Porphyrin-sensitized solar cells
Lu-Lin
Li
and
Eric Wei-Guang
Diau
*
Department of Applied Chemistry and Institute of Molecular Science, National Chiao Tung University, Hsinchu 30010, Taiwan. E-mail: diau@mail.nctu.edu.tw; Fax: +886-3-5723764; Tel: +886-3-5131524
First published on 1st October 2012
Abstract
Nature has chosen chlorophylls in plants as antennae to harvest light for the conversion of solar energy in complicated photosynthetic processes. Inspired by natural photosynthesis, scientists utilized artificial chlorophylls – the porphyrins – as efficient centres to harvest light for solar cells sensitized with a porphyrin (PSSC). After the first example appeared in 1993 of a porphyrin of type copper chlorophyll as a photosensitizer for PSSC that achieved a power conversion efficiency of 2.6%, no significant advance of PSSC was reported until 2005; beta-linked zinc porphyrins were then reported to show promising device performances with a benchmark efficiency of 7.1% reported in 2007. Meso-linked zinc porphyrin sensitizers in the first series with a push–pull framework appeared in 2009; the best cell performed comparably to that of a N3-based device, and a benchmark 11% was reported for a porphyrin sensitizer of this type in 2010. With a structural design involving long alkoxyl chains to envelop the porphyrin core to suppress the dye aggregation for a push–pull zinc porphyrin, the PSSC achieved a record 12.3% in 2011 with co-sensitization of an organic dye and a cobalt-based electrolyte. The best PSSC system exhibited a panchromatic feature for light harvesting covering the visible spectral region to 700 nm, giving opportunities to many other porphyrins, such as fused and dimeric porphyrins, with near-infrared absorption spectral features, together with the approach of molecular co-sensitization, to enhance the device performance of PSSC. According to this historical trend for the development of prospective porphyrin sensitizers used in PSSC, we review systematically the progress of porphyrins of varied kinds, and their derivatives, applied in PSSC with a focus on reports during 2007–2012 from the point of view of molecular design correlated with photovoltaic performance.
 Lu-Lin Li | Lu-Lin Li is an Assistant Research Fellow in National Chiao Tung University (Taiwan). She received her PhD in Chemistry from National Chung Hsing University, Taichung, Taiwan, in 2006. After two years as a post-doctoral fellow at Center of Nanoscience and Nanotechnology in NCHU, she joined NCTU as a postdoctoral fellow with Prof. Diau in 2008. Her main research interest is fabrication and characterization of novel functional materials for dye-sensitized solar cells, with a focus on using electrochemical methods to synthesize nanostructured materials relevant for solar energy conversion, and to characterize their interfacial properties using impedance spectroscopy. |
 Eric Wei-Guang Diau | Eric Diau is a Professor in Department of Applied Chemistry of NCTU (Taiwan). He received his PhD in Chemistry from NTHU (Taiwan) in 1991 and was a postdoctoral fellow with Prof. Ahmed Zewail at Caltech (1997–2001). He is interested in studying relaxation dynamics of organic and bio-organic molecules in solutions, as well as interfacial electron transfer and energy transfer dynamics in many dye/semiconductor systems. Starting from 2006, he devoted to the developments of novel functional materials for dye-sensitized solar cells (DSSC), with a focus on design, synthesis, processing and characterization of photosensitizers and nanostructured materials for DSSC applications. He has published more than 120 peer-reviewed papers. |
1. Introduction
Dye-sensitized solar cells (DSSC) have attracted much attention because they present a highly promising alternative to conventional photovoltaic devices based on silicon.1–5Fig. 1 shows a schematic diagram revealing the operation of a typical DSSC device, which consists of a dye-sensitized mesoporous working electrode (TiO2, anode) and a counter electrode (Pt-coated, cathode), and has an electrolyte (iodine-based or cobalt complexes, redox mediator) filling the space between the anode and the cathode. Upon light illumination, excited electrons in the LUMO level of a sensitizer are rapidly injected into the conduction band (CB) of TiO2, and then transferred to the platinized counter electrode through exterior electric circuits. The holes are reduced by the redox couple, either I−/I3− or Co2+/Co3+, and the oxidized dye thereby regenerated by the I− species (or a Co2+ complex) to produce the I3− species (or a Co3+ complex). The diffusion of the oxidized species, the I3− or Co3+ complex, to the surface of the cathode completes the circuit.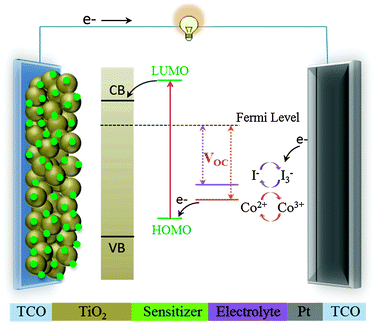 | ||
| Fig. 1 Schematic representation of the composition and the operating principle of a DSSC. | ||
In nanocrystalline TiO2 solar cells sensitized with a dye, efficiencies of conversion of light to electric power (η) greater than 11% have been obtained with polypyridyl ruthenium complexes.6–11 The cost, rarity and environmental issues of ruthenium complexes limit, however, their wide application, and encourage exploration of cheaper and safer sensitizers. Scientists have made tremendous efforts in seeking new and efficient sensitizers suitable for practical use in DSSC.8–44Fig. 2 shows the evolution of photovoltaic performances of DSSC from 1991 to 2012 based on three types of potential photosensitizers being widely investigated.5 For the ruthenium-based sensitizers, the efficiency of the N3 dye reached 10.0% already since 1993;2 the benchmark performance of the N3/N719 family was reported to be 11.2% since 2005,6 and the record efficiency of the C106 dye reached 11.7% reported in 2010.10 The efficiencies of metal-free organic dyes were reported to be 9–10% during recent five years with the best-performed organic dye (C219) reaching η = 10.3%.33 The trend in the performance progress of metal-free organic sensitizers seems to reach a bottleneck for their further development. However, recent developments on porphyrin-based solar cells exhibit a promising advance with the progress curve showing a feature of exponential rise. Moreover, porphyrin sensitizers have drawn great interest because of their excellent light-harvesting function mimicking photosynthesis.16–24,36–44
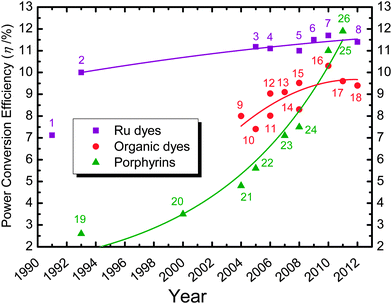 | ||
Fig. 2 Efficiency progressing records of DSSC from 1991 to 2012 on the basis of three representative sensitizers labelled 1–8 for Ru-based complexes ( ), 9–18 for organic dyes ( ), 9–18 for organic dyes ( ) and 19–26 for porphyrin dyes ( ) and 19–26 for porphyrin dyes ( ). The labelled sensitizers may have an alternative given name or a specific code given by the authors: (1) trinuclear Ru dye,1 (2) N3,2 (3) N3,6 (4) N719,7 (5) C101,25 (6) CYC-B11,8 (7) C106,10 (8) black dye,11 (9) indoline dye,26 (10) NKX-2677,27 (11) JK2,28 (12) D149,29 (13) TA-St-CA,30 (14) MK-2,31 (15) D205,32 (16) C219,33 (17) Y123,34 (18) C218,35 (19) Cu-2-a-oxymesoisochlorin,36 (20) TCPP,37 (21) Zn-1a,38 (22) Zn-3,39 (23) GD2,40 (24) tda-2b-bd-Zn,41 (25) YD-2,42 and (26) YD2-oC8.24 ). The labelled sensitizers may have an alternative given name or a specific code given by the authors: (1) trinuclear Ru dye,1 (2) N3,2 (3) N3,6 (4) N719,7 (5) C101,25 (6) CYC-B11,8 (7) C106,10 (8) black dye,11 (9) indoline dye,26 (10) NKX-2677,27 (11) JK2,28 (12) D149,29 (13) TA-St-CA,30 (14) MK-2,31 (15) D205,32 (16) C219,33 (17) Y123,34 (18) C218,35 (19) Cu-2-a-oxymesoisochlorin,36 (20) TCPP,37 (21) Zn-1a,38 (22) Zn-3,39 (23) GD2,40 (24) tda-2b-bd-Zn,41 (25) YD-2,42 and (26) YD2-oC8.24 | ||
In the photosynthetic cores of bacteria and plants, solar energy is collected at chromophores based on porphyrin;23 the captured radiant energy is converted efficiently to chemical energy. Inspired by this efficient energy transfer in naturally occurring photosynthetic reaction centres, numerous porphyrins have been designed and synthesized for DSSC applications.16–22 The intrinsic advantages of porphyrin-based dyes are their rigid molecular structures with large absorption coefficients in the visible region and their many reaction sites, i.e., four meso and eight β positions, available for functionalization: fine tuning of the optical, physical, electrochemical and photovoltaic properties of porphyrins thus becomes feasible. In particular, advances in optimization of the device performance for a zinc porphyrin sensitizer (YD2-oC8) co-sensitized with an organic dye (Y123) using a cobalt-based electrolyte to enhance photovoltage of the device (as demonstrated in Fig. 1) attained an unprecedented power conversion efficiency of η = 12.3%,24 which is superior to devices based on Ru complexes,6–10 stimulating investigation of the development of further porphyrin sensitizers to promote the device performance of porphyrin-sensitized solar cells (PSSC). In this report, we systematically review the recent development of PSSC and introduce a strategy to design potential porphyrin sensitizers for highly efficient DSSC applications.
2. Porphyrins with varied linkers
In DSSC, an anchoring group is required at the edge of the dye to link the dye with the semiconductor through a chemical bond. A carboxylic acid is so far considered to be the best binding group for porphyrins,43 but other promising anchoring groups such as 8-hydroxylquinoline (HQ) have been reported.44The structure of a porphyrin exhibits two major points – four meso-positions and eight β-positions, as shown in Fig. 3, that can serve to functionalize one or multiple linkers containing carboxylic acids or HQ substitutes as anchoring groups that attach to the surface of TiO2. In what follows we introduce the historical development of PSSC beginning with β-linked porphyrin sensitizers because we learned that lesson first from natural porphyrins and chlorophylls.
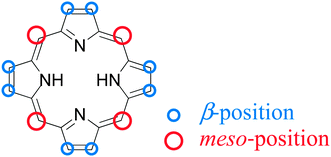 | ||
| Fig. 3 Typical structure of a porphyrin showing the four meso- and the eight β-positions to be functionalized for porphyrin-sensitized solar cells. | ||
2.1 Linkers at β-positions
When Kay and Grätzel36 reported in 1993 the first PSSC based on a copper chlorophyllin sensitizer with efficiency η = 2.6%, a Ru-based DSSC had already attained η = 10%.2 In the succeeding decade there were almost no progress in developing β-linked porphyrins for PSSC until 2004: Officer, Grätzel and their co-workers38,43 reported zinc porphyrins in a series of which a promising sensitizer (Zn-1a, Fig. 4a) attained η = 4.8%.38 They subsequently investigated 13 prospective zinc porphyrins and reported the best one (Zn-3, Fig. 4b) to attain η = 5.6% in the presence of co-adsorbent chenodeoxycholic acid (CDCA).39 In 2007, the same groups reported porphyrin sensitizers in another series with the best one (GD2, Fig. 4c) reaching η = 7.1% for PSSC,40 opening a great opportunity for the development of various porphyrin sensitizers to enhance the efficiency of a DSSC.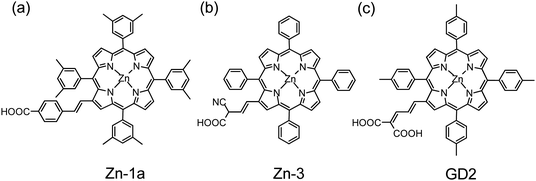
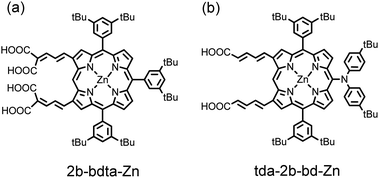
The porphyrin sensitizers shown in Fig. 4 were designed to have a π-conjugated link at the β-position of the porphyrin ring; the best dye (GD2) featured a malonic acid as an anchoring group to enhance the electronic coupling of the dye with the surface of TiO2. Such a concept to design β-functionalized porphyrin sensitizers was then applied by Kim and co-workers,41 who demonstrated that zinc porphyrin 2b-bdta-Zn (Fig. 5a) with two equivalent π-conjugated malonic-acid linkers effectively enhanced the efficiency of electron injection and retarded charge recombination. Kim and co-workers45 reported the β-functionalized zinc porphyrins with a diarylamino group for which the porphyrin coded as tda-2b-bd-Zn (Fig. 5b) exhibits the best performance with η = 7.5% that is comparable to the performance of a N3 dye (η = 7.7%) under the same conditions.
2.2 Linkers at meso-positions
The concept for the design of meso-ethynyl linked porphyrins was first given by Anderson46 and Therien.47 Although Cherian and Wamser37 reported in 2000 the first meso-substituted PSSC with η ∼ 3.5% based on a free-base porphyrin, no significant progress was made for porphyrin sensitizers of this type until 2007: Galoppini and co-workers48 reported tetrachelated zinc porphyrins with four meta-substituted linkers at four meso-positions of the porphyrin to suppress dye aggregation; Lindsey, Meyer and their co-workers49 reported meso-substituted porphyrin and chlorin derivatives to extend the absorption spectra to larger wavelengths; Imahori and co-workers50 examined the meso-substituted porphyrins with five-membered heteroaromatic linkers. The strategy of Galoppini et al.48,49 was to use a phenylethynyl (PE) unit as a bridging moiety to control the distance between the sensitizer and the semiconductor to retard charge recombination, which works well for pyrene51 and Ru complexes52 in model systems, but this approach failed when three or four PE units were combined to form a long bridge at one meso-position of a zinc porphyrin because of serious dye aggregation involved in porphyrins with a long PE linker.53–55To solve the aggregation problem of porphyrins, 3,5-di-tert-butylphenyl groups were introduced at the meso-positions of the porphyrin ring. Based on this molecular design, Yeh, Diau and co-workers56 reported meso-substituted zinc porpyrin derivatives, for which YD0 (Fig. 6a) served as a reference compound. With the diarylamino group attached at the meso-position of the porphyrin core, YD1 (Fig. 6b) attained η = 6.0%, which was comparable to that of a N3 dye under the same experimental conditions (η = 6.1%).56 The superlative cell performance of YD1 reflects its extraordinarily great short-circuit current density (JSC) that arises from the large efficiency of conversion of incident photons to current (IPCE) broadly extending beyond 700 nm. The electron donor in YD1 plays a role not only spectrally to extend the absorption to a region of greater wavelength but also spatially pushing the excited electrons toward TiO2 for an improved separation of charge. The appearance of porphyrins with a push–pull framework such as YD1 indicates the arrival of the era of PSSC.
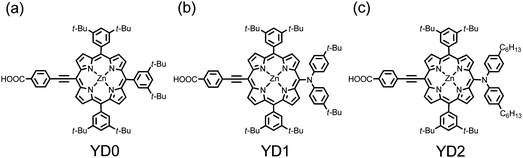 | ||
| Fig. 6 Molecular structures of (a) YD0, (b) YD1 and (c) YD2.57 | ||
Yeh and Diau57,58 designed another promising push–pull porphyrin, YD2 (Fig. 6c) based on the structure of YD1 in that two tert-butyl groups in the diarylamino substituent were replaced with two long alkyl chains to improve the thermal and photochemical stability of a device. This design mimics the strategy applied in an amphiphilic ruthenium polypyridyl sensitizer (Z907) that has shown excellent stability toward water-induced desorption under both thermal stress and light soaking.59 The device performance of YD2 was further improved to η = 6.8%, slightly better than that of YD1 (η = 6.5%) but slightly less than that of N719 (η = 7.3%);57 the electron-donating feature of amino substituents in YD1 and YD2 seems to be responsible for the VOC value being larger than that of the reference cell (YD0).57,58 Using GD2 as an example, Mozer et al.60 noted that the smaller VOC of PSSC is due to the decreased electron lifetime related to a more rapid recombination of electrons with I3− ions. The observed VOC of YD1 and YD2 larger than that of YD0 was thus expected to be due to a diminished recombination between I3− and conduction-band electrons, because I3− might be attached to the positively charged diarylamino moiety far from the TiO2 surface in the former case.57
In 2010, the device performance of YD2 was further improved by Grätzel and co-workers,42 giving JSC/mA cm−2 = 18.6, VOC/V = 0.77, FF = 0.764, and η = 10.9%, which lasted as a record for PSSC until 2011.
2.3 Multiple donor groups at the meso-positions
Because the diarylamino group plays a key role in promoting the device performance of YD2, the effects of the substituted number and position of electron-donating groups (EDG) became an interesting subject to test. Accordingly, Yeh and Diau61 reported push–pull zinc porphyrins with more than one EDG at the meso-positions of the porphyrin ring and found that YD2 was still the best sensitizer for PSSC. Almost at the same period Imahori and co-workers62 designed other pull–pull porphyrins with zero, one and two EDG, in which the two EDG are located at meso-positions of porphyrin to form a cis or a trans isomer. The same conclusion was made by Imahori that the mono-substituted porphyrin with a structure similar to that of YD2 gives the best device performance.2.4 Thiophene substitutes in the linkers
The thiophene group has been widely utilized in ruthenium-based8–10 and metal-free organic solar cells12–15 to enhance the absorption coefficient of the dye and red-shift its absorption spectrum. This concept has been adopted for porphyrin-based DSSC. Thiophene units were used in both β-linked63 and meso-linked64 porphyrin sensitizers to give efficiencies 4.0% and 5.1%, respectively. The IPCE results indicated that increasing the number of thiophene units does not extend the spectra; the spectral edge reaches ∼650 nm in both cases. Hung and co-workers65 found that the number of thiophene units substituted at the meso-position has a negative effect on device performance. Unlike photosensitizers of other types, thiophene substitutes play a lesser role in promoting the efficiencies of PSSC.3. Porphyrins with extended spectral feature
The most viable way to enhance JSC is to harvest a broader region of the solar spectrum. In general, porphyrins show a Soret band at 400–450 nm and Q bands at 500–650 nm. To extend the absorption of porphyrin dyes to the near infrared region, the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels must decrease, for which purpose there are two approaches: one is to introduce a highly conjugated π-extended chromophore coupled with the porphyrin ring, and another is to make fused or dimeric porphyrins.3.1 Porphyrins functionalized with π-extended chromophores
The early results39,41 indicated that the π-conjugation in the β-substituted porphyrins has a limited effect to extend the absorption spectra to greater wavelength. The best strategy to extend the π-conjugation is thus to functionalize the target porphyrin at the meso-positions. A promising chromophore is the acene family, for which the π-conjugation can be effectively extended with an increasing number of aromatic rings. As a result, Lin and Diau66 designed acenes, from benzene to pentacene, as π-extended chromophores to couple to the porphyrin core through the linkage of the acenyl–ethynyl group. Among those porphyrins, the anthracene-functionalized porphyrin (LAC-3, Fig. 7a) showed the best performance, which reached ∼80% of a N3-based device under the same conditions; the tetracene-functionalized porphyrin (LAC-4) featured a broad IPCE spectrum extending to ∼800 nm, but the IPCE values were too small (<50%) to give a satisfactory performance; the pentacene-substituted porphyrin (LAC-5) even showed an extended absorption spectrum beyond 900 nm, but almost no photocurrents were produced.66 When Lin and Diau67,68 further designed cyclic aromatic substituents attached at the meso-position of the macrocycle opposite to the anchoring group, they found that the fluorene-functionalized porphyrin (LD22, Fig. 7b) featured an impressive device performance η = 8.1%,67 and the pyrene-functionalized porphyrin (LD4, Fig. 7c) attained η = 10.1%,68 which was superior to that of a N719 dye (η = 9.3%) under the same conditions. The superior photovoltaic performance of the LD4-based PSSC was attributed to its enhanced ability to harvest light with the IPCE action spectrum covering the entire visible spectral region and extending beyond 800 nm, which outperforms N719. VOC of device LD4 was much smaller than that of cell N719, because of a much smaller electron lifetime of porphyrin-based solar cells than that of N719 cells.60VOC of LD4 was even smaller than that of YD2, which might be rationalized in that electron interception is more efficient for LD4 than for YD2 because of improved charge separation for the latter.57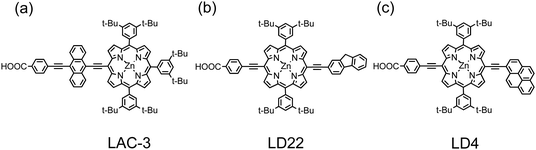
Based on the structure of YD2, Yeh and Diau69 designed the acenyl–ethynyl unit as a link in porphyrins YD11–YD13. As shown in Fig. 8a–c, the bridge between ethyne and carboxyl groups is varied from phenylene for YD11 to naphthylene for YD12, and to anthracenylene for YD13. Without an added scattering layer, the photovoltaic performances of both YD11 and YD12 exhibited performance superior relative to that of N719 dye with JSC of the two promising porphyrin-based devices being significantly greater than those of N719 devices, making the overall efficiencies of power conversion of YD11 (η = 6.7%) and YD12 (η = 6.8%) outperform that of the N719 device (η = 6.1%).69 YD11 (an analogue of YD2) and YD12 are thus two green sensitizers remarkable for their outstanding cell performances relative to that of N719 without an added scattering layer for light-penetrable DSSC applications. When the TiO2 films (∼10 μm) were covered with a scattering layer, the cell performance of N719 significantly improved to η = 7.3%, whereas the performances of the porphyrin dyes increased only marginally (η = 6.8% and 7.0% for YD11 and YD12, respectively).69 These results indicate that a substantial increase in JSC for the N719 device is a key factor for the improvement of the cell performance with the addition of a scattering layer.70 Based on those observations, the involvement of the partially allowed triplet MLCT states of ruthenium complexes was concluded to be responsible for the enhanced efficiency in the red shoulder of the IPCE spectrum of N719, whereas the effect of spin–orbit coupling in zinc porphyrins was insufficient for the S0 → T1 transitions to occur; the additional scattering layer provided no improvement in the IPCE spectra of YD11–YD13 beyond the Q-band absorptions.69
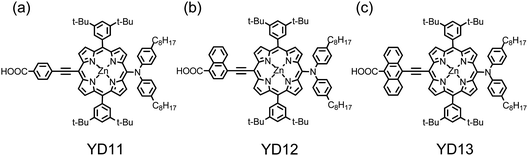 | ||
| Fig. 8 Molecular structures of (a) YD11, (b) YD12 and (c) YD13.69 | ||
Functionalized chromophore anthracene plays an important role to extend the π-conjugation in LAC-3 for an enhanced overall device performance,66 but anthracene in YD13 with a link shorter than that in LAC-3 exhibited a notable effect to deteriorate significantly the device performance of YD13.69 Results obtained from femtosecond measurements of fluorescence decay indicate that the presence of the anthracene group in the bridge from YD13 to TiO2 did not hamper the rate of interfacial electron transfer for the observed small injection yield of YD13; rather, it was the anthracene-induced rapid relaxation of intermolecular energy due to dye aggregation that gave the poor device performance of YD13, which was also evident in experiments in the absence and presence of co-adsorbent CDCA.69
Apart from the cyclic aromatic hydrocarbon systems introduced above, perylene is a promising functional chromophore to modify a porphyrin sensitizer for these two reasons: the strong absorptions of the perylene derivatives in the spectral region 500–650 nm71 are complementary to those of porphyrins, and a device made of a push–pull perylene anhydride sensitizer yields an impressive performance, η = 6.8%.72 Efforts to integrate the perylene chromophores into porphyrin sensitizers showed, however, negative effects for the improvement of device performance of PSSC,73–75 mainly because the co-planar structural nature between perylene and porphyrin moieties caused a serious problem of dye aggregation.
3.2 Fused porphyrins
According to many above examples, porphyrins and their derivatives are promising photosensitizers for DSSC because of their intense absorption in the Soret and Q bands to harvest solar energy efficiently over a broad spectral region, but the existence of a gap between the Soret and Q bands in mono-porphyrins limits their cell performance. One strategy to improve the light-harvesting ability of the sensitizer is to fuse a chromophore with a porphyrin to make a π-elongated macrocycle for PSSC. This idea became mature and practical when Imahori and co-workers76–78 reported their first examples, of which a device made of a naphthalene-based meso-β-edge fused zinc porphyrin (fused-Zn-1, Fig. 9a) attained η = 4.1% (5.0% under co-sensitization), which was improved by half relative to the reference cell with an unfused porphyrin.76,77 Such an unsymmetrical π-elongation was achieved by the same group79 to construct two quinoxaline-based β–β′-edge fused zinc porphyrins, of which a fused porphyrin with one anchoring group (ZnQMA, Fig. 9b) exhibited η = 5.2%, attaining 80% performance of a N719 device under the same conditions. Compare the IPCE spectra of the two fused porphyrins: even though the fused-Zn-1 dye involves a broad light-harvesting feature extending the IPCE action spectrum to nearly 800 nm, a large gap in the middle of the spectrum limits the growth of photocurrents to an optimal condition. In contrast, the IPCE spectrum of the ZnQMA dye extends to only ∼700 nm, but an effective electronic coupling between quinoxaline and porphyrin moieties diminishes the gap between the Soret and Q bands of the spectrum, leading to improved JSC and a performance to that of fused-Zn-1.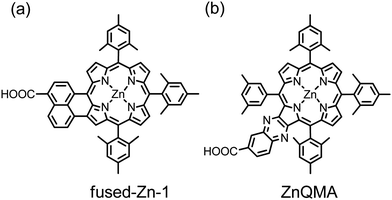
Wang and Wu75 designed and synthesized two perylene anhydride fused nickel porphyrin sensitizers (WW1 in Fig. 10a and WW2 in 10b) to extend the light absorption towards the near-infrared (∼1000 nm) region. As those fused porphyrins suffer from dye aggregation, the IPCE values did not exceed 30%, which was unable to make the devices attain a notable performance (η ∼ 1.3%). Yeh and Diau80 designed and synthesized two fused porphyrins (YDD2 in Fig. 10c and YDD3 in Fig. 10d), but they exhibited poor cell performance. For YDD2, the absorption spectrum extends even beyond 1200 nm, but essentially no photocurrent was observed because the energy level of LUMO was substantially lower than the conduction band edge of TiO2. For YDD3, although a small response was observed in the IPCE action spectrum corresponding to the contribution of broad bands I and II of the fused porphyrin, nearly no response was observed for the broad band III in region 700–900 nm.80 WW2 and YDD3 are, nevertheless, two interesting panchromatic porphyrin sensitizers with the potential to extend the light-harvesting ability toward the near-infrared region for PSSC.
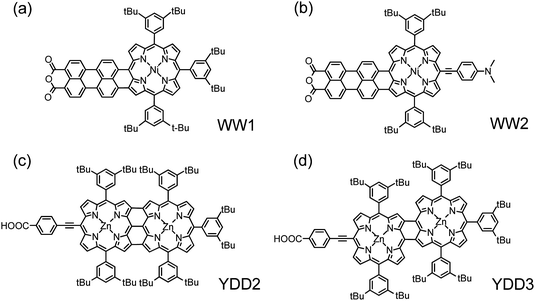 | ||
| Fig. 10 Molecular structures of fused perylene–porphyrins (a) WW1 and (b) WW2;75 fused porphyrin dimers (c)YDD2 and (d)YDD3.80 | ||
3.3 Dimeric porphyrins
Another strategy to improve light-harvesting ability of the sensitizer is to combine two porphyrin moieties through a chemical bond. The first attempt using dimeric porphyrins as potential sensitizers for PSSC was made in 2009 by Officer and co-workers;81 they reported the photovoltaic properties of porphyrin dimers comprising two monoporphyrin units connected in either a linear anti or a 90° syn fashion. The dimeric porphyrin dyes exhibited light-harvesting efficiencies slightly improved relative to the corresponding monomeric porphyrin dyes when the devices were fabricated on a thin TiO2 film (thickness ∼3 μm), but the effect of π-conjugation for the red shift of the IPCE spectra due to porphyrin dimerization was small as the link between the two porphyrins was made at the β-position.The first effort to link the two porphyrins at the meso-position was made in 2009 by Kim and Osuka;82 their poly-ethanediol (PEG)-modified dimer with two β-substituted linkers similar to those of the tda-2b-bd-Zn monomer (Fig. 5b) showed the best device performance of the various dimers, giving an overall efficiency η = 4.2%. Similarly, the link between two porphyrins with only a single CC bond showed no effect of significant spectral shift to enhance the light-harvesting power for the porphyrin dimers.
Based on the structure of YD0 (Fig. 6a), Yeh and Diau80 designed two porphyrin dimers, YDD0 (Fig. 11a) and YDD1 (Fig. 11b). Similar to the results of Kim and Osuka,82 the spectrum of YDD1 exhibited a spectral feature only slightly red-shifted relative to that of the monomeric porphyrin YD0, but, because of effective excitonic coupling between the two nearly perpendicular porpyrin units in YDD1, the gap shown in the IPCE spectrum of YD0 was completely filled in the spectrum of YDD1, making JSC of the YDD1 device become much greater than that of the YD0 device. With the link of an ethynyl group between the two porphyrin units, YDD0 showed split Soret bands in the range of 400–500 nm, and red shifts and broadening of the Q bands extending to nearly 800 nm. The broad IPCE spectrum of YDD0 showed much smaller efficiency, <40%, than those of YD0 and YDD1 (∼70%) because of the co-planar structural feature of YDD0 resulting in serious dye aggregation.
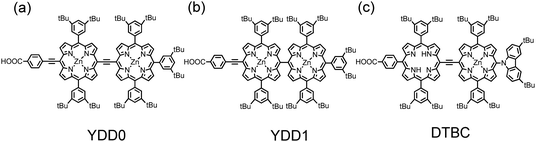
Similar to the structural design of YDD0, Segawa and co-workers83 added a carbazole EDG at the meso-position of the porphyrin edge to form a push–pull porphyrin dimer, DTBC (Fig. 11c). DTBC has an absorption spectrum similar to that of YDD0, but its IPCE spectrum showed much greater efficiency than that of YDD0. In the absence of a TBP additive, the DTBC device exhibited IPCE values up to 80%, yielding a remarkable JSC/mA cm−2 = 18.2 but also causing a poor VOC/V ∼0.4; the poor VOC was significantly improved in the presence of TBP to attain an optimized device performance η = 5.2%,83 comparable to the performance of YDD0, η = 4.1%.80
4. Strategies to suppress dye aggregation for porphyrins
The reported near-infrared dyes such as fused porphyrins75–80 and dimeric porphyrins80–83 introduced herein are particularly interesting, but the planar structural feature of those porphyrins might facilitate the formation of dye aggregates that significantly decrease the efficiency of electron injection. In the following sections we describe two plausible strategies to suppress effectively the dye aggregation for PSSC.4.1 Enveloping porphyrins with long alkoxyl chains
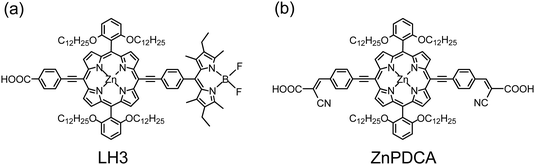
The great performance of porphyrins such as YD2,42,57,58 LD468 and YD1269 was due to their superior light-harvesting ability through introduction of an EDG or a π-extended chromophore at the meso-position of the porphyrin ring. VOC of those highly efficient porphyrin dyes was reported, however, to be significantly less than that of the commonly used ruthenium dye N719. The significantly diminished electron lifetime was reported to account for the smaller VOC of porphyrins, and I3− in the electrolyte might become attached to the positively charged Zn-center of the porphyrin core for efficient electron interception from the TiO2 surface.60 Tian and co-workers concluded that VOC can be improved on decreasing the charge recombination and increasing the efficiency of electron injection with an appropriate design of an organic dye.15 To tackle this problem for porphyrins, a new concept was introduced to design a zinc-porphyrin sensitizer with long alkoxyl chains to protect the porphyrin core for retarded charge recombination and also to decrease effectively the dye aggregation for an efficient electron injection.The examples based on such a molecular design were first given in 2010 by Hupp and co-workers;84,85 they reported porphyrin sensitizers with two phenyl groups attached at the 5,15-meso-positions bearing two dodecoxyl (–OC12H25) chains at the ortho-position of each phenyl group. Among those ortho-substituted porphyrins, LH3 (Fig. 12a)84 and ZnPDCA (Fig. 12b)85 displayed the best performance of each series, with the device made of ZnPDCA attaining efficiency η = 5.5%, comparable to that of N719 under the same conditions.85 Before Hupp's results were published, Imahori and co-workers had announced their results based on ortho-substituted porphyrins,86,87 but the alkyl chains (only methyl and ethyl groups were considered) substituted at the ortho-positions were too short to protect effectively the porphyrins from dye aggregation.88
The concept for the design of ortho-substituted porphyrins was first demonstrated by Lin, Diau, and their co-workers.88,89Fig. 13a–c show the molecular structures of porphyrins LD12, LD14 and LD16, respectively, based on this design. The photovoltaic results indicate that JSC becomes significantly improved on incorporating the π-conjugated EDG for both LD14 and LD16 with respect to LD12, and VOC of those ortho-substituted porphyrins is significantly greater than that of a para- or meta-substituted counterpart; the best devices (LD14 and LD16) attained power conversion efficiencies beyond 10%.88,89 Results from molecular-dynamics (MD) simulation85 indicate that the porphyrin core of LD14 is fully enveloped by the four dodecoxyl chains to provide more effective insulation of the dye, resulting in diminished molecular aggregation and increased solubility in non-coordinating organic solvents. The transient photoelectric results indicate that the upward shift of the TiO2 potential and the retarded charge recombination are two important factors for the enhanced VOC of the ortho-substituted porphyrins.89 The long alkoxyl chains thus play a key role to prevent the approach of I3− in the electrolyte to the surface of TiO2 so as to retard the electron interception at the electrolyte/TiO2 interface. A similar idea for the molecular design of ortho-substituted porphyrins YD20-YD22 with noticeable device performances was reported elsewhere.90
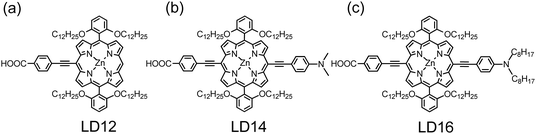
Work developed along this track led to an advance for a device made of an ortho-substituted push–pull zinc porphyrin, YD2-oC8 (Fig. 14a).24 Its design was based on YD2, for which the four tert-butyl side chains in the meso-phenyls of YD2 were replaced by four ortho-substituted alkoxyl chains so as to envelope effectively the porphyrin ring to decrease the degree of dye aggregation. The molecular design of YD2-oC8 is similar to that of LD1489 and LD1688 of which outstanding device performances were reported based on the iodide/triiodide redox electrolyte.
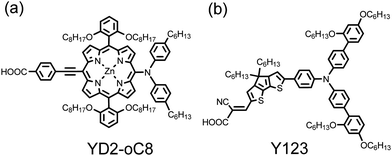
In 2011, Grätzel and co-workers24 reported an optimized performance of the YD2-oC8 device under AM1.5 one-sun irradiation: JSC/mA cm−2 = 17.7, VOC/V = 0.935, FF = 0.74, and η = 12.3%, obtained when YD2-oC8 was co-sensitized with an organic dye (Y123, Fig. 14b)34,91 on a TiO2 film of (12 + 5) μm in a cobalt-based redox electrolyte.24 The efficiency of power conversion attained η = 13.1% under 0.5-sun irradiation. Light-induced photoelectric measurements of the YD2-oC8 devices support that the enhanced VOC of the ortho-substituted devices is due to the upward shift of the TiO2 conduction band and the enhanced electron lifetimes. The long alkoxyl chains in those ortho-substituted porphyrins thus play an important role to diminish effectively the degree of dye aggregation for an improved device performance. These results24,84,85,88–90 might provide a conceptual basis for the molecular design of porphyrin sensitizers to attain even greater efficiencies of power conversion in the near future.
4.2 Co-sensitizations
Co-sensitization is an effective approach to enhance the device performance through a combination of two or more dyes with complementary spectral features sensitized on semiconductor films together, extending the light-harvesting ability so as to increase the photocurrents of the devices. Many co-sensitization systems, such as ruthenium complex plus organic dye,11,92 metal-free organic dye,93,94 phthalocyanine plus organic dye,95–97 and dye co-sensitization in separate layers,98–100 have been reported to show enhanced photovoltaic performance relative to their individual single-dye systems. Based on the organic dye systems, Robertson101 stated that co-sensitization with strongly absorbing dyes would give sufficient space on the surface of TiO2 to allow absorption of other dyes with a complementary absorption spectrum. Porphyrin sensitizers are hence perfect candidates to improve the device performance through co-sensitization.The first porphyrin/organic dye co-sensitization system was reported in 2010 by Grätzel and co-workers42 who demonstrated an instance of YD2 co-sensitized with an organic dye (D205)102 on a thin TiO2 film (2.4 μm), which showed an enhanced device performance η = 6.9% relative to those of their individual dyes, η = 5.6–5.7%. Although two mixed β-linked porphyrins served to enhance the IPCE values by 300%,103 the overall performance of the co-sensitized system was poor. For the best DSSC system with co-sensitization of YD2-oC8 with Y123 (Fig. 14),24 the enhancement in JSC was limited – 17.3 mA cm−2 with YD2-oC8 alone and 17.7 mA cm−2 with YD2-oC8 + Y123 – because the gap between the Soret and the Q bands in the IPCE spectrum of the porphyrin-alone device was small. Moreover, in not only the YD2-related systems24 but also in many other systems aforementioned,42,93–97 the values of VOC were between those of the individual single-dye sensitized devices, which limits the device performance of the co-sensitization system to improve further.
In 2011, Kim and co-workers104 reported the organic dyes (HC-A1, Fig. 15a) as hole conductors to enhance significantly the device performance of a coumarin system. For porphyrins, Kim and co-workers105 showed that the device performance of a push–pull porphyrin with the cyano-acrylic acid anchoring group co-adsorbed with HC-A1 attained η = 7.2%, which was superior to that made of a single porphyrin only (η = 4.4%) or porphyrin co-adsorbed with CDCA (η = 6.6%). The effects of HC-A1 and HC-A3 (Fig. 15b)106 on the photovoltaic performance were remarkable in improving not only JSC but also VOC to some extent. The absorptions of the hole conductors reported by Kim et al. are in the ultraviolet region,104–106 so unable to fill the gap in the IPCE spectrum between the Soret and the Q bands of a porphyrin to improve the photocurrent generation. A superior approach is to use an HC organic dye with an absorption spectrum complementary to that of a porphyrin.
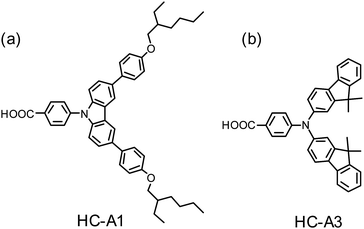
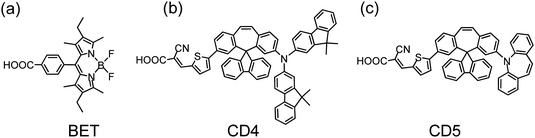
Fig. 16 shows three potential organic dyes that have served as co-sensitizers for PSSC applications. The boron dipyrromethene dye BET (Fig. 16a) belongs to the BODIPY family107 with an absorption maximum at ∼530 nm to compensate for the absorption loss of a porphyrin sensitizer; Lee and Hupp84 used a BODIPY moiety as a light-harvesting molecule connected to the meso-position of a zinc porphyrin to make LH3 (Fig. 12a) for PSSC applications; Odobel and co-workers108 used the BODIPY dye as an antenna attached to the metal centre of a zinc porphyrin derivative to form a supermolecular assembly for DSSC, and possibly also for organic photovoltaic (OPV) applications. When BET was used as a co-sensitizer with an ortho-substituted porphyrin LD12 (Fig. 13a), the gap in the IPCE spectrum of LD12 was completely filled as shown in Fig. 17a, and both JSC = 14.7 mA cm−2 and η = 7.5% were improved by ∼12% relative to the device made of LD12 alone.109 The BET-alone device exhibited a poor IPCE response that generated a small photocurrent, JSC = 0.5 mA cm−2, but co-sensitization remarkably enhanced JSC, which was much greater than the sum of the photocurrents generated from two individual devices. An energy-transfer mechanism was proposed to rationalize the observed exceptional phenomenon,109 but VOC of the LD12 + BET device was slightly less than that of the LD12 device, probably due to the lack of effective HC character for the BODIPY chromophore.
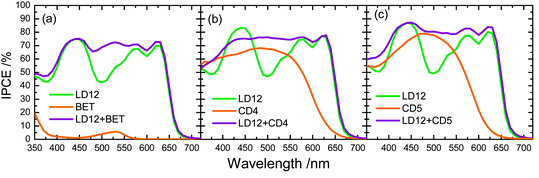 | ||
| Fig. 17 IPCE spectra of LD12 co-sensitized with (a) BET,109 (b) CD4 and (c) CD5.111 | ||
To design organic dyes with HC character and featuring absorption complementary to that of a porphyrin, Chen and co-workers110 synthesized spirally configured donor–acceptor organic dyes for DSSC applications. Among those dyes, CD4 (Fig. 16b) and CD5 (Fig. 16c) are candidates for co-sensitization with porphyrins such as LD12 (Fig. 13a). The co-sensitization of LD12 with CD4 or CD5 on a TiO2 film of thickness ∼20 μm was achieved via a stepwise approach:111 the TiO2 electrode was immersed in the LD12 solution for 3 h, and then immersed in the CD4 or CD5 solution for 2 h. The co-sensitized film was afterwards assembled into a DSSC device of sandwich type with a Pt-coated counter electrode and filled with a traditional iodide/tri-iodide electrolyte. Fig. 17b and c show the IPCE spectra of LD12 co-sensitized with CD4 and CD5, respectively. Similar to the LD12 + BET system shown in Fig. 17a, the gap of the IPCE response about 500 nm for the LD12 device was filled in the action spectra of the LD12 + CD4 or LD12 + CD5 device with the contribution of CD4 or CD5 that shows its maximum photoresponse in that spectral region. Taking CD5 as an example,111JSC of the co-sensitized device increased from 14.97 mA cm−2 to 16.74 mA cm−2 to contribute ∼12% enhancement of the overall performance. Moreover, VOC of the co-sensitized device increased from 0.711 V (LD12) and 0.689 V (CD5) to 0.736 V (LD12 + CD5), which takes advantage of the HC character for organic dyes of this series. The observed VOC upon co-sensitization was enhanced through the retarded charge recombination overwhelming the downward shift of the TiO2 potential according to the results obtained from measurements of charge extraction and intensity-modulated photovoltage spectra.111
We have shown that co-sensitization of complementary porphyrin and organic dyes produces a panchromatic spectral feature to promote the performance of PSSC. For the system LD12 + CD5,111 the IPCE spectrum extended to ∼650 nm; for the system YD2-oC8 + Y123,24 the efficiency response extended further to ∼700 nm. The near-IR dyes previously introduced75,79,80 are thus candidates to elevate the photocurrents and promote the overall cell efficiencies. Yeh and Diau112 designed a dimeric porphyrin (YDD6, Fig. 18), in which eight tert-butyl side chains in YDD0 were replaced with eight ortho-substituted isoamyloxy chains,113 and the meso-phenyl opposite the anchoring group was replaced with a diarylamino group, similar to the molecular design of YD2-oC8 (Fig. 14a) but with the YDD0 (Fig. 11a) diporphyrin core as a light-harvesting centre. The ortho-substituted alkoxyl chains in YDD6 failed to prevent dye aggregation for this porphyrin; molecular engineering of co-sensitization was implemented in a solution containing three spectrally complementary dyes – YDD6, YD2-oC8 and CD4 – at a specific molar ratio to optimize the device performance of the system. As shown in Fig. 18, the IPCE action spectrum of the co-sensitized device attained 75–80% in region 400–700 nm and 40–45% in region 700–800 nm, giving a short-circuit current density JSC = 19.28 mA cm−2, which is significantly improved over that of YD2-oC8, 16.60 mA cm−2, and that of the YD2-oC8 + CD4 co-sensitized system, 16.91 mA cm−2, because of the additional light-harvesting power in the near-infrared region without degradation of the performance in the visible region. Using an iodine-based electrolyte, the best co-sensitized system achieved a remarkable power conversion efficiency of 10.4%, which is much superior to those obtained from their individual single-dye devices and the two-dye co-sensitized systems.112 These results indicate that the suppression of dye aggregation for YDD6 in the three-dye co-sensitized system is the key to improvement of the device performance. With progress using this approach of co-sensitization, a future challenge is to seek other potential near-IR co-sensitizers with not only a light-harvesting ability extending beyond 800 nm but also decreased dye aggregation to enhance IPCE above 80%.
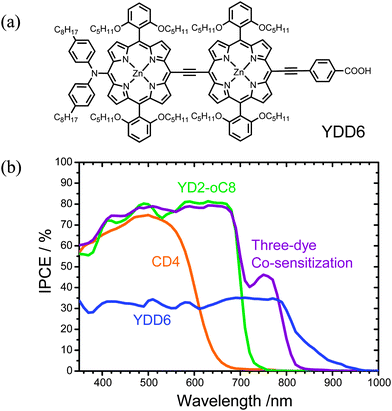 | ||
| Fig. 18 (a) Molecular structure of YDD6 and (b) IPCE spectra of CD4, YD2-oC8, YDD6 and three-dye co-sensitized systems.112 | ||
5. Summary and future perspectives
Inspired by the efficient energy transfer in naturally occurring photosynthetic reaction centers, numerous porphyrin and dimeric porphyrin sensitizers are introduced in this review of highly efficient dye-sensitized solar cells. At the current stage, the device fabricated using the push–pull zinc porphyrin, YD2-oC8, co-sensitized with Y123 on a TiO2 film and using a cobalt electrolyte has attained an unprecedented efficiency of 12.3% under standard AM 1.5 one-sun irradiation; such efficiency of power conversion has never been achieved with a ruthenium-free sensitizer. Similar to the design of YD2-oC8, ortho-substituted push–pull porphyrins such as LD14, LD16 and YD22 with extended light harvesting towards ∼750 nm are expected to yield performance at least comparable to that of YD2-oC8. Functional chromophores such as anthracene in LAC-3, tetracene in LAC-4, pentacene in LAC-5, fluorene in LD22, pyrene in LD4, BODIPY in LH3, and perylene anhydride-linked porphyrin derivatives might also play a role to improve the light-harvesting ability when they can be integrated into the framework of an ortho-substituted porphyrin to build a new PSSC. Fused porphyrins such as fused-Zn-1, WW1, WW2, YDD3, and their derivatives, porphyrin dimers such as DTBC, YDD0, YDD6 and their derivatives, are prospective photosensitizers to extend the light-harvesting power toward the near-IR spectral region. In most cases those fused and dimeric porphyrins suffered from poor device performance due to their nearly co-planar structural feature favoring the formation of dye aggregates to deteriorate significantly the device performance – a solution to solve the aggregation problem for porphyrins is to implement the approach of co-sensitization. Here we highlight the significance of molecular co-sensitization of multiple dyes with complementary absorption spectra on a semiconductor film, which would be an effective approach to enhance the light-harvesting ability and to retard the charge recombination for significant promotion of the overall performance of a PSSC. Table 1 summarizes the current status of device performance for porphyrin sensitizers or co-sensitization systems introduced in this review.| Sensitizers | Device efficiency (%) | Reference efficiency (%) | Ref. |
|---|---|---|---|
| Zn-1a | 4.8 | — | 38 |
| Zn-3 | 5.6 | — | 39 |
| GD2 | 7.1 | — | 40 |
| 2b-bdta-Zn | 3.0 | N3, 5.9 | 41 |
| tda-2b-bd-Zn | 7.5 | N3, 7.7 | 45 |
| Fused-Zn-1 | 4.1 | Zn-1, 2.8 | 76, 77 |
| ZnQMA | 5.2 | N719, 6.5 | 79 |
| WW2 | 1.4 | — | 75 |
| KS-3 | 4.0 | — | 63 |
| PZn-hT | 5.1 | N719, 8.0 | 64 |
| LAC-3 | 5.4 | N719, 6.7 | 66 |
| LAC-4 | 2.8 | N719, 6.7 | 66 |
| LD4 | 10.1 | N719, 9.3 | 68 |
| LD12 | 7.4 | — | 89 |
| LD14 | 10.2 | — | 89 |
| LD16 | 10.2 | — | 88 |
| LD22 | 8.1 | N719, 9.2 | 67 |
| YD0 | 4.5 | N719, 7.3 | 57 |
| YD1 | 6.5 | N719, 7.3 | 57 |
| YD2 | 6.8 | N719, 7.3 | 57 |
| YD2 | 11 | — | 42 |
| YD11 | 6.8 | N719, 7.3 | 69 |
| YD12 | 7.0 | N719, 7.3 | 69 |
| YD13 | 1.9 | N719, 7.3 | 69 |
| YD14 | 6.8 | YD2, 7.1 | 61 |
| YD15 | 4.2 | YD2, 7.1 | 61 |
| YD20 | 8.1 | — | 90 |
| YDD0 | 4.1 | YD0, 5.1 | 80 |
| YDD1 | 5.2 | YD0, 5.1 | 80 |
| DTBC | 5.2 | — | 83 |
| LH3 | 1.6 | — | 84 |
| ZnPDCA | 5.5 | N719, ∼6 | 85 |
| YD2-oC8 | 11.9 | YD2, 8.4 (cobalt electrolyte) | 24 |
| YD2-oC8+Y123 | 12.3 | YD2, 8.4 (cobalt electrolyte) | 24 |
| 2Flu-ZnP-CN-COOH + HC-A1 | 7.2 | N719, 8.6 | 105 |
| LD12 + BET | 7.5 | LD12, 6.7 | 109 |
| LD12 + CD5 | 9.0 | LD12, 7.5 | 111 |
| YDD6 + YD2-oC8 + CD4 | 10.4 | YD2-oC8, 8.8 | 112 |
Hardin, Snaith and McGehee114 suggested that extending the dye absorption to 830 nm might increase the efficiency of the YD2-oC8 system to 13.6%; an ultimate device performance in a cobalt-electrolyte system is estimated to be ∼19% if the total loss in potential can be decreased to 500 mV and the absorption spectrum of the dye can be extended to 920 nm. We show the absorption spectra of typical porphyrin sensitizers adsorbed on TiO2 thin films in Fig. 19, for which the spectral edges of YD0, YD2, YDD0 and YDD3 are approximately 650, 700, 800 and 950 nm, respectively; the edges of other porphyrins discussed here are also indicated. Both fused porphyrins WW2 and YDD3 have spectral edges of about 950 nm; they are perfect candidates to promote the efficiency of DSSC if suitable structural modifications and appropriate co-sensitization procedure are applied. We believe that this review provides guidance for strategies to design the best donor–acceptor porphyrin system and to apply an optimal co-sensitization for porphyrin and hole-conducting organic dyes towards a new record for the future developments and applications of DSSC.
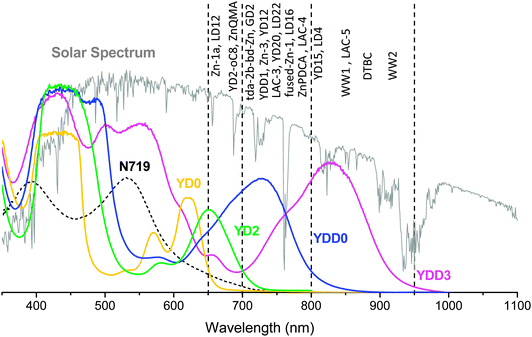 | ||
| Fig. 19 Thin-film absorption spectra of typical porphyrin sensitizers with spectral edges indicated by dashed lines at 650, 700, 800 and 950 nm for YD0, YD2, YDD0 and YDD3, respectively. The approximate positions of the spectral edges of other porphyrins reviewed in this article are indicated. | ||
Acknowledgements
National Science Council of Taiwan (NSC101-2627-M-009-007) and Ministry of Education of Taiwan under the ATU program supported this work. We are indebted to many collaborators who provided invaluable porphyrin and organic dyes for PSSC applications reported in this review article: Prof. Chen-Yu Yeh of NCHU (Taiwan) for the YD-series porphyrins, Prof. Ching-Yao Lin of NCNU (Taiwan) for the LD-series porphyrins, Prof. Chien-Tien Chen of NTHU (Taiwan) for the CD-series dyes, Prof. Chen-Hsiung Hung of Academia Sinica (Taiwan) for thienyl porphyrins, Prof. Hwan Kyu Kim of Korea University (Korea) for HC-A1 and HC-A3 dyes, and Prof. Hongshen He of SDSU (USA) for the BET dye. We thank Prof. Michael Grätzel of EPFL (Switzerland) for optimization of the device performance of PSSC and for guiding us in the direction to design new porphyrin sensitizers. We are grateful to Prof. Juan Bisquert of UJI (Spain) for many enlightening discussions on impedance spectra. We thank the researchers for their works cited in this review.References
- B. Oregan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphrybaker, E. Muller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- M. K. Nazeeruddin, P. Pechy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613–1624 CrossRef CAS.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef.
- A. Hagfeldt, G. Boschloo, L. C. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, T. Bessho and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835–16847 CrossRef CAS.
- Q. Wang, S. Ito, M. Grätzel, F. Fabregat-Santiago, I. Mora-Sero, J. Bisquert, T. Bessho and H. Imai, J. Phys. Chem. B, 2006, 110, 25210–25221 CrossRef CAS.
- C. Y. Chen, M. K. Wang, J. Y. Li, N. Pootrakulchote, L. Alibabaei, C. H. Ngoc-le, J. D. Decoppet, J. H. Tsai, C. Grätzel, C. G. Wu, S. M. Zakeeruddin and M. Grätzel, ACS Nano, 2009, 3, 3103–3109 CrossRef CAS.
- Y. M. Cao, Y. Bai, Q. J. Yu, Y. M. Cheng, S. Liu, D. Shi, F. F. Gao and P. Wang, J. Phys. Chem. C, 2009, 113, 6290–6297 CAS.
- Q. J. Yu, Y. H. Wang, Z. H. Yi, N. N. Zu, J. Zhang, M. Zhang and P. Wang, ACS Nano, 2010, 4, 6032–6038 CrossRef CAS.
- L. Y. Han, A. Islam, H. Chen, C. Malapaka, B. Chiranjeevi, S. F. Zhang, X. D. Yang and M. Yanagida, Energy Environ. Sci., 2012, 5, 6057–6060 CAS.
- N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338–2345 CrossRef CAS.
- Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903–2934 CrossRef CAS.
- A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS.
- Z. J. Ning, Y. Fu and H. Tian, Energy Environ. Sci., 2010, 3, 1170–1181 CAS.
- H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809–1818 CrossRef CAS.
- I. Radivojevic, A. Varotto, C. Farley and C. M. Drain, Energy Environ. Sci., 2010, 3, 1897–1909 CAS.
- M. V. Martínez-Díaz, G. de la Torrea and T. Torres, Chem. Commun., 2010, 46, 7090–7108 RSC.
- M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 759–792 CrossRef CAS.
- M. J. Griffith, K. Sunahara, P. Wagner, K. Wagner, G. G. Wallace, D. L. Officer, A. Furube, R. Katoh, S. Mori and A. J. Mozer, Chem. Commun., 2012, 48, 4145–4162 RSC.
- H. Imahori, T. Umeyama, K. Kurotobi and Y. Takano, Chem. Commun., 2012, 48, 4032–4045 RSC.
- M. K. Panda, K. Ladomenou and A. G. Coutsolelos, Coord. Chem. Rev., 2012, 256, 2601–2627 CrossRef CAS.
- T. Hasobe, H. Imahori, P. V. Kamat, T. K. Ahn, S. K. Kim, D. Kim, A. Fujimoto, T. Hirakawa and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 1216–1228 CrossRef CAS.
- A. Yella, H. W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS.
- F. Gao, Y. Wang, D. Shi, J. Zhang, M. K. Wang, X. Y. Jing, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 130, 10720–10728 CrossRef CAS.
- T. Horiuchi, H. Miura, K. Sumioka and S. Uchida, J. Am. Chem. Soc., 2004, 126, 12218–12219 CrossRef CAS.
- K. Hara, Z.-S. Wang, T. Sato, A. Furube, R. Katoh, H. Sugihara, Y. Dan-oh, C. Kasada, A. Shinpo and S. Suga, J. Phys. Chem. B, 2005, 109, 15476–15482 CrossRef CAS.
- S. Kim, J. K. Lee, S. O. Kang, J. Ko, J. H. Yum, S. Fantacci, F. De Angelis, D. Di Censo, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701–16707 CrossRef CAS.
- S. Ito, S. M. Zakeeruddin, R. Humphry-Baker, P. Liska, R. Charvet, P. Comte, M. K. Nazeeruddin, P. Pechy, M. Takata, H. Miura, S. Uchida and M. Grätzel, Adv. Mater., 2006, 18, 1202–1205 CrossRef CAS.
- S. Hwang, J. H. Lee, C. Park, H. Lee, C. Kim, C. Park, M.-H. Lee, W. Lee, J. Park, K. Kim, N.-G. Park and C. Kim, Chem. Commun., 2007, 4887–4889 RSC.
- Z. S. Wang, N. Koumura, Y. Cui, M. Takahashi, H. Sekiguchi, A. Mori, T. Kubo, A. Furube and K. Hara, Chem. Mater., 2008, 20, 3993–4003 CrossRef CAS.
- S. Ito, H. Miura, S. Uchida, M. Takata, K. Sumioka, P. Liska, P. Comte, P. Pechy and M. Grätzel, Chem. Commun., 2008, 5194–5196 RSC.
- W. Zeng, Y. Cao, Y. Bai, Y. Wang, Y. Shi, M. Zhang, F. Wang, C. Pan and P. Wang, Chem. Mater., 2010, 22, 1915–1925 CrossRef CAS.
- H. N. Tsao, C. Yi, T. Moehl, J.-H. Yum, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, ChemSusChem, 2011, 4, 591–594 CrossRef CAS.
- M. Xu, M. Zhang, M. Pastore, R. Li, F. De Angelis and P. Wang, Chem. Sci., 2012, 3, 976–983 RSC.
- A. Kay and M. Grätzel, J. Phys. Chem., 1993, 97, 6272–6277 CrossRef CAS.
- S. Cherian and C. C. Wamser, J. Phys. Chem. B, 2000, 104, 3624–3629 CrossRef CAS.
- M. K. Nazeeruddin, R. Humphry-Baker, D. L. Officer, W. M. Campbell, A. K. Burrell and M. Grätzel, Langmuir, 2004, 20, 6514–6517 CrossRef CAS.
- Q. Wang, W. M. Carnpbell, E. E. Bonfantani, K. W. Jolley, D. L. Officer, P. J. Walsh, K. Gordon, R. Humphry-Baker, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. B, 2005, 109, 15397–15409 CrossRef CAS.
- W. M. Campbell, K. W. Jolley, P. Wagner, K. Wagner, P. J. Walsh, K. C. Gordon, L. Schmidt-Mende, M. K. Nazeeruddin, Q. Wang, M. Grätzel and D. L. Officer, J. Phys. Chem. C, 2007, 111, 11760–11762 CAS.
- J. K. Park, H. R. Lee, J. P. Chen, H. Shinokubo, A. Osuka and D. Kim, J. Phys. Chem. C, 2008, 112, 16691–16699 CAS.
- T. Bessho, S. M. Zakeeruddin, C. Y. Yeh, E. W. G. Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6646–6649 CrossRef CAS.
- W. M. Campbell, A. K. Burrell, D. L. Officer and K. W. Jolley, Coord. Chem. Rev., 2004, 248, 1363–1379 CrossRef CAS.
- H. He, A. Gurung and L. Si, Chem. Commun., 2012, 48, 5910–5912 RSC.
- M. Ishida, S. W. Park, D. Hwang, Y. B. Koo, J. L. Sessler, D. Y. Kim and D. Kim, J. Phys. Chem. C, 2011, 115, 19343–19354 CAS.
- T. E. O. Screen, K. B. Lawton, G. S. Wilson, N. Dolney, R. Ispasoiu, T. Goodson Iii, S. J. Martin, D. D. C. Bradley and H. L. Anderson, J. Mater. Chem., 2001, 11, 312–320 RSC.
- V. Lin, S. DiMagno and M. Therien, Science, 1994, 264, 1105–1111 CAS.
- J. Rochford, D. Chu, A. Hagfeldt and E. Galoppini, J. Am. Chem. Soc., 2007, 129, 4655–4665 CrossRef CAS.
- J. R. Stromberg, A. Marton, H. L. Kee, C. Kirmaier, J. R. Diers, C. Muthiah, M. Taniguchi, J. S. Lindsey, D. F. Bocian, G. J. Meyer and D. Holten, J. Phys. Chem. C, 2007, 111, 15464–15478 CAS.
- S. Eu, S. Hayashi, T. Umeyama, A. Oguro, M. Kawasaki, N. Kadota, Y. Matano and H. Imahori, J. Phys. Chem. C, 2007, 111, 3528–3537 CAS.
- O. Taratula, J. Rochford, P. Piotrowiak, E. Galoppini, R. A. Carlisle and G. J. Meyer, J. Phys. Chem. B, 2006, 110, 15734–15741 CrossRef CAS.
- C. C. Clark, G. J. Meyer, Q. Wei and E. Galoppini, J. Phys. Chem. B, 2006, 110, 11044–11046 CrossRef CAS.
- C.-Y. Lin, C.-F. Lo, L. Luo, H.-P. Lu, C.-S. Hung and E. W.-G. Diau, J. Phys. Chem. C, 2009, 113, 755–764 CAS.
- L. Luo, C. J. Lin, C. Y. Tsai, H. P. Wu, L. L. Li, C. F. Lo, C. Y. Lin and E. W. G. Diau, Phys. Chem. Chem. Phys., 2010, 12, 1064–1071 RSC.
- L. Y. Luo, C. J. Lin, C. S. Hung, C. F. Lo, C. Y. Lin and E. W. G. Diau, Phys. Chem. Chem. Phys., 2010, 12, 12973–12977 RSC.
- C. W. Lee, H. P. Lu, C. M. Lan, Y. L. Huang, Y. R. Liang, W. N. Yen, Y. C. Liu, Y. S. Lin, E. W. G. Diau and C. Y. Yeh, Chem.–Eur. J., 2009, 15, 1403–1412 CrossRef CAS.
- H. P. Lu, C. Y. Tsai, W. N. Yen, C. P. Hsieh, C. W. Lee, C. Y. Yeh and E. W. G. Diau, J. Phys. Chem. C, 2009, 113, 20990–20997 CAS.
- C.-P. Hsieh, H.-P. Lu, C.-L. Chiu, C.-W. Lee, S.-H. Chuang, C.-L. Mai, W.-N. Yen, S.-J. Hsu, E. W.-G. Diau and C.-Y. Yeh, J. Mater. Chem., 2010, 20, 1127–1134 RSC.
- P. Wang, S. M. Zakeeruddin, J. E. Moser, M. K. Nazeeruddin, T. Sekiguchi and M. Grätzel, Nat. Mater., 2003, 2, 402–407 CrossRef CAS.
- A. J. Mozer, P. Wagner, D. L. Officer, G. G. Wallace, W. M. Campbell, M. Miyashita, K. Sunahara and S. Mori, Chem. Commun., 2008, 4741–4743 RSC.
- S. L. Wu, H. P. Lu, H. T. Yu, S. H. Chuang, C. L. Chiu, C. W. Lee, E. W. G. Diau and C. Y. Yeh, Energy Environ. Sci., 2010, 3, 949–955 CAS.
- H. Imahori, Y. Matsubara, H. Iijima, T. Umeyama, Y. Matano, S. Ito, M. Niemi, N. V. Tkachenko and H. Lemmetyinen, J. Phys. Chem. C, 2010, 114, 10656–10665 CAS.
- V. A. Nuay, D.-H. Kim, S.-H. Lee and J.-J. Ko, Bull. Korean Chem. Soc., 2009, 30, 2871–2872 CrossRef CAS.
- Y. Liu, N. Xiang, X. Feng, P. Shen, W. Zhou, C. Weng, B. Zhao and S. Tan, Chem. Commun., 2009, 2499–2501 RSC.
- R. Ambre, K.-B. Chen, C.-F. Yao, L. Luo, E. W.-G. Diau and C.-H. Hung, J. Phys. Chem. C, 2012, 116, 11907–11916 CAS.
- C.-Y. Lin, Y.-C. Wang, S.-J. Hsu, C.-F. Lo and E. W.-G. Diau, J. Phys. Chem. C, 2010, 114, 687–693 CAS.
- C. H. Wu, T. Y. Pan, S. H. Hong, C. L. Wang, H. H. Kuo, Y. Y. Chu, E. W. Diau and C. Y. Lin, Chem. Commun., 2012, 48, 4329–4331 RSC.
- C.-L. Wang, Y.-C. Chang, C.-M. Lan, C.-F. Lo, E. Wei-Guang Diau and C.-Y. Lin, Energy Environ. Sci., 2011, 4, 1788–1795 CAS.
- H. P. Lu, C. L. Mai, C. Y. Tsia, S. J. Hsu, C. P. Hsieh, C. L. Chiu, C. Y. Yeh and E. W. G. Diau, Phys. Chem. Chem. Phys., 2009, 11, 10270–10274 RSC.
- S. Ito, T. N. Murakami, P. Comte, P. Liska, C. Grätzel, M. K. Nazeeruddin and M. Grätzel, Thin Solid Films, 2008, 516, 4613–4619 CrossRef CAS.
- T. Edvinsson, C. Li, N. Pschirer, J. Schöneboom, F. Eickemeyer, R. Sens, G. Boschloo, A. Herrmann, K. Müllen and A. Hagfeldt, J. Phys. Chem. C, 2007, 111, 15137–15140 CAS.
- C. Li, J.-H. Yum, S.-J. Moon, A. Herrmann, F. Eickemeyer, N. G. Pschirer, P. Erk, J. Schöneboom, K. Müllen, M. Grätzel and M. K. Nazeeruddin, ChemSusChem, 2008, 1, 615–618 CrossRef CAS.
- H. Y. Chen, H. P. Lu, C. W. Lee, S. H. Chuang, E. W. G. Diau and C. Y. Yeh, J. Chin. Chem. Soc., 2010, 57, 1141–1146 CAS.
- C.-W. Lee, H.-P. Lu, N. M. Reddy, H.-W. Lee, E. W.-G. Diau and C.-Y. Yeh, Dyes Pigm., 2011, 91, 317–323 CrossRef CAS.
- C. Jiao, N. Zu, K.-W. Huang, P. Wang and J. Wu, Org. Lett., 2011, 13, 3652–3655 CrossRef CAS.
- M. Tanaka, S. Hayashi, S. Eu, T. Umeyama, Y. Matano and H. Imahori, Chem. Commun., 2007, 2069–2071 RSC.
- S. Hayashi, M. Tanaka, H. Hayashi, S. Eu, T. Umeyama, Y. Matano, Y. Araki and H. Imahori, J. Phys. Chem. C, 2008, 112, 15576–15585 CAS.
- S. Hayashi, Y. Matsubara, S. Eu, H. Hayashi, T. Umeyama, Y. Matano and H. Imahori, Chem. Lett., 2008, 37, 846–847 CrossRef CAS.
- S. Eu, S. Hayashi, T. Umeyama, Y. Matano, Y. Araki and H. Imahori, J. Phys. Chem. C, 2008, 112, 4396–4405 CAS.
- C. L. Mai, W. K. Huang, H. P. Lu, C. W. Lee, C. L. Chiu, Y. R. Liang, E. W. Diau and C. Y. Yeh, Chem. Commun., 2010, 46, 809–811 RSC.
- A. J. Mozer, M. J. Griffith, G. Tsekouras, P. Wagner, G. G. Wallace, S. Mori, K. Sunahara, M. Miyashita, J. C. Earles, K. C. Gordon, L. C. Du, R. Katoh, A. Furube and D. L. Officer, J. Am. Chem. Soc., 2009, 131, 15621–15623 CrossRef CAS.
- J. K. Park, J. P. Chen, H. R. Lee, S. W. Park, H. Shinokubo, A. Osuka and D. Kim, J. Phys. Chem. C, 2009, 113, 21956–21963 CAS.
- Y. Liu, H. Lin, J. T. Dy, K. Tamaki, J. Nakazaki, D. Nakayama, S. Uchida, T. Kubo and H. Segawa, Chem. Commun., 2011, 47, 4010–4012 RSC.
- C. Y. Lee and J. T. Hupp, Langmuir, 2010, 26, 3760–3765 CrossRef CAS.
- C. Y. Lee, C. X. She, N. C. Jeong and J. T. Hupp, Chem. Commun., 2010, 46, 6090–6092 RSC.
- H. Imahori, S. Hayashi, H. Hayashi, A. Oguro, S. Eu, T. Umeyama and Y. Matano, J. Phys. Chem. C, 2009, 113, 18406–18413 CAS.
- S. Mathew, H. Iijima, Y. Toude, T. Umeyama, Y. Matano, S. Ito, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, J. Phys. Chem. C, 2011, 115, 14415–14424 CAS.
- C.-L. Wang, C.-M. Lan, S.-H. Hong, Y.-F. Wang, T.-Y. Pan, C.-W. Chang, H.-H. Kuo, M.-Y. Kuo, E. W.-G. Diau and C.-Y. Lin, Energy Environ. Sci., 2012, 5, 6933–6940 CAS.
- Y. C. Chang, C. L. Wang, T. Y. Pan, S. H. Hong, C. M. Lan, H. H. Kuo, C. F. Lo, H. Y. Hsu, C. Y. Lin and E. W. Diau, Chem. Commun., 2011, 47, 8910–8912 RSC.
- T. Ripolles-Sanchis, B. C. Guo, H. P. Wu, T. Y. Pan, H. W. Lee, S. R. Raga, F. Fabregat-Santiago, J. Bisquert, C. Y. Yeh and E. W. Diau, Chem. Commun., 2012, 48, 4368–4370 RSC.
- A. Dualeh, F. De Angelis, S. Fantacci, T. Moehl, C. Yi, F. Kessler, E. Baranoff, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. C, 2011, 116, 1572–1578 Search PubMed.
- R. Y. Ogura, S. Nakane, M. Morooka, M. Orihashi, Y. Suzuki and K. Noda, Appl. Phys. Lett., 2009, 94, 073308 CrossRef.
- Y. Chen, Z. Zeng, C. Li, W. Wang, X. Wang and B. Zhang, New J. Chem., 2005, 29, 773–776 RSC.
- D. Kuang, P. Walter, F. Nuesch, S. Kim, J. Ko, P. Comte, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, Langmuir, 2007, 23, 10906–10909 CrossRef CAS.
- J. J. Cid, J. H. Yum, S. R. Jang, M. K. Nazeeruddin, E. M. Ferrero, E. Palomares, J. Ko, M. Grätzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358–8362 CrossRef CAS.
- J. N. Clifford, A. Forneli, H. J. Chen, T. Torres, S. T. Tan and E. Palomares, J. Mater. Chem., 2011, 21, 1693–1696 RSC.
- M. Kimura, H. Nomoto, N. Masaki and S. Mori, Angew. Chem., Int. Ed., 2012, 51, 4371–4374 CrossRef CAS.
- H. Choi, S. Kim, S. O. Kang, J. J. Ko, M. S. Kang, J. N. Clifford, A. Forneli, E. Palomares, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 8259–8263 CrossRef CAS.
- F. Inakazu, Y. Noma, Y. Ogomi and S. Hayase, Appl. Phys. Lett., 2008, 93, 093304 CrossRef.
- K. Lee, S. W. Park, M. J. Ko, K. Kim and N. G. Park, Nat. Mater., 2009, 8, 665–671 CrossRef CAS.
- N. Robertson, Angew. Chem., Int. Ed., 2008, 47, 1012–1014 CrossRef CAS.
- D. Kuang, S. Uchida, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 1923–1927 CrossRef CAS.
- M. J. Griffith, A. J. Mozer, G. Tsekouras, Y. Dong, P. Wagner, K. Wagner, G. G. Wallace, S. Mori and D. L. Officer, Appl. Phys. Lett., 2011, 98, 163502 CrossRef.
- B. J. Song, H. M. Song, I. T. Choi, S. K. Kim, K. D. Seo, M. S. Kang, M. J. Lee, D. W. Cho, M. J. Ju and H. K. Kim, Chem.–Eur. J., 2011, 17, 11115–11121 CrossRef CAS.
- M. S. Kang, S. H. Kang, S. G. Kim, I. T. Choi, J. H. Ryu, M. J. Ju, D. Cho, J. Y. Lee and H. K. Kim, Chem. Commun., 2012, 48, 9349–9351 RSC.
- H. M. Song, K. D. Seo, M. S. Kang, I. T. Choi, S. K. Kim, Y. K. Eom, J. H. Ryu, M. J. Ju and H. K. Kim, J. Mater. Chem., 2012, 22, 3786–3794 RSC.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS.
- J. Warnan, Y. Pellegrin, E. Blart and F. Odobel, Chem. Commun., 2012, 48, 675–677 RSC.
- M. Shrestha, L. P. Si, C. W. Chang, H. S. He, A. Sykes, C. Y. Lin and E. W. G. Diau, J. Phys. Chem. C, 2012, 116, 10451–10460 CAS.
- W. S. Chao, K. H. Liao, C. T. Chen, W. K. Huang, C. M. Lan and E. W. Diau, Chem. Commun., 2012, 48, 4884–4886 RSC.
- C.-M. Lan, H.-P. Wu, T.-Y. Pan, C.-W. Chang, W.-S. Chao, C.-T. Chen, C.-L. Wang, C.-Y. Lin and E. W.-G. Diau, Energy Environ. Sci., 2012, 5, 6460–6464 CAS.
- H.-P. Wu, Z.-W. Ou, T.-Y. Pan, C.-M. Lan, W.-K. Huang, H.-W. Lee, N. M. Reddy, C.-T. Chen, W.-S. Chao, C.-Y. Yeh and E. W.-G. Diau, Energy Environ. Sci., 2012 10.1039/c2ee22870j.
- M. P. Nikiforov, U. Zerweck, P. Milde, C. Loppacher, T. H. Park, H. T. Uyeda, M. J. Therien, L. Eng and D. Bonnell, Nano Lett., 2008, 8, 110–113 CrossRef CAS.
- B. E. Hardin, H. J. Snaith and M. D. McGehee, Nat. Photonics, 2012, 6, 162–169 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |