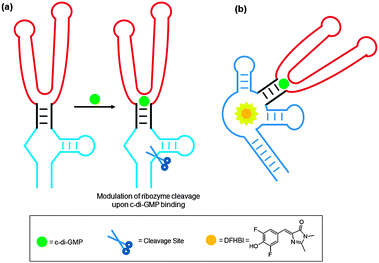 |
| | Fig. 28 Nanomolar detection of c-di-GMP using an aptamer tethered to (a) a hammerhead self-cleaving ribozyme and (b) spinach RNA. | |
3. (p)ppGpp
In bacteria two unusual nucleotides, guanosine tetraphosphate (ppGpp) and guanosine pentaphosphate (pppGpp) (collectively known as (p)ppGpp) are produced as signaling “alarmone” compounds in response to nutrient (amino acid,165 phosphates,166 fatty acids,167 carbon,168 and iron169) starvation. Stress response coordinated by (p)ppGpp ultimately leads to a reduction in the growth rate of cells and is referred to as “stringent response”.165 In addition to bacteria, stringent response has also been observed in plants.1703.1 Metabolism of (p)ppGpp
In E. coli and other beta-proteobacteria and gamma-proteobacteria, the cellular concentration of ppGpp is governed by two homologous proteins, RelA, which is a monofunctional synthase, and SpoT, which is a bifunctional hydrolase-synthase enzyme (Scheme 1, Fig. 29a).171,172 Most other bacteria express a single RelA/SpoT homolog (referred to as RSH proteins for Rel-Spo homolog).173–175 RelA uses GTP and ATP to generate pppGpp, which is then converted to ppGpp. On the other hand, SpoT or RSH, can synthesize both ppGpp and pppGpp. Moreover, they are also responsible for the hydrolysis of ppGpp and pppGpp to either GDP or pyrophosphate (PPi), or GTP and PPi respectively. Domains within the N-terminal region of RSH proteins are responsible for both the synthesis and hydrolysis of (p)ppGpp. Regulation of both these mutually antagonistic activities of the protein is crucial for cell survival.176 This regulation is orchestrated by both the N- and C-terminal regions of the protein, and involves binding of the protein to ‘A’ sites of ribosomes leading to sensing of lack of tRNA aminoacylation due to amino acid-starvation (Fig. 29a).165 An analogous mechanism for SpoT-sensing of fatty acid starvation has been discovered and involves binding of unacylated acyl carrier proteins (ACPs) to SpoT serving as triggers for stringent response (Fig. 29a).167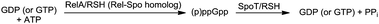 |
| | Scheme 1 Synthesis and degradation of (p)ppGpp. | |
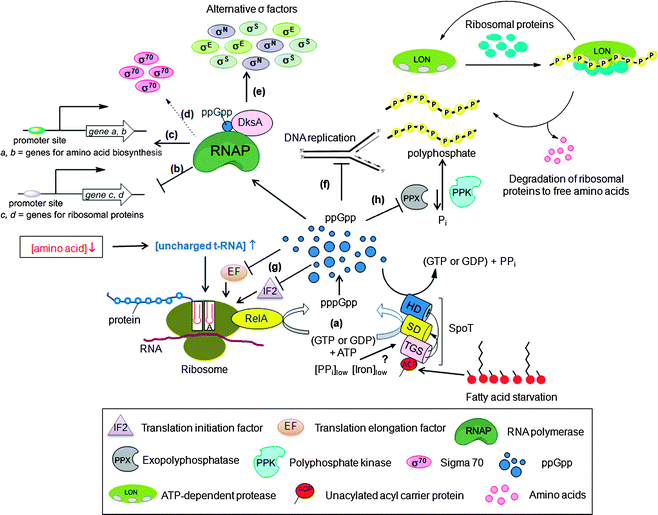 |
| | Fig. 29 (p)ppGpp signal transduction in E. coli turned on upon bacterial starvation. (a) Amino acid starvation sensed by the accumulation of uncharged tRNA molecules in the ribosomal A site triggers RelA mediated synthesis of (p)ppGpp from GTP (or GDP) and ATP.165 When amino acids become available, (p)ppGpp is degraded by the hydrolase (HS) domain of the bifunctional protein SpoT.171,172 During fatty acid starvation, acyl carrier protein (ACP) binds to the TGS domain of SpoT to allosterically activate (p)ppGpp synthesis at the synthase domain (SD).167 In contrast to amino acids or fatty acid starvation, phosphate166 or iron deprivation169 results in the activation of SpoT to make more (p)ppGpp; (b) ppGpp/DksA mediated inhibition of transcription of ribosomal promoters through direct interaction with RNA polymerase (direct inhibition); (c) ppGpp/DksA modulated activation of transcription of genes involved in amino acid biosynthesis (direct activation); (d) ppGpp and DksA indirect activation of transcription of stress-induced genes by releasing RNAP from vegetative σ-factor, σ70; (e) facilitating the interaction of RNAP with alternative σ-factors (σE, σS and σN) (indirect regulation); (f) ppGpp mediated inhibition of replication initiation; (g) ppGpp-modulated translation by binding and inhibiting the activity of initiation factor (IF2) and elongation factors (EF); (h) In response to amino acid starvation, (p)ppGpp inhibits the activity of exopolyphosphatase (PPX), resulting in polyphosphate accumulation that binds to ribosomal proteins and Lon protease to generate free amino acids. | |
3.2 (p)ppGpp signaling
(p)ppGpp regulates bacterial physiology via disparate mechanisms. It affects several biochemical processes in the cell including transcription (Fig. 29b–e), translation (Fig. 29g), DNA replication (Fig. 29f), and generates virulence factors by interacting with other signaling nucleotides.(p)ppGpp affect transcription by both directly regulating RNA polymerase (RNAP) or indirectly by interacting with σ-factors and by controlling the activity of transcription factors that regulate virulence genes. Direct regulation of RNAP involves its binding to (p)ppGpp (Fig. 29b and c). The effects of this interaction between RNAP and (p)ppGpp is modulated by the DksA protein, which binds to RNAP and enhances the effect of (p)ppGpp.177,178 Indirect transcription regulation by (p)ppGpp involves activation of the expression of many stress-induced genes by releasing RNAP from σ70-dependent promoters (Fig. 29d), and facilitating the use of alternative σ factors (such as σS, σE and σN) (Fig. 29e). One mechanism of indirect transcription regulation by (p)ppGpp involves modulation of the binding of σ-factors to RNAP—an event that initiates transcription of genes that are important for the biosynthesis of proteins, DNA and lipids. For example, (p)ppGpp inhibits binding of RNAP to strong σ70-dependent promoters allowing RNAP to bind to other σ-factors that direct RNAP to transcribe genes useful for dealing with stress conditions.174 Another mechanism of indirect effects of (p)ppGpp on transcription involves enhancement of the stability of σ-factors such as σS by induction of the expression of anti-adaptor proteins IraP179 and IraD,180 which prevent degradation of σS by not allowing it to be directed to the proteasome. A third example of indirect transcriptional regulation by (p)ppGpp include the increased activity of σE (RpoE) due to the (p)ppGpp-mediated degradation of RseA, a regulatory protein that inhibits the activity of σE.181
In addition to inhibiting translation by regulating transcription of protein synthesis apparatus, namely, tRNA, rRNA, and ribosomal proteins, (p)ppGpp directly inhibits the activity of the translation elongation factors EF-Tu and EF-G (Fig. 29g).165 Additionally, (p)ppGpp also interacts with the translation initiation factor IF2 by binding to the same site as GTP and inhibits translation initiation by preventing IF2-dependent formation of the 30S translation initiation complex, thereby inhibiting translation (Fig. 29g).182
In E. coli, (p)ppGpp inhibit replication initiation (Fig. 29f).183,184 In B. subtilis, replication elongation is inhibited by (p)ppGpp irrespective of the position of the replication forks along the chromosome.185 In S. typhimurium, (p)ppGpp promotes the dimerization of the protein, SlyA, which controls the transcription of genes essential for the virulence of the bacteria.186 Another interesting example of the role of (p)ppGpp in virulence is that it binds to PigR, a transcription factor that is essential for the growth of F. tularensis in macrophages. This binding controls PigR-mediated expression of virulence genes of the bacteria.187 An interesting strategy by which (p)ppGpp controls virulence is by regulating mRNA half-life in actinomycetes. Specifically, (p)ppGpp inhibits the enzyme polynucleotide phosphorylase (PNPase), which is responsible for RNA degradation, thereby enhancing RNA stability.188,189
In addition to the functions of (p)ppGpp mentioned above, (p)ppGpp plays an important part in the regulation of the acid stress response in E. coli by interacting with and modulating the activity of the enzyme lysine decarboxylase, inducible (LdcI).190 LdcI is induced during acid stress and serves to control the cellular acidity by increasing the cytoplasmic pH by consuming protons as it catalyses decarboxylation of L-lysine to cadaverine and CO2190,191 During extreme acid stress, the LdcI–ppGpp interaction is not sufficient to inhibit the enzyme thereby resulting in an increase in the cytoplasmic pH through normal mechanism. During mild acid or neutral conditions under amino acid starvation, however, (p)ppGpp binds LdcI and inhibit its activity, preserving the pool of cytoplasmic lysine. Another example where (p)ppGpp directly interacts with an enzyme and modulates its function is the (p)ppGpp-mediated regulation of exophosphatase (PPX), an enzyme that degrades polyphosphate, which is a polyphosphate energy source of bacteria and is a signaling molecule.192 During stress (p)ppGpp inhibits exopolyphosphatase (Fig. 29h). As a result, there is increased concentration of polyphosphate available in cells to bind to ribosomal proteins and to the Lon protease, which results in generation of free amino acids, thus enabling the cells to cope with amino acid starvation.193
3.3 (p)ppGpp and QS
Both ppGpp and QS signaling have evolved to respond to the environment of the bacteria and the two pathways appear to intersect (Fig. 30). Bally and co-workers found that in P. aeruginosa, overexpression of the ppGpp synthase gene, relA, led to an increased production of AI-1 (3-oxo-C12 HSL and C4 HSL).194 Storey and co-workers also demonstrated that at low Mg2+ concentration, RelA enhanced the production of AI-1 but inhibited the production of 2-heptyl-3-hydroxy-4(1H)-quinolone (PQS).195 The molecular details that underlie the effects of RelA on QS autoinducer production in P. aeruginosa remain poorly understood.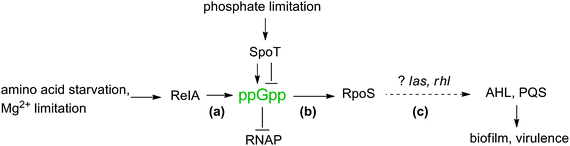 |
| | Fig. 30 Connection between ppGpp and QS (AHL and PQS mediated) in P. aeruginosa. (a) ppGpp is generated in response to stringent conditions which results in (b) increased production of sigma factor RpoS, and (c) changes in the levels of AHL and PQS resulting in regulation of biofilm formation and virulence. The link may be dependent on las and rhl, although this hypothesis remains unproven. | |
The causal agent of crown gall disease, A. tumefaciens, utilizes 3-oxo-C8 HSL as a QS signaling molecule. Zhang and co-workers revealed that upon either carbon or nitrogen starvation, the production of (p)ppGpp results in abolishment of the expression of AttM, an AHL-lactonase which degrades AI-1 molecules, thereby resulting in AHL accumulation.196
It has also been suggested that QS-dependent (mediated by AI-1) activation of exoenzyme synthesis requires the accumulation of ppGpp in the phytopathogen, E. carotovora subsp. atroseptica.197 Burne's group found that in relA knockout strains of S. mutans, the expression of luxS, which is responsible for the biosynthesis of AI-2, is significantly increased whereas biofilm formation is drastically reduced.198 Studies at the molecular level are needed to shed light on the relationship between relA and luxS, to understand how stringent response and AI-2 based QS regulate each other.
3.4 Detection of (p)ppGpp
(p)ppGpp is found in bacterial cells at a concentration of around 50 μM.199 One of the methods for the detection of (p)ppGpp in bacteria involves radiolabeling methods in which bacteria are grown in media containing 32PO4. The resulting radiolabeled (p)ppGpp is then detected by thin-layer chromatography.200 This method is not ideal as large quantities of inorganic phosphate could be mistaken for (p)ppGpp in this assay. In a method developed by Hamagishi and co-workers, a radioimmunoassay was used. This involved a competition between 3H-labeled (p)ppGpp with non-labeled (p)ppGpp from lysed cells.201,202 This method circumvented radiolabeling of intracellular (p)ppGpp, but required an extensive and nontrivial column chromatographic step to separate (p)ppGpp from more common nucleotides such as pGpp and ppGp. More recent methods have utilized high performance liquid chromatography to isolate (p)ppGpp.170,203 A fluorescence-based method developed by Hong and co-workers utilizes a selective fluorescent probe to detect (p)ppGpp.204 Two equivalents of the probe ligates to both pyrophosphate moieties of the (p)ppGpp, leading to a π-stacked pyrene excimer which can be monitored by fluorescence spectrometry at 430 nm (Fig. 31).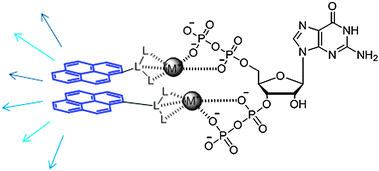 |
| | Fig. 31 Fluorescent detection of ppGpp using a π-stacked pyrene eximer. | |
4. C-di-AMP
Cyclic-di-adenosine monophosphate (c-di-AMP) was recently discovered in B. subtilis, revealing that bacteria utilize a more extensive array of nucleotide-based second messengers than previously appreciated (Scheme 2).2,14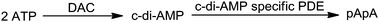 |
| | Scheme 2 Synthesis and degradation of c-di-AMP. DAC: di-adenylate cyclase, PDE: cyclic nucleotide phosphodiesterases. | |
4.1 C-di-AMP synthases
C-di-AMP is synthesized by the condensation of two molecules of ATP, a process that is catalyzed by c-di-AMP synthase proteins. One of the most well-studied c-di-AMP synthase proteins is DisA, also known as DAC (diadenylate cyclase).2 DisA is an octameric DNA integrity scanning protein, which scans the chromosome for DNA double strand breaks.205 The monomer of DisA contains a helix–hairpin–helix (HhH), which is a non-specific DNA-binding domain and a DAC domain (Fig. 32). The c-di-AMP synthesis activity of the DAC domain is allosterically inhibited by the binding of damaged DNA to the HhH domain on the DisA octamer.206 Therefore, it appears that DNA breakage is sensed by the HhH domain of the protein, which results in suppression of the di-adenylate cyclase activity of the protein which in turn results in physiologically useful downstream effects such as delay of sporulation in case of B. subtilis.206 This observation suggests that c-di-AMP has an important role in the initiation of sporulation in B. subtilis and it acts as a signaling molecule that couples DNA integrity with sporulation.2,205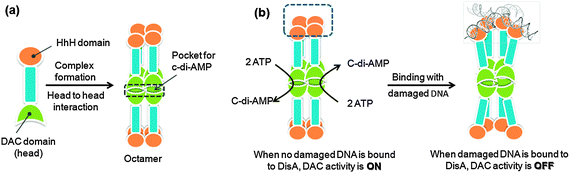 |
| | Fig. 32 Model for the mechanism of DisA in checkpoint control. DisA synthesizes c-di-AMP in the absence of chromosomal damage.2 (a) Monomer DisA is associated to octamer by head to head interaction of DAC domain (which is called DUF147 domain). This DAC domain interaction makes a pocket for c-di-AMP; (b) when damaged DNA (branched DNA) is not bound to DisA octamer, the DAC activity of DisA is ON, resulting in the synthesis of c-di-AMP from two molecules ATPs. When damaged DNA is bound to DisA octamer, the synthesis is inhibited (allosteric inhibition from damaged DNA). (Adapted from ref. 2 and ref. 206 with permission. Copyright 2008, Elsevier (ref. 2) and 2008, American Association for the Advancement of Science (ref. 206).) | |
In addition to DisA, there are two other proteins, YbbP and YojJ that are present in B. subtilis that possibly possess DAC activity.206 Whereas YbbP is predicted to be membrane-localized, YojJ is a cytosolic protein.206 In addition to B. subtilis, other bacteria also have DAC proteins. For example, in S. aureus, a DAC protein called DacA has been discovered.207
4.2 C-di-AMP hydrolases
In addition to the inhibition of c-di-AMP synthesis, binding of damaged DNA to DisA has another effect, which is enhancement of the expression levels of the c-di-AMP hydrolase enzyme, YybT (also called GdpP).205 YybT contains a DHH/DHHA1 domain, which possesses cyclic phosphodiesterase (PDE) activity, and it catalyses the conversion of c-di-AMP to 5′-pApA-3′.208 YybT also contains a PAS domain209 which recruits heme as a cofactor, enabling it to sense changes induced by light, redox potential, and oxygen (refer Section 2.7). Interestingly YybT is inhibited by ppGpp.208 This observation gives rise to the hypothesis that when the cell enters a stringent condition (which triggers enhanced production of ppGpp), YybT is turned off. Another intriguing observation is that when YybT is overexpressed, bacteria demonstrate increased sensitivity towards β-lactam antibiotics.210 Since β-lactam antibiotics function by inhibiting bacterial cell wall biosynthesis, this observation suggests an important role of c-di-AMP in cell wall homeostasis.2104.3 C-di-AMP signaling
C-di-AMP plays a role in the delay in sporulation in B. subtilis upon DNA damage, described above (Section 4.1). In S. aureus, c-di-AMP is implicated in processes that allow the cell to handle extreme membrane and cell wall stress, caused by reduction of the cell wall polymer, lipoteichoic acid (LTA).207 When LTA becomes limiting, it has been observed that S. aureus contain elevated concentrations of c-di-AMP. It is not entirely clear if this signifies a role of c-di-AMP in tolerating the stress.4.4 C-di-AMP and QS
So far, little is known about the connection between quorum sensing and c-di-AMP signal transduction pathway. As the molecular details of c-di-AMP in several bacteria become available, it is likely that connections between c-di-AMP and QS will come to light.5. cAMP and cGMP
The cyclic nucleotides, cAMP and cGMP, are universally used as second messengers in eukaryotic cells in intracellular signal transduction pathways. In higher organisms, they play important roles in cellular processes such as vision, electrolyte homeostasis, and smooth muscle relaxation by modulating the activity of protein kinases211 and ion channels.212,213 Recently, it has been discovered that these cyclic mononucleotides are also found in bacteria where they regulate important processes.214,2155.1 Metabolism of cAMP and cGMP in bacteria
Adenylyl and guanylyl cyclases catalyze the synthesis of cAMP and cGMP respectively (Scheme 3), both in prokaryotes and eukaryotes. They represent the Class III nucleotidyl cyclases family. The catalytic domain Cya2 of the nucleotidyl cyclase from Cyanobacterium S. sp. PCC6803, the only known bacterial guanylyl cyclase, has higher specific activity for synthesizing cGMP than cAMP.216 Cya2 and all eukaryotic guanylyl cyclases are closely related to type III adenylyl cyclases, and, in fact, mutations in just a few residues can change substrate specificities of these cyclases.217,218 The hydrolysis of cAMP and cGMP is catalyzed by cAMP- and cGMP-specific phosphodiesterases respectively (Scheme 3). |
| | Scheme 3 Syntheses and degradations of cAMP and cGMP. | |
5.2 cAMP signaling in bacteria
The transcription factor, cAMP-receptor protein (Crp), also called catabolite gene activator protein (Cap), is one of the best studied cAMP-receptor proteins in bacteria.219–221 This homodimeric protein controls the transcription of more than 100 promoters. Crp is activated by the allosteric binding of cAMP to its N-terminal region which triggers a conformational change in the tertiary structure of the protein.222 This change induces binding of the C-terminal DNA-binding domain of Crp to sequence-specific DNA sites in or near the promoter sequences, enabling transcription through direct protein–protein interactions with RNA polymerase.223 One well-studied example of the role of the cAMP–Crp complex is the mediation of ‘glucose response’ or catabolite repression in E. coli.221,224 At low glucose concentrations, the cAMP–Crp complex activates the lac operon, which encodes proteins that allow lactose to be used as a secondary carbon source. During catabolite repression (high glucose concentration), however, cAMP concentration decreases, thereby resulting in the inhibition of the expression of lac operon.225 Furthermore, cAMP also appears to play a central role in biofilm-regulation, type III-secretion, and virulence gene-expression in many bacteria.226 For example, during glucose limitation in V. cholerae, cAMP–Crp regulates biofilm formation both directly by down-regulating CdgA, a protein that produces c-di-GMP, and indirectly by up-regulating genes required for the biosynthesis of HapR, the major QS regulator.227,228 In P. aeruginosa, under low Ca2+ concentration, cAMP-Vfr (a Crp homolog) was found to regulate the expression of the type III secretion system (T3SS) by regulating the transcription of exoenzyme S synthesis regulatory protein (ExsA).229 More recently, an interesting but complex example of cAMP mediated virulence regulation has been demonstrated in M. tuberculosis that involves the participation of 10 active adenylyl cyclases (ACs).230 Moreover, cAMP signaling in M. tuberculosis involves the binding of cAMP to two transcription factors, Cmr (regulate intra-macrophage gene expression)231 and Crp (regulate virulence),232 and one protein acetylase, Rv0998 (regulate lysine acetylation).233 To complicate matters further, M. tuberculosis possesses ten putative cAMP binding proteins in addition to its complex array of ACs.2305.3 cAMP and QS
cAMP plays a major role in the regulation of quorum sensing. For example, in V. cholerae, the cAMP–Crp complex is required for the biosynthesis of CAI-1 via activation of cqsA, and it also indirectly affects the expression of QS regulator HapR through Fis and RpoS (Fig. 33).228,234 An interesting example where cAMP and QS collectively accomplish a common goal is in the V. cholerae O1 strains where they work in concert to enable the production of Hcp, an important component of type VI secretion system (T6SS).235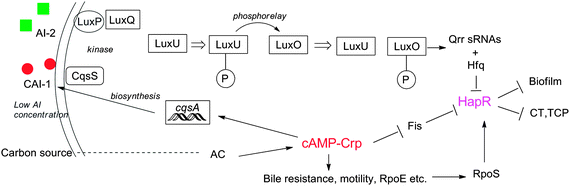 |
| | Fig. 33 Relationship between cAMP and QS in V. cholerae. | |
cAMP–Crp complex serves a central role in integrating external signals from CAI-1, AI-2 and carbon-starvation via adenylate cyclase (AC). In addition, cAMP–Crp reduces HapR production by repressing Fis at low cell density, and promotes HapR production at high cell density by interacting with the stress regulator RpoS. It also enhances bile resistance, motility and promotes the biosynthesis of RpoE, an alternative sigma factor.
In P. aeruginosa, Vfr, a Crp homolog, regulates the expression of virulence-related genes and also genes belonging to the las QS system (AI-1 controlled).236 Interestingly, Vfr was observed to regulate the expression of the lasR promoter in an cAMP-independent manner.236 It has been demonstrated that cAMP is involved in the interaction between the AI-1 and AI-2-mediated QS networks in E. coli.237 Other examples of crosstalk between cAMP signaling and QS include a report by the Bassler group that reveals a relationship between the internalization of AI-2 and the cAMP–Crp complex,238 and Bentley group's demonstration that cAMP–CRP affects both the synthesis and uptake of extracellular AI-2 in E. coli.239
5.4 cGMP signaling in bacteria
Although a considerably large body of work on cAMP signaling in bacteria has been performed, the biology of cGMP in bacteria remains largely unexplored. Early efforts to identify a cGMP-signaling machinery and understand the role of this nucleotide in bacterial signal transduction resulted in contradictory conclusions.215 For example, it was originally thought that cGMP plays a role in chemotaxis of E. coli, however, these experiments were deemed irreproducible.240 A fresh lease of life was provided to the field in 2000 with the identification of the gene cya2, which encodes guanylyl cyclase (GC) in the S. sp. PCC6803.241 Furthermore, deletion of the cya2 gene was shown to reduce cGMP levels exclusively, and had no impact on the cAMP levels. This finding was seminal as it uncovered a bona fide cGMP biosynthesis machinery in bacteria, and was in contrast to a previous study in E. coli that found a concomitant decrease in both cAMP and cGMP levels upon deletion of potential GC genes.242 Subsequent structure determination and biochemical experiments confirmed that Cya2 is indeed a guanylyl cyclase.216 Although the role of cGMP signaling in bacteria remains poorly understood, a recent report from Bauer's group showed that a R. centenum strain that contains the deletion of the guanylyl cyclase gene is deficient in cyst formation and when supplemented with cGMP regains its cyst forming ability.2435.5 cGMP and QS
cGMP was recently discovered in bacteria as a signaling molecule so the details of this particular second messenger are only beginning to emerge. It will not be surprising if in future, a link is found between cGMP signaling and quorum sensing in bacteria.5.6 Detection of cGMP and cAMP
Since the biology of cAMP and cGMP signaling in higher order organisms has been extensively studied, detection methods of these nucleotides in eukaryotic cells is extremely well developed.244,245 Detection methodologies include column chromatography, HPLC, ELISA and fluorescence resonance energy transfer (FRET). Several current methodologies utilize FRET-based approaches, which involve labeling proteins with suitable fluorescent probes. FRET-based approaches are suitable to detect physiological concentrations of these nucleotides (the physiological range of cAMP is 0.1–100 μM). Upon binding the nucleotides, these proteins undergo a conformational change, resulting in a change in the distance between two fluorophores, resulting in a change in the efficiency of FRET between them (Fig. 34). Since the initial demonstration of FRET-based sensors by Tsien and co-workers,246 significant technical advances have occurred, most notably the FRET detection method developed by Jalink and co-workers, which allows for both fluorescence lifetime imaging (FLIM) and sensitized emission (SE).247 Although most of these detection methods have been developed for detecting cAMP and cGMP in eukaryotes, they should be amenable for studying cAMP and cGMP in bacterial cells as well. |
| | Fig. 34 FRET based detection of second messengers. | |
6. Overlap in the various signaling pathways
6.1 Connectivity between signaling molecules in V. cholerae227,248,249
Fig. 35.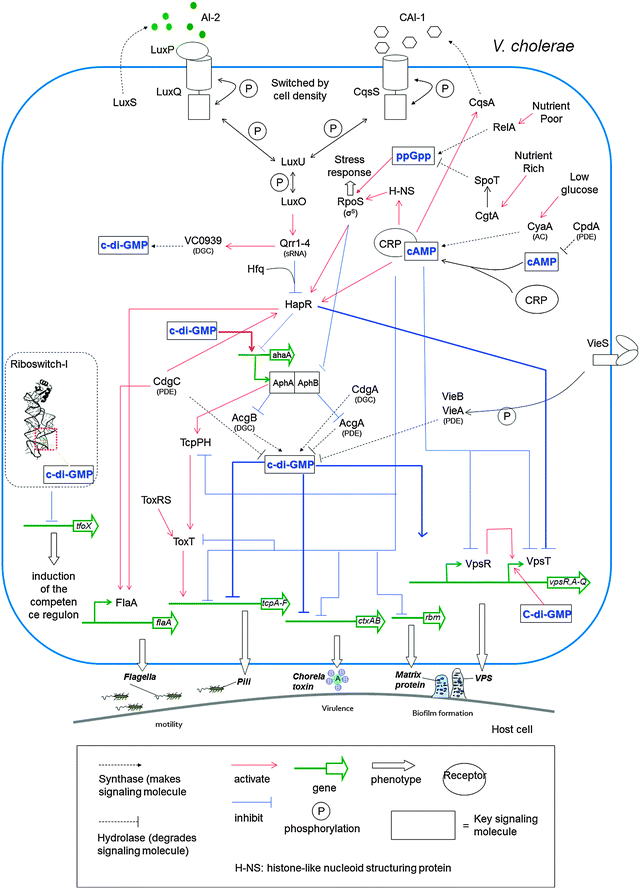 |
| | Fig. 35 When cell density is low, HapR (master regulator of virulence gene and biofilm formation) activity is reduced via mRNA degradation.250 At high cell density, HapR is not degraded and is activated by CRP–cAMP complex.227 This CRP–cAMP also activates CqsA, which produces CAI-1.234 The CRP–cAMP complex inhibits VpsR and VpsT, which activate vps gene. C-di-GMP inhibits the expression of the ctx and tcp genes and activates vps expression.248 Also c-di-GMP binds to riboswitch, which is upstream of the tfoX gene.94 | |
6.2 Connectivity between signaling molecules in E. coli251
Fig. 36.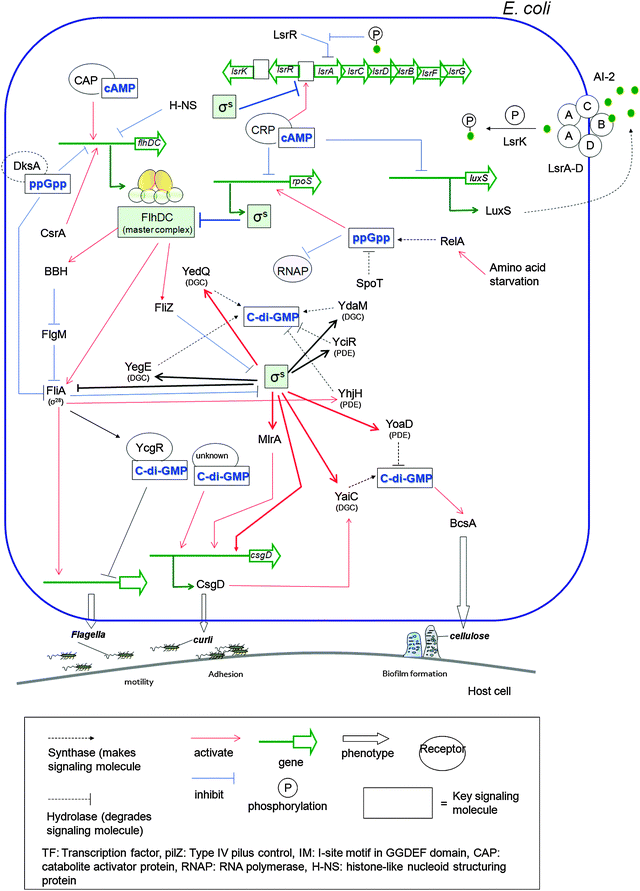 |
| | Fig. 36 FlhDC is a motility master complex, which controls motility and σs is adhesive curli fimbriae regulator. These two systems regulate diverse processes.251 YegE and YedQ have DGC activity and YhjH is a PDE. C-di-GMP binds to YcgR (which has PilZ domain66 and negatively control the flagella phenotype). Also c-di-GMP positively controls csgD via binding to unknown effector. σS controls YaiC (DGC) and YoaD (PDE) when the bacteria enter stationary phase. BcsA is a cellulose synthase. FliZ works as timing factor and regulate the expression of certain genes that are controlled by σS (ex. mlrA and ydaM genes). MlrA activates csgD transcription. Expression of flhDC is affected by cAMP–catabolite activator protein (CAP).252 Also cAMP–cAMP receptor protein (CRP) inhibits rpoS transcription, which produces σS.253,254 cAMP–CRP complex also inhibits transcription of luxS, which makes LuxS (AI-2 synthase) and increase the transcription of lsr gene expression (lsrA, B, C, D, F, G) which are involved in uptake of AI-2.239 (p)ppGpp is needed to reach higher rpoS expression levels255 and affect RNAP (RNA polymerase).256 Also ppGpp/DksA inhibits flhDC promoter and FliA (σ28).257 | |
6.3 Connectivity between signaling molecules in B. subtilis
Fig. 37.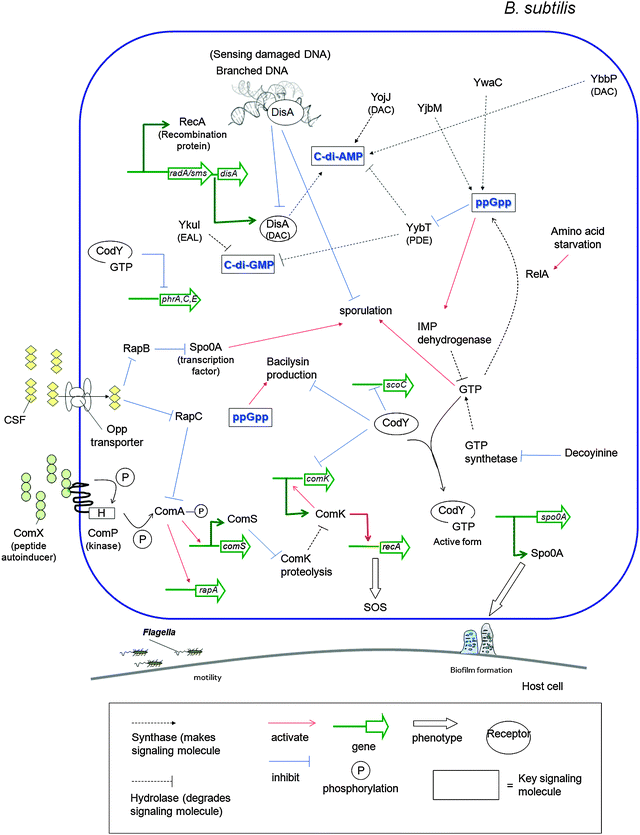 |
| | Fig. 37 C-di-AMP is synthesized by DisA.208 Also c-di-AMP is synthesized by YbbP and YojJ. DisA is involved in signaling DNA damage. RecA is a recombination protein.258 YybT, containing DHH/DHHA1 domain, hydrolyzes c-di-AMP and c-di-GMP to generate pApA and pGpG respectively.208 CodY is a GTP-binding protein.259 ComK binds comK promoter directly to activate comK expression.260 ComK activate recA, which is SOS gene.261 RelA protein reduces intracellular GTP levels.262 Bacilysin is produced by B. subtilis as peptide antibiotics.263 Bacilysin production is inhibited by CodY.264,265B. subtilis uses peptide autoinducers (ComX and CSF), which regulate competence and sporulation processes.266,267 ComX interacts with ComP sensor kinase to get phosphorylated, which in turn phosphorylates ComA. Phosphorylated ComA activates comS transcription. ComS inhibits ComK proteolysis. Second autoinducer CSF (competence and sporulation factor) inhibits RapC, which is inhibitor of phosphorylated ComA. RapB dephosphorylate Spo0A. The phosphorylated Spo0A regulates sporulation and biofilm formation.268,269 | |
7. Nucleotides as potential drug
7.1 C-di-GMP as an anti-biofilm drug
As already stated in this review, increased intracellular concentration of c-di-GMP facilitates biofilm formation in many bacteria and so it would be expected that the addition of c-di-GMP to bacterial cultures would cause biofilm formation. Yet, several reports have revealed that the addition of c-di-GMP to bacterial cells resulted in a biofilm attenuation. For example, Karaolis and co-workers demonstrated that the in vitro addition of c-di-GMP (>20 μM) to S. aureus, reduced biofilm formation by over 50%.270 Significantly, in vivo experiments also confirmed that c-di-GMP could be used to inhibit S. aureus biofilm formation in mice.271 This observation can be accounted for by the immunomodulatory activity of c-di-GMP. Wu and co-workers also found that the addition of c-di-GMP (200 μM) to S. mutans inhibited biofilm formation by over 50%.272 In an interesting study, Ohta and co-workers demonstrated that addition of low micromolar concentrations of c-di-GMP (<0.2 μM) to an S. aureus strain that lacked the ability to make c-di-GMP (a GGDEF domain mutant) induced biofilm formation whereas at higher c-di-GMP concentration (20 and 200 μM), biofilm formation by S. aureus and P. aeruginosa were suppressed.273 These observations clearly contradict our current understanding of the role of c-di-GMP in biofilm formation. One explanation for this unexpected observation is that externally added c-di-GMP is unable to enter bacterial cells, and the observed biofilm attenuation is due to binding of c-di-GMP to an extracellular receptor. Further work is clearly needed to explain these intriguing observations.7.2 C-di-GMP as an anticancer reagent
In 1995, Amikan and co-workers showed that extracellular c-di-GMP could enter Molt 4 cells and bind to p21ras.274 In a follow-up paper, the same group showed that addition of c-di-GMP to CD4+ Jurkat cells induced the expression of CD4 receptors to a level of up to 6.3 fold higher than in control cells.275 C-di-GMP markedly decreased cell division and blocked the cell cycle at the S phase. Because cGMP, GMP and GTP did not replicate these effects, it was concluded that the effects seen upon the addition of c-di-GMP to Jurkat cells were c-di-GMP specific.More than a decade after Amikan's important observations, Karaolis and co-workers showed that c-di-GMP arrests both basal and growth factor-stimulated proliferation of human colon cancer cells H508, whereas cGMP and 5′-GMP were not as effective.276 Since c-di-GMP did not affect acetylcholine- and EGF-induced activation of the p44/42 MAPK signaling cascade, it is unlikely that c-di-GMP inhibited cell proliferation by disrupting ligand–receptor interactions. Instead, it is more likely that c-di-GMP blocked cell cycle as Amikam showed in Jurkat cells.276
7.3 Cyclic (c-di-GMP, c-di-AMP, c-di-IMP) and linear nucleotides (pppGpp, ppGpp) as immune modulators
Recent studies have shown that c-di-GMP is a “danger” signal recognized by eukaryotic cells277 and therefore the immunomodulatory and immunostimulatory properties of c-di-GMP make it a potential vaccine adjuvant.278–280 The mechanism of the immune modulation by c-di-GMP is not yet clear, but some progress has been made towards revealing the mechanism by which c-di-GMP stimulates the host immune response. McWhirter and co-workers showed some evidence for c-di-GMP inducing host type I interferon response in cytosol via induction of TBK1, IRF3, nuclear factor NFκB and MAP kinases, but the sensor of c-di-GMP was not identified.281 Vance and co-workers found that a single nucleotide mutation of STimulator of INterferon Genes (STING) failed to produce type I interferons (IFNs) in a murine model upon L. monocytogenes infection. STING, also called MITA, MYPS and ERIS, is a transmembrane protein located at endoplasmic reticulum.282 Analysis of isolated macrophages revealed that STING is required for the type I interferon response to both c-di-GMP and c-di-AMP.283 Recently they showed further evidence that STING could selectively bind c-di-GMP (but not GMP, cGMP, ppGpp), demonstrating that STING functioned as a direct sensor of cyclic dinucleotides (Fig. 38).284 |
| | Fig. 38 Mechanism of c-di-GMP/c-di-AMP induced innate immune response in macrophage. (a) The bacteria are taken by the macrophage via endocytosis; (b) phagosome and lysosome merge into a big phagolysosome; (c) the bacteria are digested by the enzymes; (d, e) big fragments of bacteria are kept inside of the phagolysosome and eventually discharged by exocytosis; (f) c-di-GMP and c-di-AMP are secreted into the cytosol with the help of multidrug efflux pumps;281,285 (g) upon c-di-GMP/c-di-AMP binding, STING dimer associates with Tank-binding kinase 1(TBK1); (h, i) TBK1 activates IFN regulatory factor 3 (IRF3) and IRF7, ultimately stimulates the type I interferon gene expression.283,284 | |
C-di-AMP has also been found to trigger type I interferon in the cytosol of host immune cells. c-di-AMP is secreted into the cytosol of host by intracellular pathogens, via the multidrug efflux pumps and induced the innate immune responses (Fig. 38).285 Another member of the cyclic di-nucleotide family, c-di-IMP (which is not a natural signaling molecule), showed potent adjuvant property.286 Co-administration of c-di-IMP led to significant immune responses, such as higher immunoglobulin IgG in sera. ppGpp and pppGpp also have immunomodulatory activities and the co-administration of these nucleotides with antigen (HBsAg) generated fast and strong host immune responses.287
8. Synthesis of nucleotide signaling molecules and analogs
8.1 C-di-GMP synthesis
It is close to half a century since Khorana's group observed the formation of cyclic dinucleotides during the synthesis of polynucleotides.288,289 In the 80s, Dennis and Jones developed a phosphotriester method to make both homo- and heterocyclic dinucleotides.290,291 Subsequently there have been refinements of the earlier synthesis of c-di-GMP and now the most commonly used strategies to make cyclic dinucleotides are either solid phase (see Scheme 4)98,292 or solution phase (see Scheme 5).138,293–295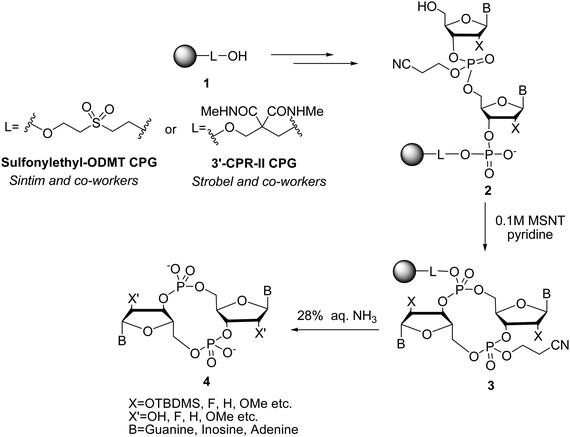 |
| | Scheme 4 Solid-phase synthesis strategy of c-di-GMP. | |
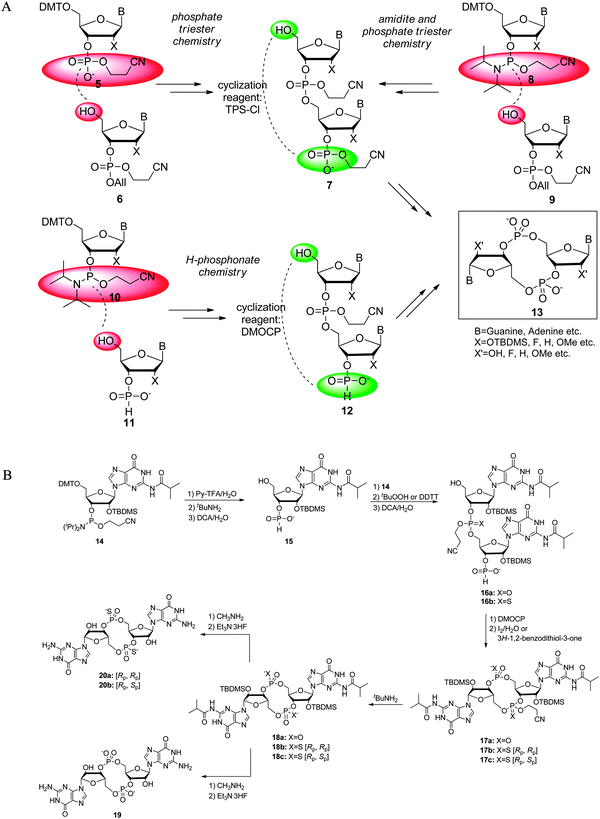 |
| | Scheme 5 (A) Solution-phase synthesis strategies to prepare c-di-GMP, including phosphate triester291,293 and H-phosphonate chemistry.138,295 Red background: functional groups that are involved in the linear coupling step (one step before cyclization); green backgroud: functional groups that are involved in the cyclization step. TPS-Cl: 2,4,6-triisopropylbenzenesulfonyl chloride. DMOCP: 2-chloro-5,5-dimethyl-1,3,2-dioxaphosphorinane 2-oxide. (B) Details of H-phosphonate chemistry used by Jones to synthesize c-di-GMP and thiophosphate analogs.299 DCA: dichloroacetic acid; DDTT: 3-((dimethylaminomethylidene)-amino)-3H-1,2,4-dithiazole-5-thione. (Adapted from ref. 299 with permission. Copyright 2010, American Chemical Society.) | |
In the mid-90s the synthesis of cyclic RNAs on a solid support was reported by Piccialli.296 However, Piccialli's synthesis was only amenable to synthesis of cyclic nucleotides that contained cytosine. In 1997, Pedroso and co-workers reported an improved solid phase synthesis method that did not require a particular nucleobase to be present in the cyclic nucleotide.297 Kool described a very interesting and simple solid phase synthesis of cyclic nucleotides containing phosphorothiate linkages.298 Inspired by these earlier works, Sintim and co-workers developed a simple solid-phase synthesis of c-di-GMP using an automated DNA synthesizer (Scheme 4). In this synthesis, sulfonylethyl-ODMT CPG was used and two different phosphoramidites (p-methoxyphosphoramidite and cyanoethyl phosphate protected phosphoramidite) were chosen and coupled to the solid support.292 After the selective deprotection on the phosphate group of the first coupled phosphoramidite, cyclization was carried out by using 0.1 M 1-mesitylenesulfonyl-3-nitro-1, 2, 4-triazole (MSNT) in pyridine. Jones and Strobel96,98 later used a modified solid-phase synthesis strategy to make several c-di-GMP analogs (see Scheme 4).
Solid-phase synthesis is relatively straightforward; however, it is not the method of choice to make c-di-GMP on a gram scale. Solution-phase synthesis of c-di-GMP can be categorized into two major strategies: (a) phosphate triester chemistry291,293 and (b) H-phosphonate chemistry (Scheme 5).138,295 All these syntheses rely on air or water sensitive reagents, which makes the synthesis of c-di-GMP via these approaches non-trivial. Recently Jones reported that the entire synthesis of c-di-GMP could carried out in one flask.299 Our group has performed all of the reported solution phase syntheses of c-di-GMP and in our opinion, Jones' new method is the best solution-phase method reported to date and recommend this to groups that are interested in making gram-quantity c-di-GMP or analogs.
Enzymatic synthesis of c-di-GMP, using DGC and GTP, has also been described by several groups.300,301 Synthetic approaches that enable generation of large quantities of c-di-GMP (gram quantities) or analogs of c-di-GMP using these enzymatic approaches remain to be developed.
8.2 Chemical syntheses of cAMP, cGMP, ppGpp
The chemical syntheses of cyclic AMP and cyclic GMP were reported in the 1960s (Scheme 6). The most commonly used synthetic route towards these mononucleotides is via the phosphorylation of unprotected nucleoside,302 followed by the cyclization with N,N′-dicyclohexylcarbodiimide (DCC)303 or potassium hydroxide (see Scheme 6).304 |
| | Scheme 6 Chemical synthesis of cyclic AMP and cyclic GMP. | |
The first two examples of chemical syntheses of ppGpp were reported by Tomasz in 1974305 and Hecht in 1975 (Scheme 7).306 A common theme used in these syntheses was the protection of the 2′-OH (see transformation 23 to 24 in Scheme 7), followed by the addition of pyrophosphate groups to the 3′- and 5′-hydroxyl groups and a final removal of the 2′-protecting group to give compound 25 in Scheme 7.
 |
| | Scheme 7 Chemical synthesis of ppGpp.305,306 | |
8.3 Synthetic analogs of c-di-GMP and ppGpp
Considering the vital role of signaling molecules in bacterial physiology and virulence, analogs of these compounds have been synthesized270,271,307,308 and are being considered as antibacterial agents. Moreover, the enzymes that are involved in the biosynthesis of these nucleotide second messengers (c-di-GMP, c-di-AMP and ppGpp) in bacteria are not present in humans and therefore analogs that inhibit these enzymes could potentially disrupt biofilm formation and virulence factors production in bacteria. The caveat though is that these analogs would have to be cell permeable and so far this issue has not been adequately addressed. Structures of c-di-GMP and ppGpp analogs that have been synthesized and the respective binding, inhibitory constants or IC50s with various binding proteins are shown in Tables 5–9. These values indicate that modifications of both c-di-GMP or ppGpp lead to analogs that have lower affinity for the binding receptors, compared to c-di-GMP or ppGpp.
Table 5 C-di-GMP analogs with phosphate or 2′ modification
| Name | Modification | Kinetic and thermodynamic constants |
|---|
| IC50 was determined by competing radiolabeled c-di-GMP (0.8 nM) with various analog concentrations. The protein concentrations are as follows: Alg44 = 2.5 μM, WspR = 10 μM, RocR = 2.5 μM.100 |
|---|
| C-di-GMP | | IC50: 7.69 μM (Alg44)a,100 |
| IC50: 21 nM (RocR)a,100Km: 0.25 μM (PDE-A),309 relative Ki: 1.0 (PDE-A)309 60% inhibition (PDE-A)309 |
| IC50: 48.91 μM (WspR)a,100 ∼20% inhibition (Slr1143)308 |
| Class-I riboswitch Kd: 2.2 ± 0.8 nM,100 class-II Kd: 2.2 ± 0.2 nM100 |
| Endo-(S)-c-di-GMP |  | IC50: 81.53 μM (Alg44)a,100 IC50: 140 nM (RocR)a,100 IC50: >100 μM (WspR)a,100 |
| 2′-Biotin-endo-(S)-c-di-GMP | 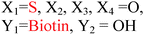 | IC50: >100 μM (Alg44)a,100 IC50: 1.88 μM (RocR)a,100 IC50: >100 μM (WspR)a,100 |
| 2′-Biotin-AHC-c-di-GMP | 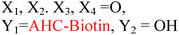 | IC50: >82.41 μM (Alg44)a,100 IC50: 2.73 μM (RocR)a,100 IC50: >100 μM (WspR)a,100 |
| cGp(S)Gp (S-diastereomer) | 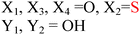 | 55% inhibition (PDE-A),309 relative Ki: 0.3 (PDE-A)309 |
| cGp(S)Gp (R-diastereomer) | 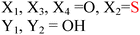 | 15% inhibition (PDE-A),309 relative Ki: 3.2 (PDE-A)309 |
| C-(RpSp)di-Gps | 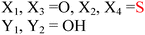 | Class-I riboswitch Kd: 750 ± 63 nM,98 class-II Kd: 4.0 ± 0.8 nM98 |
| C-(RpRp)di-Gps | 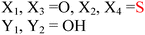 | Class-I riboswitch Kd: 150 ± 33 nM,98 class-II Kd: 3.6 ± 0.6 nM98 |
| C-dGpGp | 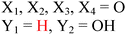 | 55% inhibition (PDE-A),309 relative Ki: 0.3 (PDE-A),309 class-I riboswitch Kd: 51 ± 5.4 nM98 |
| C-di-dGMP | 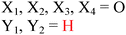 | 50% inhibition (PDE-A),309 relative Ki: 1.0 (PDE-A),309 class-I riboswitch Kd: 2600 ± 88 nM,98 class-II Kd: 11 ± 1.2 nM98 |
| 2′-O-methyl c-di-GMP | 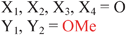 | Class-I riboswitch Kd: ∼60000 nM,96 class-II Kd: 4.3 ± 0.7 nM96 |
| C-2′-OMe-G-GMP | 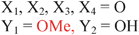 | Class-I riboswitch Kd: 200 ± 31 nM98 |
| c-di-2′F-GMP | 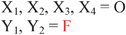 | Class-I riboswitch Kd: 56 ± 9.0 nM class-II Kd: 5.8 ± 1.0 nM98 |
Table 6 C-di-GMP analogs with base modifications
| Name | Modification | Kinetic constant |
|---|
| C-di-IMP | Base = inosine, inosine | 15% Inhibition (PDE-A),309 relative Ki: 4.6 (PDE-A),309 ∼60% inhibition (Slr1443),308 class-I riboswitch Kd: 1100 ± 210 nM,98 Class-II Kd: 4.1 ± 0.4 nM98 |
| C-di-UMP | Base = uracil, uracil | Relative Ki: 180 (PDE-A),309 ∼−5% inhibition (Slr1443)308 |
| C-GMP-UMP | Base = guanine, uracil | Relative Ki: 180 (PDE-A),309 ∼−30% inhibition (Slr1443)308 |
| C-IMP-UMP | Base = inosine, uracil | ∼25% Inhibition (Slr1443)308 |
| C-IMP-GMP | Base = inosine, guanine | 50% Inhibition (PDE-A),309 relative Ki: 1.6 (PDE-A),309 class-I riboswitch Kd: 39.0 ± 2.4 nM98 |
| C-XMP-GMP | Base = xanthine, guanine | 55% Inhibition (PDE-A),309 relative Ki: 1.1 (PDE-A)309 |
| C-di-AMP | Base = adenine, adenine | Relative Ki: 3.2 (PDE-A),309 class-II Kd: >30,000 nM98 |
| C-di-CMP | Base = cytosine, cytosine | Relative Ki: 180 (PDE-A)309 |
| C-di-XMP | Base = xanthine, xanthine | 25% Inhibition (PDE-A),309 relative Ki: 5.7 (PDE-A)309 |
| C-CMP-GMP | Base = cytosine, guanine | Relative Ki: 15.6 (PDE-A)309 |
| C-AMP-GMP | Base = adenine, guanine | Class-I riboswitch Kd: 1600 ± 160 nM,98 class-II Kd: 271.1 ± 47.1 nM98 |
Table 7 C-di-GMP analogs with various guanine modifications
| Name | Modification | Kinetic constant |
|---|
| C-c7G-GMP | 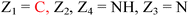 | Class-I riboswitch Kd: 1100 ± 80 nM,98 class-II Kd: 33.0 ± 2.6 nM98 |
| C-di-c7GMP | 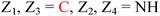 | Class-II Kd: 3500 ± 630 nM98 |
| C-N1mG-GMP | 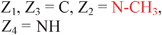 | Class-I riboswitch Kd: 420 ± 16 nM,98 class-II Kd: 160 ± 14 nM98 |
Table 8 Linear di-nucleotides
| Name | Modification | Kinetic constant |
|---|
| GpG | — | Class-I riboswitch Kd: 210 ± 24 nM99 |
| PGpG | — | Class-I riboswitch Kd: 470 ± 150 nM99 |
| GpGp | — | Class-I riboswitch Kd: 22000 ± 14000 nM99 |
Table 9 ppGpp analogs with various modifications
| Name | Modification | Kinetic constant |
|---|
| % is eye measurement. |
|---|
| ppGpp (1) | X1, X2 = O, Y3 = OH, Z1 = NH2 | Relative Ki: 9.3 (PDE-A)309 |
| ppGpp analog (2) |  | 15% Inhibitiona,310 (RelA) |
| ppGpp analog (3) | 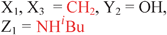 | 5% Inhibitiona,310 (RelA) |
| ppGpp analog (4) |  | 20% Inhibitiona,310 (RelA) |
| ppGpp analog (5) | 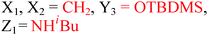 | 50% Inhibitiona,310 (RelA) |
| ppGpp analog (6) |  | 0% Inhibitiona,310 (RelA), 30% inhibitiona,310 (Relseq385) |
| ppGpp analog (7) |  | 17% Inhibitiona,310 (RelA), 80% inhibitiona,310 (Relseq385) |
| ppGpp analog (8) | 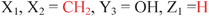 | 45% Inhibitiona,310 (RelA), 60% inhibitiona,310 (Relseq385) |
| ppGpp analog (9) | 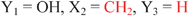 | 10% Inhibitiona,310 (RelA), 75% inhibitiona,310 (Relseq385) |
| ppGpp analog (10) | 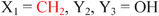 | 0% Inhibitiona,310 (RelA), 10% inhibitiona,310 (Relseq385) |
| ppGpp analog (11) | 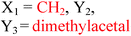 | 25% Inhibitiona,310 (RelA), 30% inhibitiona,310 (Relseq385) |
9. Conclusion
It is now widely acknowledged that both quorum sensing molecules and nucleotides play important roles in bacterial lifestyle. Quorum sensing regulation and ppGpp signaling in bacteria are better understood than c-di-GMP, c-di-AMP, cAMP and cGMP signaling. Although the proteins that regulate the intracellular concentrations of these second messengers are well characterized, the intermediary proteins and RNAs that bind to these nucleotides to regulate bacterial phenotype are far from being well characterized and the environmental cues that modulate the concentrations of these nucleotides are also not well understood. Most studies on these nucleotides have concentrated on extracellular pathogens, although recent reports have demonstrated that intracellular pathogens also utilize these nucleotides for host colonization and adaptation.281,283,285 Future work will continue to lead to the identification new binding partners of these nucleotides and provide a clearer understanding of how these nucleotides integrate with the various components to regulate the bacterial lifestyle. Another dinucleotide, pGpG (the breakdown product of c-di-GMP) has so far been treated as a benign molecule. However, pGpG and analogs has been shown to bind to c-di-GMP riboswitches at physiological concentrations, albeit with less affinity than c-di-GMP.97,311,312 This hints that pGpG could also regulate some riboswitches and even protein effectors. pGpG, being a nanoRNA could play other roles in bacteria and future directions should look at pGpG signaling. We also expect future work to provide more direct links between the various nucleotide second messengers as well as quorum sensing. The redundancy of the second messenger networks provides a great challenge to the medicinal chemist who is interested in developing small molecules to disrupt bacterial biofilm or virulence factors production. For example, how does one design a small molecule that inhibits all >50 DDGEF domain proteins in say S. oneidensis, which could potentially make c-di-GMP? Recent studies that report the discovery of small molecules that inhibit c-di-GMP signaling and quorum sensing in bacteria provide optimism for the future.313,314 We have indeed entered a golden period of small molecule control of bacterial lifestyle.154,313,315–318 Bacteria have been caught cold – increasingly, we are beginning to figure out the majority of the tactics used by these bugs. The biologists have done excellent jobs unravelling bacteria. Now the baton has to be passed on to the medicinal chemists to utilize all of these information to design the elusive anti-biofilm and anti-virulence drugs.Acknowledgements
We thank the NSF (CHE0746446), Camille Dreyfus foundation, GANN fellowship (to BTR) and the American Heart Association (to JZ; 11PRE7890040) for generous support for our program.Notes and references
- P. Ross, H. Weinhouse, Y. Aloni, D. Michaeli, P. Weinberger-Ohana, R. Mayer, S. Braun, E. de Vroom, G. A. van der Marel, J. H. van Boom and M. Benziman, Nature, 1987, 325, 279–281 CrossRef CAS
 .
. - G. Witte, S. Hartung, K. Buttner and K. P. Hopfner, Mol. Cell, 2008, 30, 167–178 CrossRef CAS .
- R. W. Bernlohr, M. K. Haddox and N. D. Goldberg, J. Biol. Chem., 1974, 249, 4329–4331 CAS .
- R. S. Makman and E. W. Sutherland, J. Biol. Chem., 1965, 240, 1309–1314 CAS .
- M. Cashel and B. Kalbacher, J. Biol. Chem., 1970, 245, 2309–2318 CAS .
- L. U. Magnusson, A. Farewell and T. Nyström, Trends Microbiol., 2005, 13, 236–242 CrossRef CAS .
- A. Camilli and B. L. Bassler, Science, 2006, 311, 1113–1116 CrossRef CAS .
- B. L. Bassler, Cell, 2002, 109, 421–424 CrossRef CAS .
- U. Jenal and J. Malone, Annu. Rev. Genet., 2006, 40, 385–407 CrossRef CAS .
- U. Römling, M. Gomelsky and M. Y. Galperin, Mol. Microbiol., 2005, 57, 629–639 CrossRef .
- R. Hengge, Nat. Rev. Microbiol., 2009, 7, 263–273 CrossRef CAS .
- M. Cashel, Annu. Rev. Microbiol., 1975, 29, 301–318 CrossRef CAS .
- H. Yan and W. Chen, Chem. Soc. Rev., 2010, 39, 2914–2924 RSC .
- M. Gomelsky, Mol. Microbiol., 2011, 79, 562–565 CrossRef CAS .
- J. Berthet, T. W. Rall and E. W. Sutherland, J. Biol. Chem., 1957, 224, 463–475 CAS .
- D. F. Ashman, R. Lipton, M. M. Melicow and T. D. Price, Biochem. Biophys. Res. Commun., 1963, 11, 330–334 CrossRef CAS .
- U. Jenal, Curr. Opin. Microbiol., 2004, 7, 185–191 CrossRef CAS .
- A. Boehm, M. Kaiser, H. Li, C. Spangler, C. A. Kasper, M. Ackermann, V. Kaever, V. Sourjik, V. Roth and U. Jenal, Cell, 2010, 141, 107–116 CrossRef CAS .
- K. Paul, V. Nieto, W. C. Carlquist, D. F. Blair and R. M. Harshey, Mol. Cell, 2010, 38, 128–139 CrossRef CAS .
- X. Fang and M. Gomelsky, Mol. Microbiol., 2010, 76, 1295–1305 CrossRef CAS .
- P. D. Newell, R. D. Monds and G. A. O'Toole, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3461–3466 CrossRef CAS .
- P. D. Newell, C. D. Boyd, H. Sondermann and G. A. O'Toole, PLoS Biol., 2011, 9, e1000587 CAS .
- S. Beyhan, L. S. Odell and F. H. Yildiz, J. Bacteriol., 2008, 190, 7392–7405 CrossRef CAS .
- S. Beyhan, A. D. Tischler, A. Camilli and F. H. Yildiz, J. Bacteriol., 2006, 188, 3600–3613 CrossRef CAS .
- B. Lim, S. Beyhan, J. Meir and F. H. Yildiz, Mol. Microbiol., 2006, 60, 331–348 CrossRef CAS .
- Z. T. Güvener and C. S. Harwood, Mol. Microbiol., 2007, 66, 1459–1473 Search PubMed .
- M. Christen, H. D. Kulasekara, B. Christen, B. R. Kulasekara, L. R. Hoffman and S. I. Miller, Science, 2010, 328, 1295–1297 CrossRef CAS .
- R. Tal, H. C. Wong, R. Calhoon, D. Gelfand, A. L. Fear, G. Volman, R. Mayer, P. Ross, D. Amikam, H. Weinhouse, A. Cohen, S. Sapir, P. Ohana and M. Benziman, J. Bacteriol., 1998, 180, 4416–4425 CAS .
- A. J. Schmidt, D. A. Ryjenkov and M. Gomelsky, J. Bacteriol., 2005, 187, 4774–4781 CrossRef CAS .
- R. P. Ryan, Y. McCarthy, M. Andrade, C. S. Farah, J. P. Armitage and J. M. Dow, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5989–5994 CrossRef CAS .
- G. B. Hecht and A. Newton, J. Bacteriol., 1995, 177, 6223–6229 CAS .
- N. De, M. V. Navarro, R. V. Raghavan and H. Sondermann, J. Mol. Biol., 2009, 393, 619–633 CrossRef CAS .
- E. Drenkard and F. M. Ausubel, Nature, 2002, 416, 740–743 CrossRef CAS .
- E. Huitema, S. Pritchard, D. Matteson, S. K. Radhakrishnan and P. H. Viollier, Cell, 2006, 124, 1025–1037 CrossRef CAS .
- Y. Qi, M. L. Chuah, X. Dong, K. Xie, Z. Luo, K. Tang and Z. X. Liang, J. Biol. Chem., 2011, 286, 2910–2917 CrossRef CAS .
- N. Bomchil, P. Watnick and R. Kolter, J. Bacteriol., 2003, 185, 1384–1390 CrossRef CAS .
- N. Ausmees, R. Mayer, H. Weinhouse, G. Volman, D. Amikam, M. Benziman and M. Lindberg, FEMS Microbiol. Lett., 2001, 204, 163–167 CrossRef CAS .
- R. Paul, S. Weiser, N. C. Amiot, C. Chan, T. Schirmer, B. Giese and U. Jenal, Genes Dev., 2004, 18, 715–727 CrossRef CAS .
- R. Paul, S. Abel, P. Wassmann, A. Beck, H. Heerklotz and U. Jenal, J. Biol. Chem., 2007, 282, 29170–29177 CrossRef CAS .
- J. G. Malone, R. Williams, M. Christen, U. Jenal, A. J. Spiers and P. B. Rainey, Microbiology, 2007, 153, 980–994 CrossRef CAS .
- B. Christen, M. Christen, R. Paul, F. Schmid, M. Folcher, P. Jenoe, M. Meuwly and U. Jenal, J. Biol. Chem., 2006, 281, 32015–32024 CrossRef CAS .
- P. C. Fineran, N. R. Williamson, K. S. Lilley and G. P. Salmond, J. Bacteriol., 2007, 189, 7653–7662 CrossRef CAS .
- C. Chan, R. Paul, D. Samoray, N. C. Amiot, B. Giese, U. Jenal and T. Schirmer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17084–17089 CrossRef CAS .
- D. A. Ryjenkov, M. Tarutina, O. V. Moskvin and M. Gomelsky, J. Bacteriol., 2005, 187, 1792–1798 CrossRef CAS .
- C. Y. Yang, K. H. Chin, M. L. Chuah, Z. X. Liang, A. H. Wang and S. H. Chou, Acta Crystallogr., 2011, D67, 997–1008 CAS .
- A. S. Seshasayee, G. M. Fraser and N. M. Luscombe, Nucleic Acids Res., 2010, 38, 5970–5981 CrossRef CAS .
- R. P. Ryan, Y. Fouhy, J. F. Lucey, L. C. Crossman, S. Spiro, Y. W. He, L. H. Zhang, S. Heeb, M. Cámara, P. Williams and J. M. Dow, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6712–6717 CrossRef CAS .
- M. Christen, B. Christen, M. Folcher, A. Schauerte and U. Jenal, J. Biol. Chem., 2005, 280, 30829–30837 CrossRef CAS .
- R. Tamayo, A. D. Tischler and A. Camilli, J. Biol. Chem., 2005, 280, 33324–33330 CrossRef CAS .
- T. R. Barends, E. Hartmann, J. J. Griese, T. Beitlich, N. V. Kirienko, D. A. Ryjenkov, J. Reinstein, R. L. Shoeman, M. Gomelsky and I. Schlichting, Nature, 2009, 459, 1015–1018 CrossRef CAS .
- A. Tchigvintsev, X. Xu, A. Singer, C. Chang, G. Brown, M. Proudfoot, H. Cui, R. Flick, W. F. Anderson, A. Joachimiak, M. Y. Galperin, A. Savchenko and A. F. Yakunin, J. Mol. Biol., 2010, 402, 524–538 CrossRef CAS .
- M. A. Larkin, G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson and D. G. Higgins, Bioinformatics, 2007, 23, 2947–2948 CrossRef CAS .
- J. Zhou, G. Yuan, J. Liu and C.-G. Zhan, Chem.–Eur. J., 2007, 13, 945–949 CrossRef CAS .
- A. L. Lovering, M. J. Capeness, C. Lambert, L. Hobley and R. E. Sockett, mBio, 2011, 2, e00163–11 CrossRef .
- R. B. Ferreira, L. C. Antunes, E. P. Greenberg and L. L. McCarter, J. Bacteriol., 2008, 190, 851–860 CrossRef CAS .
- M. Tarutina, D. A. Ryjenkov and M. Gomelsky, J. Biol. Chem., 2006, 281, 34751–34758 CrossRef CAS .
- M. Merighi, V. T. Lee, M. Hyodo, Y. Hayakawa and S. Lory, Mol. Microbiol., 2007, 65, 876–895 CrossRef CAS .
- H. Weber, C. Pesavento, A. Possling, G. Tischendorf and R. Hengge, Mol. Microbiol., 2006, 62, 1014–1034 CrossRef CAS .
- M. Y. Galperin, Environ. Microbiol., 2004, 6, 552–567 CrossRef CAS .
- F. H. Yildiz and K. L. Visick, Trends Microbiol., 2009, 17, 109–118 CrossRef CAS .
- M. Y. Galperin, A. N. Nikolskaya and E. V. Koonin, FEMS Microbiol. Lett., 2001, 203, 11–21 CrossRef CAS .
- K. M. Thormann, S. Duttler, R. M. Saville, M. Hyodo, S. Shukla, Y. Hayakawa and A. M. Spormann, J. Bacteriol., 2006, 188, 2681–2691 CrossRef CAS .
- M. Collin and R. Schuch, Bacterial Sensing and Signaling, Karger Publishers, New York, 2009 Search PubMed .
- F. Shang, T. Xue, H. Sun, L. Xing, S. Zhang, Z. Yang, L. Zhang and L. Sun, Infect. Immun., 2009, 77, 2849–2856 CrossRef CAS .
- R. Simm, M. Morr, A. Kader, M. Nimtz and U. Römling, Mol. Microbiol., 2004, 53, 1123–1134 CrossRef CAS .
- D. A. Ryjenkov, R. Simm, U. Römling and M. Gomelsky, J. Biol. Chem., 2006, 281, 30310–30314 CrossRef CAS .
- J. T. Pratt, R. Tamayo, A. D. Tischler and A. Camilli, J. Biol. Chem., 2007, 282, 12860–12870 CrossRef CAS .
- D. Amikam and M. Y. Galperin, Bioinformatics, 2006, 22, 3–6 CrossRef CAS .
- A. Duerig, S. Abel, M. Folcher, M. Nicollier, T. Schwede, N. Amiot, B. Giese and U. Jenal, Genes Dev., 2009, 23, 93–104 CrossRef CAS .
- T. Petters, X. Zhang, J. Nesper, A. Treuner-Lange, N. Gomez-Santos, M. Hoppert, U. Jenal and L. Søgaard-Andersen, Mol. Microbiol., 2012, 84, 147–165 CrossRef CAS .
- S. Rakshe, M. Leff and A. M. Spormann, Appl. Environ. Microbiol., 2011, 77, 2196–2198 CrossRef CAS .
- J. R. Tuckerman, G. Gonzalez and M. A. Gilles-Gonzalez, J. Mol. Biol., 2011, 407, 633–639 CrossRef CAS .
- J. W. Hickman and C. S. Harwood, Mol. Microbiol., 2008, 69, 376–389 CrossRef CAS .
- K. H. Chin, Y. C. Lee, Z. L. Tu, C. H. Chen, Y. H. Tseng, J. M. Yang, R. P. Ryan, Y. McCarthy, J. M. Dow, A. H. Wang and S. H. Chou, J. Mol. Biol., 2010, 396, 646–662 CrossRef CAS .
- J. L. Leduc and G. P. Roberts, J. Bacteriol., 2009, 191, 7121–7122 CrossRef CAS .
- J. J. Wilksch, J. Yang, A. Clements, J. L. Gabbe, K. R. Short, H. Cao, R. Cavaliere, C. E. James, C. B. Whitchurch, M. A. Schembri, M. L. Chuah, Z. X. Liang, O. L. Wijburg, A. W. Jenney, T. Lithgow and R. A. Strugnell, PLoS Pathog., 2011, 7, e1002204 CAS .
- N. Sudarsan, E. R. Lee, Z. Weinberg, R. H. Moy, J. N. Kim, K. H. Link and R. R. Breaker, Science, 2008, 321, 411–413 CrossRef CAS .
- E. R. Lee, J. L. Baker, Z. Weinberg, N. Sudarsan and R. R. Breaker, Science, 2010, 329, 845–848 CrossRef CAS .
- R. A. Alm, A. J. Bodero, P. D. Free and J. S. Mattick, J. Bacteriol., 1996, 178, 46–53 CAS .
- J. Benach, S. S. Swaminathan, R. Tamayo, S. K. Handelman, E. Folta-Stogniew, J. E. Ramos, F. Forouhar, H. Neely, J. Seetharaman, A. Camilli and J. F. Hunt, EMBO J., 2007, 26, 5153–5166 CrossRef CAS .
- J. Ko, K. S. Ryu, H. Kim, J. S. Shin, J. O. Lee, C. Cheong and B. S. Choi, J. Mol. Biol., 2010, 398, 97–110 CrossRef CAS .
- J. Habazettl, M. G. Allan, U. Jenal and S. Grzesiek, J. Biol. Chem., 2011, 286, 14304–14314 CrossRef CAS .
- M. Christen, B. Christen, M. G. Allan, M. Folcher, P. Jenö, S. Grzesiek and U. Jenal, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4112–4117 CrossRef CAS .
- C. Baraquet, K. Murakami, M. R. Parsek and C. S. Harwood, Nucleic Acids Res., 2012, 40, 7207–7218 CrossRef CAS .
- P. V. Krasteva, J. C. Fong, N. J. Shikuma, S. Beyhan, M. V. Navarro, F. H. Yildiz and H. Sondermann, Science, 2010, 327, 866–868 CrossRef CAS .
- N. J. Shikuma, J. C. Fong and F. H. Yildiz, PLoS Pathog., 2012, 8, e1002719 CAS .
- J. G. Johnson, C. N. Murphy, J. Sippy, T. J. Johnson and S. Clegg, J. Bacteriol., 2011, 193, 3453–3460 CrossRef CAS .
- M. V. Navarro, P. D. Newell, P. V. Krasteva, D. Chatterjee, D. R. Madden, G. A. O'Toole and H. Sondermann, PLoS Biol., 2011, 9, e1000588 CAS .
- S. Abel, P. Chien, P. Wassmann, T. Schirmer, V. Kaever, M. T. Laub, T. A. Baker and U. Jenal, Mol. Cell, 2011, 43, 550–560 CrossRef CAS .
- V. T. Lee, J. M. Matewish, J. L. Kessler, M. Hyodo, Y. Hayakawa and S. Lory, Mol. Microbiol., 2007, 65, 1474–1484 CrossRef CAS .
- M. V. Navarro, N. De, N. Bae, Q. Wang and H. Sondermann, Structure, 2009, 17, 1104–1116 CrossRef CAS .
- B. I. Kazmierczak, M. B. Lebron and T. S. Murray, Mol. Microbiol., 2006, 60, 1026–1043 CrossRef CAS .
- R. R. Breaker, Cold Spring Harbor Perspect. Biol., 2012, 4, a003566 CrossRef CAS .
- N. Kulshina, N. J. Baird and A. R. Ferré-D'Amaré, Nat. Struct. Mol. Biol., 2009, 16, 1212–1217 CAS .
- K. D. Smith, S. V. Lipchock, T. D. Ames, J. Wang, R. R. Breaker and S. A. Strobel, Nat. Struct. Mol. Biol., 2009, 16, 1218–1223 CAS .
- K. D. Smith, C. A. Shanahan, E. L. Moore, A. C. Simon and S. A. Strobel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7757–7762 CrossRef CAS .
- K. D. Smith, S. V. Lipchock, A. L. Livingston, C. A. Shanahan and S. A. Strobel, Biochemistry, 2010, 49, 7351–7359 CrossRef CAS .
- C. A. Shanahan, B. L. Gaffney, R. A. Jones and S. A. Strobel, J. Am. Chem. Soc., 2011, 133, 15578–15592 CrossRef CAS .
- K. D. Smith, S. V. Lipchock and S. A. Strobel, Biochemistry, 2012, 51, 425–432 CrossRef CAS .
- Y. Luo, J. Zhou, S. K. Watt, V. T. Lee, T. K. Dayie and H. O. Sintim, Mol. BioSyst., 2012, 8, 772–778 RSC .
- N. Sommerfeldt, A. Possling, G. Becker, C. Pesavento, N. Tschowri and R. Hengge, Microbiology, 2009, 155, 1318–1331 CrossRef CAS .
- Y. Qi, F. Rao, Z. Luo and Z. X. Liang, Biochemistry, 2009, 48, 10275–10285 CrossRef CAS .
- X. Wan, J. R. Tuckerman, J. A. Saito, T. A. Freitas, J. S. Newhouse, J. R. Denery, M. Y. Galperin, G. Gonzalez, M. A. Gilles-Gonzalez and M. Alam, J. Mol. Biol., 2009, 388, 262–270 CrossRef CAS .
- J. C. Whitney, K. M. Colvin, L. S. Marmont, H. Robinson, M. R. Parsek and P. L. Howell, J. Biol. Chem., 2012, 287, 23582–23593 CrossRef CAS .
- E. Bordeleau, E. Brouillette, N. Robichaud and V. Burrus, Environ. Microbiol., 2010, 12, 510–523 CrossRef CAS .
- I. D. Hay, U. Remminghorst and B. H. Rehm, Appl. Environ. Microbiol., 2009, 75, 1110–1120 CrossRef CAS .
- A. L. Chang, J. R. Tuckerman, G. Gonzalez, R. Mayer, H. Weinhouse, G. Volman, D. Amikam, M. Benziman and M. A. Gilles-Gonzalez, Biochemistry, 2001, 40, 3420–3426 CrossRef CAS .
- M. Sivaneson, H. Mikkelsen, I. Ventre, C. Bordi and A. Filloux, Mol. Microbiol., 2011, 79, 1353–1366 CrossRef CAS .
- Q. Wu and K. H. Gardner, Biochemistry, 2009, 48, 2620–2629 CrossRef CAS .
- H. Kulasakara, V. Lee, A. Brencic, N. Liberati, J. Urbach, S. Miyata, D. G. Lee, A. N. Neely, M. Hyodo, Y. Hayakawa, F. M. Ausubel and S. Lory, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2839–2844 CrossRef .
- N. Liu, Y. Xu, S. Hossain, N. Huang, D. Coursolle, J. A. Gralnick and E. M. Boon, Biochemistry, 2012, 51, 2087–2099 CrossRef CAS .
- F. Tao, Y. W. He, D. H. Wu, S. Swarup and L. H. Zhang, J. Bacteriol., 2010, 192, 1020–1029 CrossRef CAS .
- M. Fazli, A. O'Connell, M. Nilsson, K. Niehaus, J. M. Dow, M. Givskov, R. P. Ryan and T. Tolker-Nielsen, Mol. Microbiol., 2011, 82, 327–341 CrossRef CAS .
- M. Y. Galperin, J. Bacteriol., 2006, 188, 4169–4182 CrossRef CAS .
- T. Schirmer and U. Jenal, Nat. Rev. Microbiol., 2009, 7, 724–735 CrossRef CAS .
- J. R. Tuckerman, G. Gonzalez, E. H. Sousa, X. Wan, J. A. Saito, M. Alam and M. A. Gilles-Gonzalez, Biochemistry, 2009, 48, 9764–9774 CrossRef CAS .
- H. K. Carlson, R. E. Vance and M. A. Marletta, Mol. Microbiol., 2010, 77, 930–942 CAS .
- N. Barraud, D. Schleheck, J. Klebensberger, J. S. Webb, D. J. Hassett, S. A. Rice and S. Kjelleberg, J. Bacteriol., 2009, 191, 7333–7342 CrossRef CAS .
- I. Kolodkin-Gal, D. Romero, S. Cao, J. Clardy, R. Kolter and R. Losick, Science, 2010, 328, 627–629 CrossRef CAS .
- T. Kanazawa, S. Ren, M. Maekawa, K. Hasegawa, F. Arisaka, M. Hyodo, Y. Hayakawa, H. Ohta and S. Masuda, Biochemistry, 2010, 49, 10647–10655 CrossRef CAS .
- M. Gjermansen, M. Nilsson, L. Yang and T. Tolker-Nielsen, Mol. Microbiol., 2010, 75, 815–826 CrossRef CAS .
- S. An, J. Wu and L.-H. Zhang, Appl. Environ. Microbiol., 2010, 76, 8160–8173 CrossRef CAS .
- P. D. Newell, S. Yoshioka, K. L. Hvorecny, R. D. Monds and G. A. O'Toole, J. Bacteriol., 2011, 193, 4685–4698 CrossRef CAS .
- O. Kirillina, J. D. Fetherston, A. G. Bobrov, J. Abney and R. D. Perry, Mol. Microbiol., 2004, 54, 75–88 CrossRef CAS .
- Y.-C. Sun, A. Koumoutsi, C. Jarrett, K. Lawrence, F. C. Gherardini, C. Darby and B. J. Hinnebusch, PLoS One, 2011, 6, e19267 CAS .
- V. M. Delgado-Nixon, G. Gonzalez and M. A. Gilles-Gonzalez, Biochemistry, 2000, 39, 2685–2691 CrossRef CAS .
- B. J. Nablo, T. Y. Chen and M. H. Schoenfisch, J. Am. Chem. Soc., 2001, 123, 9712–9713 CrossRef CAS .
- N. Barraud, D. J. Hassett, S.-H. Hwang, S. A. Rice, S. Kjelleberg and J. S. Webb, J. Bacteriol., 2006, 188, 7344–7353 CrossRef CAS .
- N. Barraud, M. V. Storey, Z. P. Moore, J. S. Webb, S. A. Rice and S. Kjelleberg, Microbiol. Biotechnol., 2009, 2, 370–378 CrossRef CAS .
- I. Schmidt, P. J. M. Steenbakkers, H. J. M. op den Camp, K. Schmidt and M. S. M. Jetten, J. Bacteriol., 2004, 186, 2781–2788 CrossRef CAS .
- M. L. Falsetta, A. G. McEwan, M. P. Jennings and M. A. Apicella, Infect. Immun., 2010, 78, 2320–2328 CrossRef CAS .
- B. M. Henares, K. E. Higgins and E. M. Boon, ACS Chem. Biol., 2012, 7, 1331–1336 CrossRef CAS .
- M. A. van der Horst and K. J. Hellingwerf, Acc. Chem. Res., 2004, 37, 13–20 CrossRef CAS .
- A. Möglich, X. Yang, R. A. Ayers and K. Moffat, Annu. Rev. Plant Biol., 2010, 61, 21–47 CrossRef .
- G. A. James, D. R. Korber, D. E. Caldwell and J. W. Costerton, J. Bacteriol., 1995, 177, 907–915 CAS .
- R. D. Monds, P. D. Newell, R. H. Gross and G. A. O'Toole, Mol. Microbiol., 2007, 63, 656–679 CrossRef CAS .
- M. Egli, R. V. Gessner, L. D. Williams, G. J. Quigley, G. A. van der Marel, J. H. van Boom, A. Rich and C. A. Frederick, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 3235–3239 CrossRef CAS .
- Z. Zhang, B. L. Gaffney and R. A. Jones, J. Am. Chem. Soc., 2004, 126, 16700–16701 CrossRef CAS .
- Z. Zhang, S. Kim, B. L. Gaffney and R. A. Jones, J. Am. Chem. Soc., 2006, 128, 7015–7024 CrossRef CAS .
- J. Wang, J. Zhou, G. P. Donaldson, S. Nakayama, L. Yan, Y. F. Lam, V. T. Lee and H. O. Sintim, J. Am. Chem. Soc., 2011, 133, 9320–9330 CrossRef CAS .
- M. Gentner, M. G. Allan, F. Zaehringer, T. Schirmer and S. Grzesiek, J. Am. Chem. Soc., 2012, 134, 1019–1029 CrossRef CAS .
- S. Nakayama, K. Roelofs, V. T. Lee and H. O. Sintim, Mol. BioSyst., 2012, 8, 726–729 RSC .
- S. Nakayama, I. Kelsey, J. X. Wang, K. Roelofs, B. Stefane, Y. L. Luo, V. T. Lee and H. O. Sintim, J. Am. Chem. Soc., 2011, 133, 4856–4864 CrossRef CAS .
- S. Nakayama, I. Kelsey, J. X. Wang and H. O. Sintim, Chem. Commun., 2011, 47, 4766–4768 RSC .
- I. Kelsey, S. Nakayama and H. O. Sintim, Bioorg. Med. Chem. Lett., 2012, 22, 881–885 CrossRef CAS .
- Addition of intercalators to c-di-GMP at room temperature causes the disappearances of both the c-di-GMP and intercalator peaks. When heated to 60 °C, partial recovery of the peaks occurs. We attribute this to a probable polymer formation. C-di-GMP-intercalator crystals obtained by us failed to diffract.
- P. Travascio, Y. Li and D. Sen, Chem. Biol., 1998, 5, 505–517 CrossRef CAS .
- S. Nakayama, J. Wang and H. O. Sintim, Chem.–Eur. J., 2011, 17, 5691–5698 CrossRef CAS .
- P. Wassmann, C. Chan, R. Paul, A. Beck, H. Heerklotz, U. Jenal and T. Schirmer, Structure, 2007, 15, 915–927 CrossRef CAS .
- C. Chan, R. Paul, D. Samoray, N. C. Amiot, B. Giese, U. Jenal and T. Schirmer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17084–17089 CrossRef CAS .
- W. C. Fuqua, S. C. Winans and E. P. Greenberg, J. Bacteriol., 1994, 176, 269–275 CAS .
- S. Atkinson and P. Williams, J. R. Soc., Interface, 2009, 6, 959–978 CrossRef CAS .
- D. G. Davies and C. N. H. Marques, J. Bacteriol., 2009, 191, 1393–1403 CrossRef CAS .
- G. D. Geske, J. C. O'Neill and H. E. Blackwell, Chem. Soc. Rev., 2008, 37, 1432–1447 RSC .
- M. H. Sturme, M. Kleerebezem, J. Nakayama, A. D. Akkermans, E. E. Vaugha and W. M. de Vos, Antonie van Leeuwenhoek, 2002, 81, 233–243 CrossRef CAS .
- C. A. Lowery, T. J. Dickerson and K. D. Janda, Chem. Soc. Rev., 2008, 37, 1337–1346 RSC .
- C. M. Waters and B. L. Bassler, Annu. Rev. Cell Dev. Biol., 2005, 21, 319–346 CrossRef CAS .
- G. Kovacikova and K. Skorupski, Mol. Microbiol., 2002, 46, 1135–1147 CrossRef CAS .
- G. Kovacikova, W. Lin and K. Skorupski, Mol. Microbiol., 2005, 57, 420–433 CrossRef CAS .
- A. Ueda and T. K. Wood, PLoS Pathog., 2009, 5, e1000483 Search PubMed .
- E. V. Kozlova, B. K. Khajanchi, J. Sha and A. K. Chopra, Microb. Pathog., 2011, 50, 213–223 CrossRef CAS .
- R. Simm, M. Morr, U. Remminghorst, M. Andersson and U. Romling, Anal. Biochem., 2009, 386, 53–58 CrossRef CAS .
- H. Gu, K. Furukawa and R. R. Breaker, Anal. Chem., 2012, 84, 4935–4941 CrossRef CAS .
- S. Nakayama, Y. Luo, J. Zhou, T. K. Dayie and H. O. Sintim, Chem. Commun., 2012, 48, 9059–9061 RSC .
- M. Cashel, D. Gentry, V. Hernandez and D. Vinella, The stringent response, in Escherichia coli and Salmonella: cellular and molecular biology, ASM Press, 2nd edn, 1996 Search PubMed .
- B. Spira, N. Silberstein and E. Yagil, J. Bacteriol., 1995, 177, 4053–4058 CAS .
- A. Battesti and E. Bouveret, Mol. Microbiol., 2006, 62, 1048–1063 CrossRef CAS .
- K. Lapouge, M. Schubert, F. H. Allain and D. Haas, Mol. Microbiol., 2008, 67, 241–253 CrossRef CAS .
- D. Vinella, C. Albrecht, M. Cashel and R. D'Ari, Mol. Microbiol., 2005, 56, 958–970 CrossRef CAS .
- K. Takahashi, K. Kasai and K. Ochi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4320–4324 CrossRef CAS .
- J. Justesen, T. Lund, F. S. Pedersen and N. O. Kjeldgaard, Biochimie, 1986, 68, 715–722 CrossRef CAS .
- H. Xiao, M. Kalman, K. Ikehara, S. Zemel, G. Glaser and M. Cashel, J. Biol. Chem., 1991, 266, 5980–5990 CAS .
- K. Potrykus and M. Cashel, Annu. Rev. Microbiol., 2008, 62, 35–51 CrossRef CAS .
- M. Jishage, K. Kvint, V. Shingler and T. Nyström, Genes Dev., 2002, 16, 1260–1270 CrossRef CAS .
- Z. D. Dalebroux, S. L. Svensson, E. C. Gaynor and M. S. Swanson, Microbiol. Mol. Biol. Rev., 2010, 74, 171–199 CrossRef CAS .
- T. Hogg, U. Mechold, H. Malke, M. Cashel and R. Hilgenfeld, Cell, 2004, 117, 57–68 CrossRef CAS .
- B. J. Paul, M. M. Barker, W. Ross, D. A. Schneider, C. Webb, J. W. Foster and R. L. Gourse, Cell, 2004, 118, 311–322 CrossRef CAS .
- B. J. Paul, M. B. Berkmen and R. L. Gourse, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7823–7828 CrossRef CAS .
- A. Bougdour and S. Gottesman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12896–12901 CrossRef CAS .
- H. Merrikh, A. E. Ferrazzoli and S. T. Lovett, J. Bacteriol., 2009, 191, 7436–7446 CrossRef CAS .
- S. E. Ades, I. L. Grigorova and C. A. Gross, J. Bacteriol., 2003, 185, 2512–2519 CrossRef CAS .
- P. Milon, E. Tischenko, J. Tomsic, E. Caserta, G. Folkers, A. La Teana, M. V. Rodnina, C. L. Pon, R. Boelens and C. O. Gualerzi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13962–13967 CrossRef CAS .
- G. Schreiber, E. Z. Ron and G. Glaser, Curr. Microbiol., 1995, 30, 27–32 CrossRef CAS .
- A. Levine, F. Vannier, M. Dehbi, G. Henckes and S. J. Séror, J. Mol. Biol., 1991, 219, 605–613 CrossRef CAS .
- J. D. Wang, G. M. Sanders and A. D. Grossman, Cell, 2007, 128, 865–875 CrossRef CAS .
- G. Zhao, N. Weatherspoon, W. Kong, R. Curtiss and Y. Shi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20924–20929 CrossRef CAS .
- J. C. Charity, L. T. Blalock, M. M. Costante-Hamm, D. L. Kasper and S. L. Dove, PLoS Pathog., 2009, 5, e1000641 Search PubMed .
- L. Siculella, F. Damiano, R. di Summa, S. M. Tredici, R. Alduina, G. V. Gnoni and P. Alifano, Mol. Microbiol., 2010, 77, 716–729 CrossRef CAS .
- M. L. Gatewood and G. H. Jones, J. Bacteriol., 2010, 192, 4275–4280 CrossRef CAS .
- U. Kanjee, I. Gutsche, E. Alexopoulos, B. Zhao, M. El Bakkouri, G. Thibault, K. Liu, S. Ramachandran, J. Snider, E. F. Pai and W. A. Houry, EMBO J., 2011, 30, 931–944 CrossRef CAS .
- B. Zhao and W. A. Houry, Biochem. Cell Biol., 2010, 88, 301–314 CrossRef CAS .
- A. Kuroda, H. Murphy, M. Cashel and A. Kornberg, J. Biol. Chem., 1997, 272, 21240–21243 CrossRef CAS .
- A. Kuroda, K. Nomura, R. Ohtomo, J. Kato, T. Ikeda, N. Takiguchi, H. Ohtake and A. Kornberg, Science, 2001, 293, 705–708 CrossRef CAS .
- C. van Delden, R. Comte and A. M. Bally, J. Bacteriol., 2001, 183, 5376–5384 CrossRef CAS .
- D. L. Erickson, J. L. Lines, E. C. Pesci, V. Venturi and D. G. Storey, Infect. Immun., 2004, 72, 5638–5645 CrossRef CAS .
- H.-B. Zhang, C. Wang and L.-H. Zhang, Mol. Microbiol., 2004, 52, 1389–1401 CrossRef CAS .
- J. Wang, N. Gardiol, T. Burr, G. P. C. Salmond and M. Welch, J. Bacteriol., 2007, 189, 7643–7652 CrossRef CAS .
- J. A. C. Lemos, T. A. Brown and R. A. Burne, Infect. Immun., 2004, 72, 1431–1440 CrossRef CAS .
- M. Kajitani and A. Ishihama, J. Biol. Chem., 1984, 259, 1951–1957 CAS .
- R. J. Heath, S. Jackowski and C. O. Rock, J. Biol. Chem., 1994, 269, 26584–26590 CAS .
- Y. Hamagishi, T. Oki, H. Tone and T. Inui, J. Biochem., 1980, 88, 1785–1792 CAS .
- Y. Hamagishi, A. Yoshimoto and T. Oki, Arch. Microbiol., 1981, 130, 134–137 CrossRef CAS .
- M. Fischer, T. P. Zimmerman and S. A. Short, Anal. Biochem., 1982, 121, 135–139 CrossRef CAS .
- H. W. Rhee, C. R. Lee, S. H. Cho, M. R. Song, M. Cashel, H. E. Choy, Y. J. Seok and J. I. Hong, J. Am. Chem. Soc., 2008, 130, 784–785 CrossRef CAS .
- Y. Oppenheimer-Shaanan, E. Wexselblatt, J. Katzhendler, E. Yavin and S. Ben-Yehuda, EMBO Rep., 2011, 12, 594–601 CrossRef CAS .
- U. Romling, Sci. Signaling, 2008, 1, pe39 CrossRef .
- R. M. Corrigan, J. C. Abbott, H. Burhenne, V. Kaever and A. Gründling, PLoS Pathog., 2011, 7, e1002217 CAS .
- F. Rao, R. Y. See, D. W. Zhang, D. C. Toh, Q. Ji and Z. X. Liang, J. Biol. Chem., 2010, 285, 473–482 CrossRef CAS .
- F. Rao, Q. Ji, I. Soehano and Z. X. Liang, J. Bacteriol., 2011, 193, 1543–1551 CrossRef CAS .
- Y. Luo and J. D. Helmann, Mol. Microbiol., 2012, 83, 623–639 CrossRef CAS .
- S. H. Francis and J. D. Corbin, Crit. Rev. Clin. Lab. Sci., 1999, 36, 275–328 CrossRef CAS .
- K. A. Lucas, G. M. Pitari, S. Kazerounian, I. Ruiz-Stewart, J. Park, S. Schulz, K. P. Chepenik and S. A. Waldman, Pharmacol. Rev., 2000, 52, 375–414 CAS .
- U. B. Kaupp and R. Seifert, Physiol. Rev., 2002, 82, 769–824 CAS .
- J. L. Botsford and J. G. Harman, Microbiol. Rev., 1992, 56, 100–122 CAS .
- J. U. Linder, Mol. Cell Biochem., 2010, 334, 215–219 CrossRef CAS .
- A. Rauch, M. Leipelt, M. Russwurm and C. Steegborn, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15720–15725 CrossRef CAS .
- R. K. Sunahara, A. Beuve, J. J. Tesmer, S. R. Sprang, D. L. Garbers and A. G. Gilman, J. Biol. Chem., 1998, 273, 16332–16338 CrossRef CAS .
- M. H. Ryu, O. V. Moskvin, J. Siltberg-Liberles and M. Gomelsky, J. Biol. Chem., 2010, 285, 41501–41508 CrossRef CAS .
- S. Busby and R. H. Ebright, J. Mol. Biol., 1999, 293, 199–213 CrossRef CAS .
- J. G. Harman, Biochim. Biophys. Acta, 2001, 1547, 1–17 CrossRef CAS .
- A. Kolb, S. Busby, H. Buc, S. Garges and S. Adhya, Annu. Rev. Biochem., 1993, 62, 749–795 CrossRef CAS .
- I. T. Weber and T. A. Steitz, J. Mol. Biol., 1987, 198, 311–326 CrossRef CAS .
- B. de Crombrugghe, S. Busby and H. Buc, Science, 1984, 224, 831–838 CAS .
- J. L. Botsford, Microbiol. Rev., 1981, 45, 620–642 CAS .
- B. Görke and J. Stülke, Nat. Rev. Microbiol., 2008, 6, 613–624 CrossRef .
- M. R. Diaz, J. M. King and T. L. Yahr, Front Microbiol., 2011, 2, 89 CAS .
- J. C. N. Fong and F. H. Yildiz, J. Bacteriol., 2008, 190, 6646–6659 CrossRef CAS .
- W. Liang, A. Pascual-Montano, A. J. Silva and J. A. Benitez, Microbiology, 2007, 153, 2964–2975 CrossRef CAS .
- M. C. Wolfgang, V. T. Lee, M. E. Gilmore and S. Lory, Dev. Cell, 2003, 4, 253–263 CrossRef CAS .
- L. A. McCue, K. A. McDonough and C. E. Lawrence, Genome Res., 2000, 10, 204–219 CrossRef CAS .
- M. A. Gazdik, G. Bai, Y. Wu and K. A. McDonough, Mol. Microbiol., 2009, 71, 434–448 CrossRef CAS .
- G. Bai, L. A. McCue and K. A. McDonough, J. Bacteriol., 2005, 187, 7795–7804 CrossRef CAS .
- S. Nambi, N. Basu and S. S. Visweswariah, J. Biol. Chem., 2010, 285, 24313–24323 CrossRef CAS .
- W. Liang, S. Z. Sultan, A. J. Silva and J. A. Benitez, FEBS Lett., 2008, 582, 3744–3750 CrossRef CAS .
- T. Ishikawa, P. K. Rompikuntal, B. Lindmark, D. L. Milton and S. N. Wai, PLoS One, 2009, 4, e6734 Search PubMed .
- E. L. Fuchs, E. D. Brutinel, A. K. Jones, N. B. Fulcher, M. L. Urbanowski, T. L. Yahr and M. C. Wolfgang, J. Bacteriol., 2010, 192, 3553–3564 CrossRef CAS .
- X. Zhou, X. Meng and B. Sun, Cell Res., 2008, 18, 937–948 CrossRef CAS .
- K. B. Xavier and B. L. Bassler, J. Bacteriol., 2005, 187, 238–248 CrossRef CAS .
- L. Wang, Y. Hashimoto, C.-Y. Tsao, J. J. Valdes and W. E. Bentley, J. Bacteriol., 2005, 187, 2066–2076 CrossRef CAS .
- R. A. Black, A. C. Hobson and J. Adler, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 3879–3883 CrossRef CAS .
- J. A. G. Ochoa de Alda, G. Ajlani and J. Houmard, J. Bacteriol., 2000, 182, 3839–3842 CrossRef CAS .
- M. Shibuya, Y. Takebe and Y. Kaziro, Cell, 1977, 12, 521–528 CrossRef CAS .
- J. N. Marden, Q. Dong, S. Roychowdhury, J. E. Berleman and C. E. Bauer, Mol. Microbiol., 2011, 79, 600–615 CrossRef CAS .
- M. Berrera, G. Dodoni, S. Monterisi, V. Pertegato, I. Zamparo and M. Zaccolo, Handb. Exp. Pharmacol., 2008, 186, 285–298 CrossRef CAS .
- P. M. Schmidt, Handb. Exp. Pharmacol., 2009, 191, 195–228 CrossRef CAS .
- S. R. Adams, A. T. Harootunian, Y. J. Buechler, S. S. Taylor and R. Y. Tsien, Nature, 1991, 349, 694–697 CrossRef CAS .
- J. B. Klarenbeek, J. Goedhart, M. A. Hink, T. W. J. Gadella and K. Jalink, PloS One, 2011, 6, e19170 CAS .
- P. A. Cotter and S. Stibitz, Curr. Opin. Microbiol., 2007, 10, 17–23 CrossRef CAS .
- B. Lim, S. Beyhan and F. H. Yildiz, J. Bacteriol., 2007, 189, 717–729 CrossRef CAS .
- B. K. Hammer and B. L. Bassler, J. Bacteriol., 2009, 191, 169–177 CrossRef CAS .
- C. Pesavento, G. Becker, N. Sommerfeldt, A. Possling, N. Tschowri, A. Mehlis and R. Hengge, Genes Dev., 2008, 22, 2434–2446 CrossRef CAS .
- O. Soutourina, A. Kolb, E. Krin, C. Laurent-Winter, S. Rimsky, A. Danchin and P. Bertin, J. Bacteriol., 1999, 181, 7500–7508 CAS .
- R. Lange and R. Henggearonis, Genes Dev., 1994, 8, 1600–1612 CrossRef CAS .
- C. Ueguchi, N. Misonou and T. Mizuno, J. Bacteriol., 2001, 183, 520–527 CrossRef CAS .
- B. Spira, X. Hu and T. Ferenci, Microbiology, 2008, 154, 2887–2895 CrossRef CAS .
- K. Kvint, C. Hosbond, A. Farewell, O. Nybroe and T. Nystrom, Mol. Microbiol., 2000, 35, 435–443 CrossRef CAS .
- J. J. Lemke, T. Durfee and R. L. Gourse, Mol. Microbiol., 2009, 74, 1368–1379 CrossRef CAS .
- S. T. Lovett, DNA Repair, 2006, 5, 1421–1427 CrossRef CAS .
- T. Inaoka and K. Ochi, J. Bacteriol., 2002, 184, 3923–3930 CrossRef CAS .
- P. Serror and A. L. Sonenshein, J. Bacteriol., 1996, 178, 5910–5915 CAS .
- B. J. Haijema, D. van Sinderen, K. Winterling, J. Kooistra, G. Venema and L. W. Hamoen, Mol. Microbiol., 1996, 22, 75–85 CrossRef CAS .
- T. Inaoka, K. Takahashi, M. Ohnishi-Kameyama, M. Yoshida and K. Ochi, J. Biol. Chem., 2003, 278, 2169–2176 CrossRef CAS .
- J. E. Walker and E. P. Abraham, Biochem. J., 1970, 118, 563–570 CAS .
- T. Inaoka, G. Wang and K. Ochi, J. Bacteriol., 2009, 191, 7367–7371 CrossRef CAS .
- M. Ratnayake-Lecamwasam, P. Serror, K. W. Wong and A. L. Sonenshein, Genes Dev., 2001, 15, 1093–1103 CrossRef CAS .
- R. Magnuson, J. Solomon and A. D. Grossman, Cell, 1994, 77, 207–216 CrossRef CAS .
- J. M. Solomon, B. A. Lazazzera and A. D. Grossman, Genes Dev., 1996, 10, 2014–2024 CrossRef CAS .
- J. D. Quisel, W. F. Burkholder and A. D. Grossman, J. Bacteriol., 2001, 183, 6573–6578 CrossRef CAS .
- M. A. Hamon and B. A. Lazazzera, Mol. Microbiol., 2001, 42, 1199–1209 CrossRef CAS .
- D. K. R. Karaolis, M. H. Rashid, R. Chythanya, W. Luo, M. Hyodo and Y. Hayakawa, Antimicrob. Agents Chemother., 2005, 49, 1029–1038 CrossRef CAS .
- E. Brouillette, M. Hyodo, Y. Hayakawa, D. K. R. Karaolis and F. Malouin, Antimicrob. Agents Chemother., 2005, 49, 3109–3113 CrossRef CAS .
- W. Yan, T. Qu, H. Zhao, L. Su, Q. Yu, J. Gao and B. Wu, Microbiol. Res., 2010, 165, 87–96 CrossRef CAS .
- Y. Ishihara, M. Hyodo, Y. Hayakawa, T. Kamegaya, K. Yamada, A. Okamoto, T. Hasegawa and M. Ohta, FEMS Microbiol. Lett., 2009, 301, 193–200 CrossRef CAS .
- D. Amikam, O. Steinberger, T. Shkolnik and Z. Ben-Ishai, Biochem. J., 1995, 311, 921–927 CAS .
- O. Steinberger, Z. Lapidot, Z. Ben-Ishai and D. Amikam, FEBS Lett., 1999, 444, 125–129 CrossRef CAS .
- D. K. R. Karaolis, K. Cheng, M. Lipsky, A. Elnabawi, J. Catalano, M. Hyodo, Y. Hayakawa and J. P. Raufman, Biochem. Biophys. Res. Commun., 2005, 329, 40–45 CrossRef CAS .
- W. Chen, R. KuoLee and H. Yan, Vaccine, 2010, 28, 3080–3085 CrossRef CAS .
- D.-L. Hu, K. Narita, M. Hyodo, Y. Hayakawa, A. Nakane and D. K. R. Karaolis, Vaccine, 2009, 27, 4867–4873 CrossRef CAS .
- T. Ebensen, K. Schulze, P. Riese, M. Morr and C. A. Guzmán, Clin. Vaccine Immunol., 2007, 14, 952–958 CrossRef CAS .
- H. Yan, R. KuoLee, K. Tram, H. Qiu, J. Zhang, G. B. Patel and W. Chen, Biochem. Biophys. Res. Commun., 2009, 387, 581–584 CrossRef CAS .
- S. M. McWhirter, R. Barbalat, K. M. Monroe, M. F. Fontana, M. Hyodo, N. T. Joncker, K. J. Ishii, S. Akira, M. Colonna, Z. J. Chen, K. A. Fitzgerald, Y. Hayakawa and R. E. Vance, J. Exp. Med., 2009, 206, 1899–1911 CrossRef CAS .
- P. Nakhaei, J. Hiscott and R. Lin, J. Mol. Cell Biol., 2010, 2, 110–112 CrossRef CAS .
- J. D. Sauer, K. Sotelo-Troha, J. von Moltke, K. M. Monroe, C. S. Rae, S. W. Brubaker, M. Hyodo, Y. Hayakawa, J. J. Woodward, D. A. Portnoy and R. E. Vance, Infect. Immun., 2011, 79, 688–694 CrossRef CAS .
- D. L. Burdette, K. M. Monroe, K. Sotelo-Troha, J. S. Iwig, B. Eckert, M. Hyodo, Y. Hayakawa and R. E. Vance, Nature, 2011, 478, 515–518 CrossRef CAS .
- J. J. Woodward, A. T. Iavarone and D. A. Portnoy, Science, 2010, 328, 1703–1705 CrossRef CAS .
- R. Libanova, T. Ebensen, K. Schulze, D. Bruhn, M. Nörder, T. Yevsa, M. Morr and C. A. Guzmán, Vaccine, 2010, 28, 2249–2258 CrossRef CAS .
- I. Rombel, US Pat., 6 562 801, 2003 Search PubMed .
- H. Schaller and H. G. Khorana, J. Am. Chem. Soc., 1963, 85, 3841–3851 CrossRef CAS .
- Y. Lapidot and H. G. Khorana, J. Am. Chem. Soc., 1963, 85, 3857–3862 CrossRef CAS .
- C.-Y. J. Hsu and D. Dennis, Nucleic Acids Res., 1982, 10, 5637–5647 CrossRef CAS .
- C.-Y. J. Hsu, D. Dennis and R. A. Jones, Nucleosides Nucleotides, 1985, 4, 377–389 CAS .
- I. Kiburu, A. Shurer, L. Yan and H. O. Sintim, Mol. BioSyst., 2008, 4, 518–520 RSC .
- M. Hyodo and Y. Hayakawa, Bull. Chem. Soc. Jpn., 2004, 77, 2089–2093 CrossRef CAS .
- N. Amiot, K. Heintz and B. Giese, Synthesis, 2006, 4230–4236 CAS .
- H. Yan and A. L. Aguilar, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 189–204 CAS .
- L. De Napoli, A. Galeone, L. Mayol, A. Messere, D. Montesarchio and G. Piccialli, Bioorg. Med. Chem., 1995, 3, 1325–1329 CrossRef CAS .
- E. Alazzouzi, N. Escaja, A. Grandas and E. Pedroso, Angew. Chem., Int. Ed. Engl., 1997, 36, 1506–1508 CrossRef CAS .
- M. Smietana and E. T. Kool, Angew. Chem., Int. Ed., 2002, 41, 3704–3707 CrossRef CAS .
- B. L. Gaffney, E. Veliath, J. Zhao and R. A. Jones, Org. Lett., 2010, 12, 3269–3271 CrossRef CAS .
- F. Rao, S. Pasunooti, Y. Ng, W. Zhuo, L. Lim and A. W. Liu, Anal. Biochem., 2009, 389, 138–142 CrossRef CAS .
- F. Zähringer, C. Massa and T. Schirmer, Appl. Biochem. Biotechnol., 2011, 163, 71–79 CrossRef .
- M. Yoshikawa, T. Kato and T. Takenishi, Tetrahedron Lett., 1967, 8, 5065–5068 CrossRef .
- M. Smith, G. T. Drummond and H. G. Khorana, J. Am. Chem. Soc., 1961, 83, 698–706 CrossRef CAS .
- H.-G. Genieser, E. Butt, U. Bottin, W. Dostmann and B. Jastorff, Synthesis, 1989, 53–54 CrossRef CAS .
- A. Simoncsits and J. Tomasz, Biochim. Biophys. Acta, 1974, 340, 509–515 CrossRef CAS .
- J. W. Kozarich, A. C. Chinault and S. M. Hecht, Biochemistry, 1975, 14, 981–988 CrossRef CAS .
- T. Kline, S. R. Jackson, W. Deng, C. L. Verlinde and S. I. Miller, Nucleosides, Nucleotides Nucleic Acids, 2008, 27, 1282–1300 CAS .
- S. M. Ching, W. J. Tan, K. L. Chua and Y. Lam, Bioorg. Med. Chem., 2010, 18, 6657–6665 CrossRef CAS .
- P. Ross, R. Mayer, H. Weinhouse, D. Amikam, Y. Huggirat, M. Benziman, E. de Vroom, A. Fidder, P. de Paus, L. A. Sliedregt, G. A. van der Marel and J. H. van Boom, J. Biol. Chem., 1990, 265, 18933–18943 CAS .
- E. Wexselblatt, J. Katzhendler, R. Saleem-Batcha, G. Hansen, R. Hilgenfeld, G. Glaser and R. R. Vidavski, Bioorg. Med. Chem., 2010, 18, 4485–4497 CrossRef CAS .
- U. Mechold, G. Fang, S. Ngo, V. Ogryzko and A. Danchin, Nucleic Acids Res., 2007, 35, 4552–4561 CrossRef CAS .
- K. Furukawa, H. Gu, N. Sudarsan, Y. Hayakawa, M. Hyodo and R. R. Breaker, ACS Chem. Biol., 2012, 7, 1436–1443 CrossRef CAS .
- H. O. Sintim, J. A. Smith, J. Wang, S. Nakayama and L. Yan, Future Med. Chem., 2010, 2, 1005–1035 CrossRef CAS .
- V. C. Kalia and H. J. Purohit, Crit. Rev. Microbiol., 2011, 37, 121–140 CrossRef CAS .
- R. W. Huigens, J. J. Richards, G. Parise, T. E. Ballard, W. Zeng, R. Deora and C. Melander, J. Am. Chem. Soc., 2007, 129, 6966–6967 CrossRef CAS .
- J. J. Richards and C. Melander, ChemBioChem, 2009, 10, 2287–2294 CrossRef CAS .
- S. A. Rogers and C. Melander, Angew. Chem., Int. Ed., 2008, 47, 5229–5231 CrossRef CAS .
- A. E. Clatworthy, E. Pierson and D. T. Hung, Nat. Chem. Biol., 2007, 3, 541–548 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2013 |