Modulation of excimer formation of 9-(dicyano-vinyl)julolidine by the macrocyclic hosts†
Received
18th September 2012
, Accepted 26th October 2012
First published on 29th October 2012
Abstract
This article reports the alteration of the excited state photophysics of a molecular rotor, namely 9-(dicyano-vinyl)julolidine (DCVJ), which has been extensively used to report protein aggregation and protein conformational changes, by the various cavity sizes of cyclodextrin (CD) macrocyclic hosts, with the help of steady state, time-resolved fluorescence techniques. It is observed that, in the presence of α-CD, the characteristic features of both the monomer and excimer emissions of DCVJ are almost unperturbed. However, in the presence of β-CD, the excited photophysics of the molecule is significantly perturbed, and it is noted that β-CD inhibits the excimer formation drift of DCVJ by incorporation of a DCVJ monomer inside its cavity. The most striking findings are observed in the case of γ-CD. Initially, the excimer peak intensity drops and the monomer intensity increases, due to the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DCVJ/γ-CD inclusion complex formation. Above a certain concentration, another DCVJ molecule is accommodated inside the γ-CD cavity and forms an excimer, which is reflected in the intensification of the excimer peak. At higher γ-CD concentration the fluorescence intensity of the excimer shoots up, due to the formation of 2:2 host–guest complex, in which an additional γ-CD molecule provides extra stabilization to the excimer. Insight on the molecular picture of this host–guest interaction has been provided by docking studies followed by quantum chemical calculations.
:1 DCVJ/γ-CD inclusion complex formation. Above a certain concentration, another DCVJ molecule is accommodated inside the γ-CD cavity and forms an excimer, which is reflected in the intensification of the excimer peak. At higher γ-CD concentration the fluorescence intensity of the excimer shoots up, due to the formation of 2:2 host–guest complex, in which an additional γ-CD molecule provides extra stabilization to the excimer. Insight on the molecular picture of this host–guest interaction has been provided by docking studies followed by quantum chemical calculations.
Introduction
Supramolecular hosts are a good model system for nano-cavities due to their various sizes, shapes and hydrophobicities.1–7 They have been found to be particularly useful in monitoring the formation of host–guest inclusion complexes by incorporating fluorescent dyes inside their cavities.1–7 The inclusion of a fluorescent guest into a cavity is accompanied by a large change in fluorescent properties, as a consequence of an altered microenvironment.1–7 One typical example of such a supramolecular host–guest inclusion complex consists of CD and a guest fluorescent molecule.1–5 CDs are cyclic oligosaccharides with 6, 7 and 8 D-glucose units, and they have lipophilic cavities with different inner diameters (5.7, 7.0, 8.5 Å for α-, β- and γ-CD, respectively), which provide relatively hydrophobic cavities for guest binding.1–3 The most remarkable feature of CDs is their ability to form inclusion complexes with a variety of organic molecules. Although the stoichiometry of the complex is usually 1:1 in aqueous solution, a 1:2 host–guest inclusion complex is also feasible for higher cavity sizes.8–12 This property of cyclodextrins is used to control the monomer–dimer equilibrium of dyes by monitoring their absorption and emission spectral properties.8–12
DCVJ is a classic example of a molecular rotor (Scheme 1). A molecular rotor is a fluorescent molecule that has the ability to undergo an intramolecular twisting motion in the excited state.13,14 Unlike other molecular rotors, like p-(dimethylamino)benzonitrile (DMABN), DCVJ always exhibits only a single emission band, even in a polar solvent, as the energy gap between S1 and S0 in the twisted intramolecular charge transfer (TICT) state of DCVJ is almost 3 times smaller than the locally excited (LE) energy gap.13 Hence, non-radiative relaxation from TICT is preferred over radiative relaxation.13 Very recently, it has been reported that the primary mechanism of non-radiative relaxation of DCVJ is isomerisation, rather than TICT state formation.15,16 DCVJ has been extensively used in biological applications. It has been reported that fluorescence quantum yield of DCVJ increases when it binds to tubilin.17 Moreover, during tubilin to tubules aggregation, further enhancement of DCVJ quantum yield takes place.17 The transformation of actin from its monomeric G-state to its polymeric F-state is monitored with the help of the fluorescence of DCVJ.18 DCVJ has also been used as a probe for measurement of micro viscosity and free volume in phospholipids bilayers and micelles.19,20
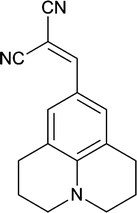 |
| | Scheme 1 Chemical structure of DCVJ. | |
Several studies have been devoted to understand the excited state properties of DCVJ in different solvents,16 polymers21 and ionic liquids.22,23 However, relatively few investigations have dealt with its excited state aggregation behaviour in water, despite the fact that dimerization/aggregation has a strong effect on the emission spectra of DCVJ, which might have significant implications for analyzing protein structure and conformation using DCVJ. Bhattacharyya et al. noticed for the first time that the emission spectra of DCVJ in water depend on concentration, and the intensity of longer wavelength emission bands increases with increasing DCVJ concentration.24 They also observed that longer wavelength emission band intensity increases with an increase in γ-CD concentration, and this was attributed to the formation of a DCVJ excimer inside the γ-CD cavity.24 However, the dynamics aspects, which provides a deeper perception of this excimer formation process, has not been addressed in their study. In this work, we continue our efforts in this direction to gain insight into the detailed picture of the excimer formation dynamics of DCVJ in the presence of α-, β- and γ-CD with the help of steady state and time-resolved fluorescence spectroscopic techniques. Our studies reveal the formation of both 1:1 and multiple inclusion complexes depending on the cavity sizes of CDs. Finally, docking, as well as some theoretical calculations, has been employed to understand the molecular picture of this host–guest interaction. We think that this kind of stoichiometry, as well as size based excimer formation drift of a molecule, might have some applications towards fluorescence on/off switches.
Experimental section
DCVJ (purity ≥97%) and all CDs were obtained from Sigma Aldrich, and used without further purification. Methanol (high-performance liquid chromatography grade) was obtained from Rankem, India chemicals. Due to low solubility of DCVJ in water, we prepared a concentrated stock solution of DCVJ in methanol. DCVJ stock solution in methanol (∼2 μL) was added to 2 ml of pH 7.0 phosphate buffer solution and sonicated for few minutes, in order to achieve a homogeneous solution of DCVJ. CD was then gradually added to the solution containing DCVJ, and the solution was gently shaken after each addition until CD was completely solubilized. Moreover, we allowed 15–20 minutes equilibration time for each addition of CD before taking measurements.
Absorption measurements were performed on a Perkin–Elmer UV-visible spectrophotometer (Lambda-45), and steady-state fluorescence spectra were recorded with a Fluorolog-3 spectrofluorometer (Horiba Jobin Yvon). All time-resolved fluorescence measurements were collected on a time correlated single photon counting (TCSPC) spectrometer (Horiba Jobin Yvon IBH, UK). The detailed description of the instrument can be found elsewhere.25–27 Briefly, in the present work a 444 nm diode laser (∼130 ps, 1 MHz repetition rate) was used as the excitation source and a MCP-PMT detector was used for collecting the fluorescence signal. The instrument response function for this experimental set-up was ∼80 ps. The analysis of lifetime was done by IBH DAS6 analysis software, in which a reconvolution procedure was used to analyze the observed decays using a proper instrument response function obtained by replacing the sample cell with a light scatterer (suspension of Ludox colloidal solution). The fluorescence decays were analyzed as a sum of exponentials as
| | 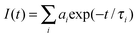 | (1) |
where
I(
t) is the time dependent fluorescence intensity, and
ai and
τi are the pre-exponential factor and the fluorescence lifetime for the
ith component of the fluorescence decay, respectively. The quality of the fits and the mono- or bi-exponential nature of the decays were evaluated by the reduced chi-square (
χ2) values and the distribution of the weighted residuals among the data channels. For a good fit, the
χ2 value was close to unity and the weighted residuals were distributed randomly among the data channels.
For time-resolved fluorescence anisotropy decay measurements, the polarized fluorescence decays for the parallel [I∥(t)] and perpendicular [I⊥(t)] emission polarizations with respect to the vertical excitation polarization were first collected at the emission maxima of the probe (either monomer peak at 510 nm or excimer peak at 570 nm). The anisotropy decay function r(t) was constructed from these I∥(t) and I⊥(t) decays using the following equation,
| | 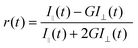 | (2) |
where
G is the correction factor for the detector sensitivity to the polarization detection of the emission. The anisotropy decay fitting was carried out using the following functional form
| | | r(t) = r0[a1r exp(− t/τ1r) + a2r exp(− t/τ2r)] | (3) |
in which
r0 is the limiting anisotropy that describes the inherent depolarization of the fluorophore and
air is the pre-exponential term that renders the fraction of the
ith rotational relaxation time,
i.e.,
τir. The quality of each fitting was judged by
χ2 values and visual inspection of the residuals.
Results and discussion
(a) Absorption spectral features of DCVJ in the absence and presence of CDs
Fig. 1 depicts the characteristic absorption profile of DCVJ in pH 7 buffer and in the presence of α-, β- and γ-CD. DCVJ exhibits an absorption maximum at 465 nm in pH 7 buffer and its absorption spectral features are almost unchanged with respect to the concentration of DCVJ. However, on gradual addition of α- and β-CD, the absorption maxima are red shifted together with an increase in absorption, and there is almost ∼6 nm bathochromic shift at higher CD concentrations. The increase in absorption, as well as the bathochromic shift in absorption spectra, indicates that some interaction is going on between the CDs (host) and DCVJ (guest). The origin of this red shift is not clear to us at this point, but it can probably be attributed to the hydrogen bonding interaction between the –OH groups of CDs and DCVJ molecule.
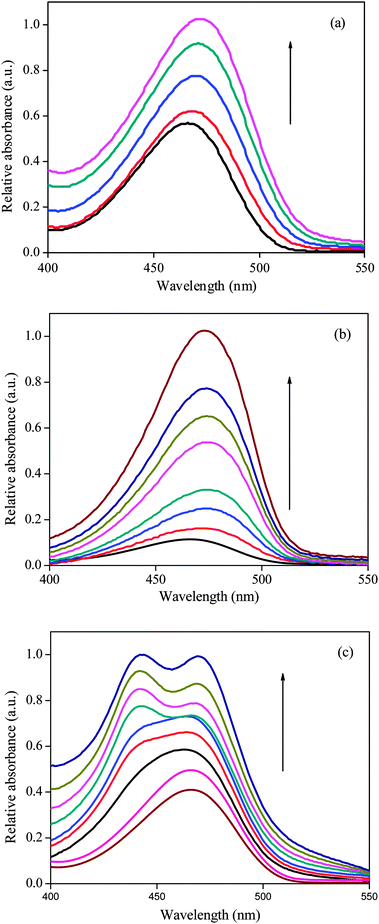 |
| | Fig. 1 Absorption spectra of DCVJ (∼12 μM) in presence of (a) α-CD (from 0 to 16 mM), (b) β-CD (from 0 to 14 mM) and (c) γ-CD (from 0 to 15.4 mM). | |
The spectral features of DCVJ in the presence of γ-CD are distinctly different from that of α- and β-CD (Fig. 1). The absorbance of DCVJ at ∼465 nm increases with incremental addition of γ-CD. Moreover, a shoulder at ∼446 nm begins to appear at relatively higher concentration of the host, and it transforms to a peak at the highest macrocyclic host concentration (≥12.5 mM). As γ-CD does not have any absorption in this region, the changes observed in the spectral features are related to the interaction of DCVJ with the γ-CD macrocycle. These notable changes in the absorption spectral feature of DCVJ at various concentrations of γ-CD indicate that more than one type of host–guest equilibrium exists in the presence of the macrocyclic host. Moreover, the interaction behaviour of DCVJ in the presence of γ-CD is quite different tfrom that of α- and β-CD, as the ∼444 nm peak is not at all present in the later macrocyles (Fig. 1). The appearance of a new peak at ∼444 nm at higher γ-CD concentration may be attributed to the formation of π-stack dimer of DCVJ molecules inside the nano-cavity of γ-CD, considering the larger cavity dimer of γ-CD. All these above mentioned observations will be further clarified by steady state, time-resolved and theoretical studies.
(b) Steady state fluorescence characteristics of DCVJ in absence and presence of CDs
The emission spectral features of DCVJ in pH 7 buffer and in the presence of CDs are depicted in Fig. 2. The emission spectrum of DCVJ in pH 7 consists of two peaks, one at 510 nm and another at 570 nm. It is observed that the 570 nm peak is absent at very low DCVJ concentration, however, the 570 nm peak starts appearing after ∼5 μM of DCVJ concentration (Fig. S1 in ESI†). At higher DCVJ concentration (>7.5 μM), the 570 nm peak intensity is significantly higher than that of 510 nm peak (Fig. S1, ESI†). Therefore, we believe that the 510 nm peak is due to the monomeric form of DCVJ, whereas the 570 nm peak may arise due to the association of dye molecules or excimer formation. The absorption spectra at higher DCVJ concentration exhibit a peak around 380 nm, indicating aggregation of dye takes place at higher concentration (Fig. S1 in ESI†). However, the emission spectra were collected during excitation at 465 nm, and there is no way that these aggregates of DCVJ are being excited at this wavelength. Therefore, we conclude that DCVJ molecules associate in the ground state by hydrophobic interaction, and these associated species are the origin of the long wavelength emission band, which is the characteristic feature of the excimer spectra. The prior association only lowers the concentration required for excimer formation, thereby excimer formation takes place even in such a low concentration. The excimer formation is further verified by the excitation spectra, in which it is found that the excitation spectra of DCVJ monitored at the two peaks (i.e. at 510 nm and 570 nm) is clearly different. Moreover, the excitation spectra monitored at 570 nm do not resemble the absorption spectrum of DCVJ in pH buffer medium. The concentration dependent emission profiles of DCVJ in water are in good agreement with that of Bhattacharyya et al.24
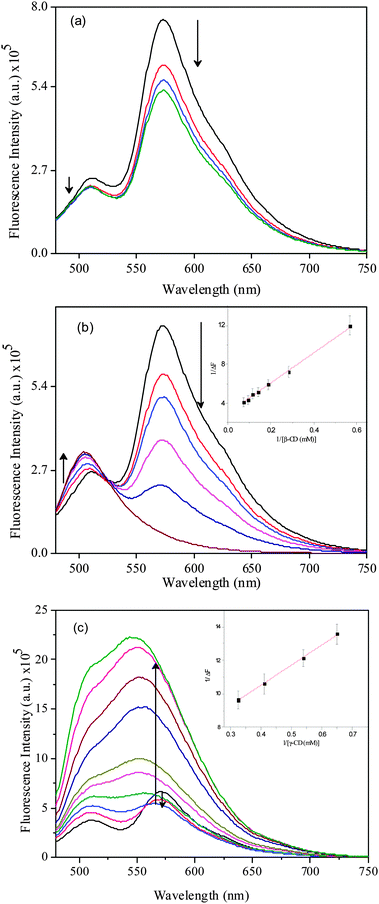 |
| | Fig. 2 Emission spectra of DCVJ (∼12 μM) in presence of (a) α-CD (0 to 16 mM), (b) β-CD (0 to 14 mM) and (c) γ-CD (0 to 15.4 mM). Inset represents the B–H plot for 1:1 inclusion complex for (b) β-CD and (c) γ-CD. | |
The emission spectral features of DCVJ in the presence of α-CD do not alter significantly, only the 570 nm peak intensity slightly reduces with incremental addition of α-CD (Fig. 2a). Here, it is worth mentioning that the width of a DCVJ molecule is ∼6.8 Å (along the julolidine group) and the length of a DCVJ molecule is ∼9.2 Å. Since the internal cavity diameter of α-CD is ∼5.7 Å,1 it is very unlikely that the julolidine moiety of DCVJ can be accommodated inside the cavity of α-CD. However, DCVJ can be partially included inside the α-CD cavity through the dicyano-vinyl part of the DCVJ molecule. Considering the fact that the major non-radiative pathway of DCVJ arises from rotation about the dicyano-vinyl group, the peak intensity at 510 nm would have increased significantly if the dicyano-vinyl part of the DCVJ molecule was encapsulated inside the cavity of α-CD. However, we have noticed that the monomer peak intensity of the DCVJ molecule remains almost constant with the increase in α-CD concentration (Fig. 2a). Therefore, we can rule out the possibility that the dicyano-vinyl group of DCVJ encapsulates inside the cavity of α-CD. The small variation in the excimer peak may be attributed to the hydrogen bonding interactions between the dicyano-vinyl group of DCVJ and –OH groups of α-CD, thereby inhibiting the excimer formation drift of DCVJ in water. The excitation spectra of DCVJ in the presence of α-CD, monitored at both monomer and excimer peak, well resembles the excitation spectrum of DCVJ in pH7 buffer (Fig. S2 and S3, ESI†). This again confirms that most of the DCVJ molecules are involved in excimer formation in solution, even in the presence of high concentration of α-CD.
The fluorescence spectra of DCVJ in the presence of β-CD display a striking feature (Fig. 2b). On incremental addition of β-CD, the intensity of the monomer peak at 510 nm increases along with a slight red shift, whereas the intensity of the excimer peak at 570 nm peak gradually reduces. At higher β-CD concentration, the excimer emission peak is almost invisible. These noteworthy changes in the emission spectral profile of DCVJ in the presence of β-CD are ascribed to the strong host–guest interaction between the macrocyclic host and the fluorophore. The significant decrease in excimer peak intensity suggests that formation of the excimer is inhibited by the inclusion of individual DCVJ molecules into the cavity of β-CD. Since the internal cavity diameter of β-CD is ∼7 Å1 and depth of the cavity is ∼8 Å1, almost a whole DCVJ molecule can be accommodated inside the cavity of β-CD. The linearity of the Benesi–Hildebrand28 plot monitored at 510 nm also supports the formation of 1:1 host–guest complexation between DCVJ and β-CD (inset of Fig. 2b). The binding constant value is determined to be 190(±15) M−1 from the linear fitting of the plot and this is certainly a high binding constant for a host–guest interaction. The increase of monomer peak intensity suggests that the non-radiative pathway, which arises due to olefinic bond rotation of DCVJ molecule of DCVJ molecule, is reduced on complexation with β-CD. Unlike α-CD, the excitation spectra of DCVJ in the presence of β-CD monitored at both monomer and excimer peaks are quite different to the excitation spectrum of dye in pH7 buffer (Fig. S2 and S3, ESI†). This observation once again supports the hypothesis that the DCVJ molecule is strongly interacting with β-CD. The above findings will be confirmed further through time-resolved fluorescence, as well as theoretical studies.
The emission spectra of DCVJ in the presence of γ-CD exhibit distinct features (Fig. 2c). It can be seen that the intensity of the monomer species is progressively enhanced with the increase in γ-CD concentration (Fig. 2c). However, excimer intensity at 570 nm initially decreases and then a significant enhancement in the excimer fluorescence is observed compared to the monomer peak, with a concomitant blue shift (Fig. 2c). At reasonably higher concentration (>3 mM), the enhancement of the excimer peak is substantially higher than that of the monomer peak. As the excimer peak becomes progressively broader, it is quite difficult to get an exact picture of the intensity pattern of each species in these complicated spectral features. Therefore, we have deconvoluted the emission spectra of DCVJ after each γ-CD addition (Fig. 3a). It can be clearly seen from the deconvoluted spectra that there is suppression of excimer formation by inclusion of DCVJ monomer in the γ-CD cavity at lower host concentration (<3 mM). The slight increase in monomer peak intensity is attributed to the reduction of non-radiative pathways of the DCVJ molecule inside the γ-CD cavity, due to the restriction imposed on the free rotation of the olefinic bond by the γ-CD cavity.15 It can also be clearly observed from the deconvoluted spectra (Fig. 3a) that, after a certain guest concentration (>3 mM), the excimer peak intensity is enhanced (Fig. 2c) significantly compared to the monomer peak. Since the internal cavity diameter of γ-CD (∼9 Å1) is large enough to accommodate two DCVJ molecules, the incorporation of two DCVJ molecules into the γ-CD cavity is possible. Therefore, the hike in intensity of the excimer peak with the increase in γ-CD concentration is ascribed to formation of an excimer inside the cavity of γ-CD, inferring that γ-CD forms a 1:2 (γ-CD:DCVJ) inclusion complex with DCVJ in this concentration range. In the titration procedure which we followed, the possibility of inclusion of two dye molecules by one γ-CD macrocycle exists until a certain concentration of guest. After that, there is a definite possibility of 2:2 inclusion complex formation between host and guest macrocycle. Moreover, when the two DCVJ molecules are trying to fit inside the cavity of γ-CD, both the molecules cannot be completely included in the host cavity. In such a case, part of the DCVJ molecule projecting out of the cavity can be encapsulated by another γ-CD. It is also noticeable that after a certain γ-CD concentration (∼7.5 mM), the extent of the increase in excimer peak intensity is higher (Fig. 3a). A large enhancement of the excimer peak is expected, as the interaction of a new γ-CD molecule with the 1:2 host–guest complex would further reduce the free rotation of the olefinic bond, which reduces the stability of the excimer. As the monomer peak intensity does not alter significantly, even though the excimer population in the solution increases (Fig. 3A), we believe that monomer fluorescence cannot be attributed to 1:1 complex, but instead to higher order complex formation (1:2 or 2:2) between γ-CD and DCVJ, in which the excited monomer does not always form an excimer. A similar type of observation was reported for α-naphthylacetate in the presence of γ-CD.29
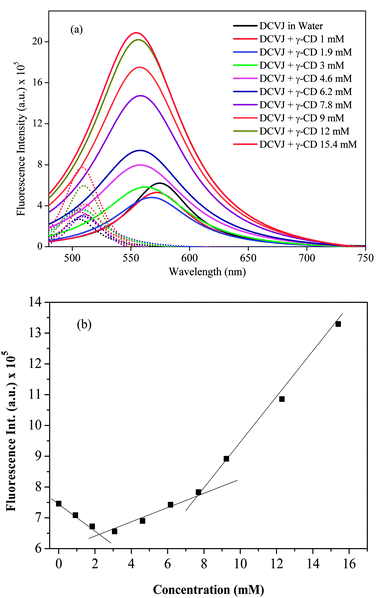 |
| | Fig. 3 (a) Deconvoluted spectra of DCVJ (∼12 μM) emission profiles in the absence and presence of γ-CD (0 to 15.4 mM). Solid lines represent the excimer, whereas dotted lines represent the monomer. Color codes for the concentration remain the same both for monomer and excimer. (b) Intensity profile of DCVJ (∼12 μM) monitored at 570 nm in the presence of different concentration of γ-CD. | |
The binding interactions between the dye and γ-CD can be further examined by monitoring the fluorescence intensity of the dye with increasing concentration of the macrocyclic host. Fig. 3b shows the fluorescence titration curve obtained by monitoring the intensity of the excimer peak in the presence of γ-CD. It is clearly noticeable that the binding curve significantly deviates from linearity, pointing to multiple inclusion complexes between host and guest. Moreover, two slope changes are clearly observed in the titration curve, one at around 2.5 mM and another slope change observed at around 8 mM. From these results, it is reasonable to assume that 1:1 complexation equilibrium between γ-CD and DCVJ exists until γ-CD concentration reaches 3 mM, and 1:2 (γ-CD:DCVJ) stoichiometric complex dominates from >3 mM to 8 mM. At higher concentration of hosts (>8 mM), domination by 2:2 (γ-CD:DCVJ) stoichiometry complex occurs. The predicted stoichiometry shift from 1:2 to 2:2 complex with an increase in γ-CD concentration is also reflected in the absorption spectra, where a second peak at ∼444 nm appears at higher γ-CD concentration (>7.7 mM). With all these observations, we believe that the changes in the absorption and emission spectra observed at higher γ-CD concentrations (>8 mM) are due to the transformation of the 1:2 inclusion complex to a 2:2 host–guest inclusion complex, which may provide better rigidity of DCVJ molecules. The increased rigidity of DCVJ molecules leads to formation of a π-stacked dimer, which shows an absorption maximum around 444 nm, and this dimer in the excited state either forms an excimer or behaves like an excited state monomer. There are numerous reports where the complexation of the fluorophore with the macrocyclic host has been proposed to be through 1:2 and 2:2 host–guest inclusion complexes, in addition to the 1:1 complex, where the size of the cavity of macrocyclic host is large.12,30–33 Considering the dimensions of DCVJ and the γ-CD cavity, it is quite likely that 1:1, 1:2 and 2:2 complexes are also formed in the present system. The fluorescence excitation spectra (see note 1, Fig. S2 and S3, ESI†), fluorescence lifetime, fluorescence anisotropy and theoretical calculation studies also support the formation of 1:1, 1:2 and 2:2 complexes between DCVJ and γ-CD in solution.
All the above observations can be explained on the basis of the following equilibria. Here, hνf indicates fluorescence from monomer, whereas hν/f indicates the fluorescence from excimer. Three different types of equilibrium exist depending on the concentration of γ-CD. Equilibrium 1 (eqn (4)) prevails at lower concentration of γ-CD (<3 mM); equilibrium 2 (eqn (6)) exists at the concentration range of >3 to ∼8 mM, whereas equilibrium 3 (eqn (9)) endures at higher γ-CD concentration (>8 mM).
| | | γ-CD + DCVJ ⇌ γ-CD:DCVJ | (4) |
| | 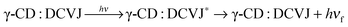 | (5) |
| | | γ-CD:DCVJ + DCVJ ⇌ γ-CD:(DCVJ)2 | (6) |
| | 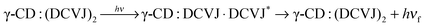 | (7) |
| | 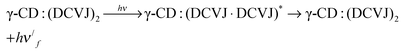 | (8) |
| | | γ-CD:(DCVJ)2 + γ-CD ⇌ γ-CD:(DCVJ)2:γ-CD | (9) |
| | 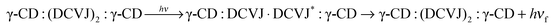 | (10) |
| | 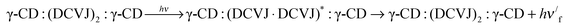 | (11) |
The association constant (K1) for the 1:1 DCVJ/γ-CD complex can be determined by monitoring the intensity contribution at 510 nm. However, it is found from the deconvoluted spectra that the excimer also contributes significantly at 510 nm. Hence, the estimated binding constant based on 510 nm intensity may not provide an accurate picture of the binding. However, to get an approximate idea of the 1:1 binding constant (K1), we have considered intensity at 510 nm up to a 3 mM concentration of γ-CD. The double reciprocal plot of 1/ΔF vs. 1/[γ-CD] yields a straight line (inset of Fig. 2c), and the K1 value has been estimated to be 450(±20) M−1. The binding curve reproduced from intensity at 570 nm exhibited deviation from a smooth curve (Fig. 3), indicating the presence of multiple stoichiometric equilibria in the system. Due to this inherent complexity, the 1:2 binding constant (K2) was hard to measure. However, an attempt was made to calculate the 2:2 binding constant (K3) from the latter part of the curve (above 8 mM of γ-CD concentration) after subtracting the value for the lower γ-CD concentration. The resulting titration was fitted with a 1:1 binding model (Fig. S4, ESI†) to obtain the approximate binding constant for the 2:2 complex as K3 = 45(±8) M−1. This is certainly a low binding constant, however, a binding constant of lower than ∼50 M−1 has been reported for the interaction of other ligands with β-CD.34,35
(c) Fluorescence lifetime features of DCVJ in absence and presence of CDs
It is well known that DCVJ belongs to the class of molecular rotors which show enhancement of fluorescence quantum yield when viscosity of the medium is increased.13,14 This has been attributed to restriction of the olefinic bond rotation imposed by the high viscosity of the microenvironment.13,14 Since the olefin bond rotation of DCVJ is expected to be severely restricted on forming an inclusion complex with CD(s), hence it is expected that the fluorescence lifetime of DCVJ should be affected by the formation of an inclusion complex with CD(s). We have therefore probed the inclusion complexes between DCVJ and CDs by monitoring the fluorescence lifetime of DCVJ in presence of CDs. The fluorescence lifetime results are summarized in Tables 1 and 2 and representative decay profiles of DCVJ (monitored at 570 nm) in the presence and absence of different kinds of CDs are shown in Fig. 4.
Table 1 Fluorescence decay parameters for DCVJ (∼12 μM) in the absence and presence of β-CD monitored (a) at 510 nm and (b) at 570 nm
| Concentration of β-CD (mM) |
τ
1 (ns) |
τ
2 (ns) |
τ
3 (ns) |
a
1
|
a
2
|
a
3
|
τ
av
(ns) |
χ
2
|
|
τ
av = (τ1 × a1 + τ2 × a2 + τ3 × a3).
|
| (a) |
| 0 |
0.060 |
3.79 |
0.343 |
0.93 |
0.04 |
0.04 |
0.200 |
1.09 |
| 1.8 |
0.048 |
3.98 |
0.308 |
0.94 |
0.02 |
0.04 |
0.168 |
1.06 |
| 5.3 |
0.017 |
3.99 |
0.139 |
0.96 |
0.01 |
0.03 |
0.054 |
1.01 |
| 8.8 |
0.038 |
4.16 |
0.223 |
0.97 |
0.01 |
0.02 |
0.09 |
1.05 |
| 14 |
0.014 |
4.19 |
0.064 |
0.95 |
0.01 |
0.04 |
0.028 |
0.96 |
| (b) |
| 0 |
1.41 |
4.42 |
— |
0.78 |
0.22 |
— |
2.07 |
0.98 |
| 1.8 |
1.84 |
4.60 |
0.525 |
0.69 |
0.09 |
0.23 |
1.79 |
0.99 |
| 5.3 |
1.55 |
4.06 |
0.12 |
0.54 |
0.13 |
0.33 |
1.40 |
1.07 |
| 8.8 |
1.22 |
3.11 |
0.057 |
0.23 |
0.15 |
0.63 |
0.772 |
1.05 |
| 14 |
0.716 |
3.94 |
0.031 |
0.01 |
0.01 |
0.98 |
0.075 |
0.96 |
Table 2 Fluorescence decay parameters for DCVJ (∼12 μM) in the absence and presence of γ-CD monitored (a) at 510 nm and (b) at 570 nm
| Concentration of γ-CD (mM) |
τ
1 (ns) |
τ
2 (ns) |
τ
3 (ns) |
a
1
|
a
2
|
a
3
|
τ
av
(ns) |
χ
2
|
|
τ
av = (τ1 × a1 + τ2 × a2 + τ3 × a3).
|
| (a) |
| 0 |
0.060 |
3.79 |
0.343 |
0.93 |
0.04 |
0.04 |
0.20 |
1.09 |
| 1 |
0.040 |
3.9 |
0.246 |
0.92 |
0.02 |
0.05 |
0.150 |
1.08 |
| 1.9 |
0.048 |
4.0 |
0.355 |
0.94 |
0.03 |
0.03 |
0.157 |
1.1 |
| (b) |
| 0 |
1.41 |
4.43 |
— |
0.78 |
0.22 |
— |
2.07 |
0.967 |
| 1.9 |
1.75 |
5.35 |
0.360 |
0.69 |
0.11 |
0.20 |
1.87 |
0.966 |
| 6.2 |
1.98 |
7.76 |
0.275 |
0.54 |
0.18 |
0.28 |
2.54 |
1.06 |
| 9.2 |
2.51 |
8.94 |
0.291 |
0.42 |
0.21 |
0.37 |
3.0 |
0.99 |
| 15.4 |
2.74 |
9.05 |
0.310 |
0.36 |
0.28 |
0.36 |
3.58 |
1.05 |
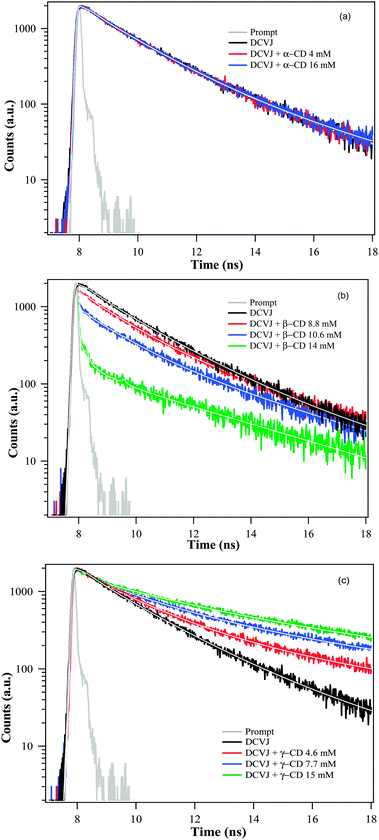 |
| | Fig. 4 Fluorescence decay overlays of DCVJ (∼12 μM) at 570 nm in presence of (a) α-CD, (b) β-CD and (c) γ-CD. | |
The fluorescence decay of DCVJ in pH7 buffer monitored at 510 nm displays tri-exponential decay with time constants of ∼60 ps (93%), 3.79 ns (4%) and 340 ps (4%). The excimer peak monitored at 570 nm gives two long lifetime components of 1.41 ns (78%) and 4.43 ns (22%). Since the DCVJ has conformational flexibility, therefore the multiexponential decay observed at 510 nm and 570 nm is partly due to the different conformational structure of the fluorophore in the monomeric or excimer state. In the presence of α-CD, the lifetime features of both monomer and excimer (Fig. 4a) remain almost the same, affirming that DCVJ encapsulation does not take place in the presence of α-CD. The decrease in monomer lifetime of DCVJ on complexation with β-CD (Table 1) can be rationalized in terms of alternations in the radiative and non-radiative decay pathways (eqn (12)) of the dye, due to their interaction with the macrocyclic host.
| | 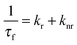 | (12) |
Since the average lifetime of the DCVJ monomer is reduced (
Table 1) and the intensity of the monomer is enhanced in the presence of β-CD (
Fig. 2b), it suggests the increase in the radiative rate constant (
kr) of DCVJ is significantly higher than the decrease in the non-radiative decay rate constant (
knr) of DCVJ inside the cavity of β-CD. The notably reduced excimer lifetime of DCVJ (monitored at 570 nm) in the presence of β-CD (
Fig. 4b,
Table 1) confirms the inclusion of DCVJ monomers inside the cavity of β-CD.
The abatement of the excimer lifetime of DCVJ at lower γ-CD concentration indicates that the DCVJ monomer encapsulates inside the γ-CD cavity (Fig. 4c, Table 2). The increased lifetime of the excimer (Fig. 4c, Table 2) at relatively higher γ-CD concentration (>3 mM) supports our conjecture of participation in 2:1 stoichiometric equilibrium between DCVJ and γ-CD. Prolonged lifetime at very high γ-CD concentration (>10 mM, Table 1) is attributable to the characteristics of the 2:2 complex between host and guest, which we have already speculated from the steady state results. The fluctuation of fluorescence lifetime of DCVJ excimer in the presence of γ-CD supports our hypothesis of there being different kinds of equilibrium depending on γ-CD concentration. The fluorescence lifetime feature of DCVJ monomer inside the γ-CD cavity can be deduced from the decay profiles monitored at 510 nm. However, it was found that the excimer also has a reasonable intensity at 510 nm after 3 mM of γ-CD concentration, when 1:2 complexation dominates (see the deconvoluted spectra in Fig. 3a). In this case, it is hard to separate the contribution of each species in the transient profiles when both of them contribute to the total decay curve. Therefore, we report the lifetime of the monomer up to ∼2 mM of γ-CD concentration, where the excimer contribution at 510 nm is negligibly small (Fig. 3a). It is found that the fluorescence lifetime of the DCVJ monomer remains almost the same inside the cavity of γ-CD (Table 2a), indicating that non-radiative rate processes of the DCVJ monomer are least affected inside the cavity of γ-CD. This insight into the molecular picture of the interaction between γ-CD and DCVJ will be highlighted in the latter part of this manuscript, based on docking studies followed by geometry optimization through quantum chemical semi-empirical methods.
Formation of host–guest inclusion complexes between dye and macrocyclic host are expected to increase hydrodynamic volume, which in turn affects the rotational relaxation of dye. With this concept, time-resolved fluorescence anisotropy measurements of DCVJ were carried out in the absence and presence of macrocyclic host. The results are summarized in Table 3 and shown in Fig. 5. The rotational relaxation of the uncomplexed dye monitored at 510 nm exhibits single exponential decay with a time-scale of 250 ps. The anisotropy decays of DCVJ monomer in the presence of α-CD almost overlap with that of the buffer, indicating that the molecules are not encapsulating inside the α-CD cavity. Hence, time-resolved anisotropy results corroborate our steady state results, in which it was found that DCVJ does not form an inclusion complex with α-CD. The anisotropy decays of DCVJ monomers in the presence of β-CD were found to be significantly slower (∼400 ps), affirming formation of inclusion complex between dye and cyclodextrin host. To monitor the rotational relaxation of the excimer inside the γ-CD, we have also probed the anisotropy decays of DCVJ at 570 nm at different γ-CD concentrations. The existence of 1:2 and 2:2 (γ-CD:DCVJ) assemblies in the presence of γ-CD are further reinforced by the rotational relaxation time (τr), monitored at 570 nm at the corresponding solution condition for the above mentioned stoichiometries, and the anisotropy decays, which give τr as ∼400 ps and ∼750 ps at the 1:2 and 2:2 stoichiometries, respectively (Table 3). To get an idea of the rotational relaxation of the monomer, we have monitored the relaxation dynamics at 510 nm and it is noticeable that the rotational relaxation of the monomer and excimer inside the cavity of γ-CD takes place over a similar time scale, e.g., the rotational relaxation of a monomer monitored at 510 nm takes place over a ∼350 ps time scale at ∼3.0 mM γ-CD concentration (at this concentration the contribution of the excimer peak at 510 nm is minute; moreover, after this concentration, excimer formation inside the γ-CD cavity starts), whereas the rotational relaxation of an excimer monitored at 570 nm occurs over a ∼400 ps time scale in the presence of ∼6 mM of γ-CD. This is justified as both the monomer and dimer are encapsulated by the γ-CD cavity, and ultimately we are getting the rotational relaxation of the γ-CD containing the monomer or excimer.
Table 3 Anisotropy decay parameters of DCVJ excimer monitored at 570 nm in presence of γ-CD
| Concentration of γ-CD (mM) |
τ
1r (ns) |
τ
2r (ns) |
a
1r
|
a
2r
|
τ
av
(ns) |
r
0
|
χ
2
|

|
| 8 |
0.160 |
3.9 |
0.28 |
0.02 |
0.400 |
0.30 |
1.05 |
| 16 |
0.170 |
3.8 |
0.21 |
0.04 |
0.750 |
0.26 |
1.04 |
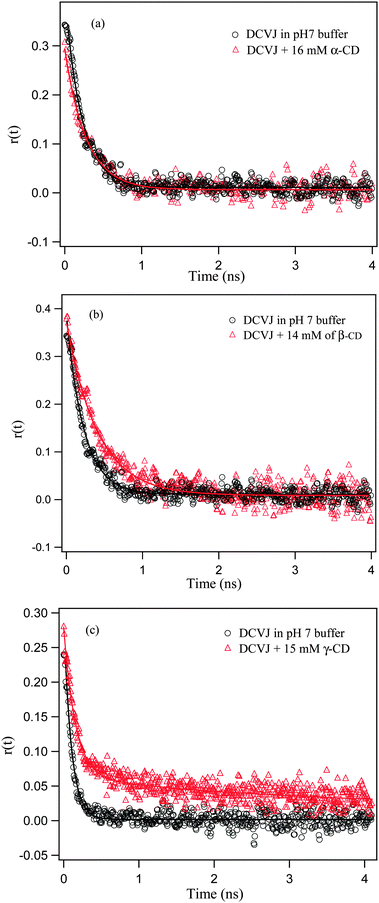 |
| | Fig. 5 Anisotropy decay overlays of DCVJ (∼12 μM) in presence of (a) α-CD (monitored at 510 nm), (b) β-CD (monitored at 510 nm), and (c) γ-CD (monitored at 570 nm). Markers are experimental observed points and solid lines are the fitting curves of the experimental results. | |
Further, the τr values of the excimer can be used to calculate the hydrodynamic diameters of the 1:1 (monitored at 510 nm), 1:2 and 2:2 inclusion complexes between DCVJ and γ-CD macrocyclic host using the following Stokes–Einstein–Debye36 relationship:
| | 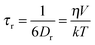 | (13) |
Here,
Dr and
η are the rotational diffusion coefficient and viscosity of the medium, respectively,
V is the hydrodynamic molecular volume of the complex, and
T is the absolute temperature. By using the above relation and assuming the viscosity of the medium is the same as that of
water, the evaluated effective hydrodynamic diameters of the complexes displayed are 14.0 Å, 14.7 Å and 18.2 Å for the 1
:
1, 1
:
2 and 2
:
2 host–guest complexes, respectively, which reasonably accounts for the molecular sizes of the complexes.
(e) Docking and quantum chemical calculations
To obtain a molecular picture of the orientation of DCVJ in the inclusion complexes with various CDs, as well as to gain insight into the stabilization achieved due to encapsulation, we have docked the ligand (DCVJ) into the CDs, followed by a quantum chemical optimization. Crystal structures of α-CD (PDB ID:2ZYM), β-CD (PDB ID:ZYN) and γ-CD (PDB ID:2ZYK) were taken as receptor and the optimized geometry of either monomeric or dimeric form of the DCVJ was taken as ligand. In all docking we have used Gasteiger charges for both the CDs and the ligand. The grid was generated on a whole receptor with grid points of 126 on each of the orthogonal directions with a spacing of 0.16 Å. Search was performed using Genetic algorithm using AutoDock (version 1.4) software.37 During docking, the receptor was kept rigid and the ligand was flexible. Following the experimental evidence, we have also tried to find 1:2 or 2:2 inclusion complex (DCVJ:γ-CD) in the case of γ-CD by docking the dimeric form of the ligand on to the host. Although experimental studies have indicated an excimer complex, we have made an approximation that the geometry of the ground state dimer would reflect that of the excimer. To create the dimeric form of ligand, we have docked one DCVJ molecule on to the other using a smaller grid spacing of 0.08 Å. The most stable DCVJ dimer complex obtained from docking was then optimized using ab initio density functional theory using WB97XD functional and polarizable continuum model of water to account for the dispersion interaction, which stabilizes the stacked geometry of the DCVJ dimer, with 6-31G* basis set in Gaussian 09.38 All the docking structures are shown in Fig. S5–S9 (ESI†), and all the docking parameters are summarized in Table S1 (ESI†). For all 1:1 inclusion complexes of three macrocyclic hosts, the best docked structures were then optimized using ab initio density functional theory using B3LYP functional with 6-31G* basis set (Fig. 6).38 The binding energy was calculated by optimizing the complex and the individual molecules, and then subtracting the energy of the individual molecules from the complex. A full ab initio quantum chemical calculation for higher inclusion complexes (1:2 or 2:2) of γ-CD is computationally prohibitive and we believe that PM6 semi-empirical method would provide at least a geometrically correct result for higher order inclusion complexes of γ-CD (Fig. 7 and 8). Unlike, ab initio density functional theory, the semi-empirical method does not provide binding energy. However, it provides the heat of formation (ΔH/kcal mol−1) for the inclusion complexes. Therefore, in the case of multiple inclusion complexes of γ-CD, instead of calculating the binding energy, we have provided the heat of formation (ΔH/kcal mol−1) of an inclusion complex by optimizing the complex and individual molecules by PM6 method in MOPAC,39 and then subtracting the heat of formation of the individual molecules from the complex. Since the docking, as well as the geometry optimization, was done without consideration of any solvent and other parameters, these geometries provide only a qualitative picture of the structures in the ground states. All the figures (Fig. 6–8 and Fig. S5–S9, ESI†) are made in UCSF Chimera.40
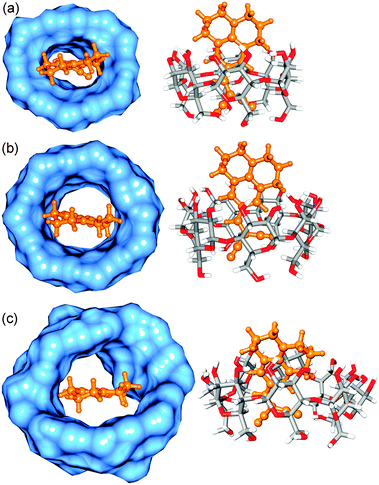 |
| | Fig. 6 Optimized structure of (a) docked α-CD:DCVJ complex (1:1), (b) docked β-CD:DCVJ complex (1:1) and (c) docked γ-CD:DCVJ complex (1:1). DCVJ is shown in ball and stick model (orange color) and CDs are shown in both surface representation (blue color) and stick model (element based color). | |
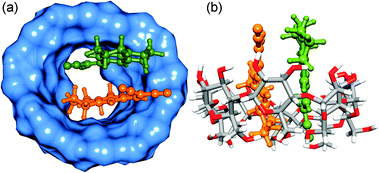 |
| | Fig. 7 Optimized structures of γ-CD:DCVJ (1:2) complex. (a) and (b) correspond to the different orientations of the same optimized complex. The DCVJs are represented as ball and stick model (orange and green colors). The rest is the same as that of Fig. 6. | |
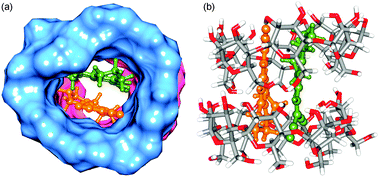 |
| | Fig. 8 Optimized structures of γ-CD:DCVJ (2:2) complex. Two cyclodextrins are shown in both surface (blue and pink colors) and stick representations. The rest is the same as that of Fig. 7. | |
Although the docking study indicates the formation of an inclusion complex between DCVJ and α-CD (Fig. 6a), the binding energy (after basis set superposition error (BSSE) correction) obtained from quantum chemical optimization method is positive (32.27 kcal mol−1), which indicates that energetically DCVJ does not prefer to be included inside the cavity of α-CD, probably due to the smaller size of the host cavity. Therefore, theoretical calculations support our experimental findings that DCVJ does not form an inclusion complex with α-CD. In the case of β-CD, the binding energy (after BSSE correction) calculated from quantum chemical optimization method is negative (−9.70 kcal mol−1), suggesting that 1:1 inclusion complex formation between DCVJ and β-CD is energetically feasible, as we have observed experimentally (Fig. 6b).
For γ-CD, the 1:1 host–guest inclusion complex exhibits energetically feasible negative binding energy (−4.62 kcal mol−1) obtained from an ab initio quantum chemical optimization method (Fig. 6c). Following the experimental evidence of formation of 1:2 inclusion complex (DCVJ:γ-CD) in the presence of γ-CD, we have docked an optimized dimer of DCVJ to a γ-CD host, and then optimized the docked structure of a 1:2 inclusion complex (γ-CD:DCVJ) by semi-empirical calculation (Fig. 7). Considering the experimental proof of formation of 2:2 complex in γ-CD, we have first created a dimeric complex of γ-CD by docking one γ-CD on to the 1:2 complex of γ-CD and DCVJ. This was done because docking another γ-CD on to an empty γ-CD resulted in an unusual interpenetrating ring structure, where a 2:2 complex would not be feasible. Subsequently, we removed the DCVJ dimer from the host and obtained a dimeric complex of γ-CD. This was followed by docking of a DCVJ dimer to obtain the best docked configuration of the 2:2 complex of γ-CD and the DCVJ complex. All the allowable torsions for the DCVJ dimer were switched off during the docking. Evaluating heat of formation from PM6 semi-empirical calculations, we confirm that 1:2 and 2:2 stoichiometries, as shown in Fig. 7 and 8, are energetically feasible by ΔHf of −25 kcal mol−1 and −67 kcal mol−1, respectively. This therefore confirms that higher stoichiometric complexes are more favourable than those of lower stoichiometry. It is also noticeable that, inside the cavity, the two DCVJ molecules are separated by ∼3.65 Å, which is in agreement with the literature, where it was observed that aromatic molecules involved in stacking interaction with approximately parallel molecular plane are separated by 3.3–3.6 Å.41 Moreover, it can also be seen from Fig. 8 that the 2:2 inclusion complex provides better rigidity to the excimer than the 1:2 complex, as free rotation of DCVJ molecules is completely restricted in the 2:2 inclusion complex, and therefore the quantum yield of excimer formation in 2:2 inclusion complex is higher than that of lower stoichiometric complexes, as observed in the experiments.
Conclusion
In this work, we report the modulation over excimer formation drift of a molecular rotor, called DCVJ, by various cavity sizes of CD macrocyclic host. The experimental and theoretical studies confirm that DCVJ does not form an inclusion complex with α-CD, due to the smaller cavity size of the host. However, β-CD, a relatively larger size of CD, accommodates one DCVJ molecule into its cavity, and therefore the excimer population of DCVJ in solution is significantly reduced. The most striking observation is made in the case of γ-CD, which has a larger cavity size. Our experimental results indicate that at lower γ-CD concentration, only one DCVJ molecule is being encapsulated by the host cavity. However, at relatively higher γ-CD concentration, two DCVJ molecules are included in the cavity of the host, and they form an excimer inside the cavity. At a very high concentration of the host, we also obtained substantial evidence for the formation of a 2:2 inclusion complex between γ-CD and DCVJ. The molecular picture of the interaction between DCVJ and CDs is supported by docking and quantum chemical calculations.
Acknowledgements
This work is partly supported by SERC, the Department of Science and Technology (DST), and Council of Scientific and Industrial Research (CSIR), Government of India. Authors are thankful to reviewers for their valuable comments and suggestions. Authors thank Director IISER-Pune for providing excellent experimental and computation facilities. Authors are thankful to Piyush Agrawal for initial help during computational calculation.
References
- J. Szejtli, Chem. Rev., 1998, 98, 1743–1754 CrossRef CAS.
- A. Douhal, Chem. Rev., 2004, 104, 1955–1976 CrossRef CAS.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS.
- V. Ramamurthy and D. F. Eaton, Acc. Chem. Res., 1988, 21, 300–306 CrossRef CAS.
- V. K. Indirapriyadharshini, P. Karunanithi and P. Ramamurthy, Langmuir, 2001, 17, 4056–4060 CrossRef CAS.
- R. Kaliappan, Y. Ling, A. E. Kaifer and V. Ramamurthy, Langmuir, 2009, 25, 8982–8992 CrossRef CAS.
- A. Baldridge, S. R. Samanta, N. Jayaraj, V. Ramamurthy and L. M. Tolbert, J. Am. Chem. Soc., 2010, 132, 1498–1499 CrossRef CAS.
- T. Yorozu, M. Hoshino and M. Imamura, J. Phys. Chem., 1982, 86, 4426–4429 CrossRef CAS.
- C. Retna Raj and R. Ramaraj, Chem. Phys. Lett., 1997, 273, 285–290 CrossRef CAS.
- T. C. Barros, K. Stefaniak, J. F. Holzwarth and C. Bohne, J. Phys. Chem. A, 1998, 102, 5639–5651 CrossRef CAS.
- C. Lee, Y. W. Sung and J. W. Park, J. Phys. Chem. B, 1999, 103, 893–898 CrossRef CAS.
- A. S. M. Dyck, U. Kisiel and C. Bohne, J. Phys. Chem. B, 2003, 107, 11652–11659 CrossRef CAS.
- M. Haidekker and E. Theodorakis, J. Biol. Eng., 2010, 4, 11 CrossRef.
- J. A. Levitt, P.-H. Chung, M. K. Kuimova, G. Yahioglu, Y. Wang, J. Qu and K. Suhling, ChemPhysChem, 2011, 12, 662–672 CrossRef CAS.
- C. Swalina and M. Maroncelli, J. Phys. Chem. C, 2009, 114, 5602–5610 Search PubMed.
- H. Jin, M. Liang, S. Arzhantsev, X. Li and M. Maroncelli, J. Phys. Chem. B, 2010, 114, 7565–7578 CrossRef CAS.
- C. E. Kung and J. K. Reed, Biochemistry, 1989, 28, 6678–6686 CrossRef CAS.
- M. Lindgren, K. Sörgjerd and P. Hammarström, Biophys. J., 2005, 88, 4200–4212 CrossRef CAS.
- S. Luka, J. Am. Chem. Soc., 1984, 106, 4386–4392 CrossRef CAS.
- C. E. Kung and J. K. Reed, Biochemistry, 1986, 25, 6114–6121 CrossRef CAS.
- A.-Y. Jee, E. Bae and M. Lee, J. Phys. Chem. B, 2009, 113, 16508–16512 CrossRef CAS.
- H. Jin, X. Li and M. Maroncelli, J. Phys. Chem. B, 2007, 111, 13473–13478 CrossRef CAS.
- A. Paul and A. Samanta, J. Phys. Chem. B, 2008, 112, 16626–16632 CrossRef CAS.
- A. Bhattacharyya, K. Bhattacharya, B. Bhattacharyya and S. Roy, Indian J. Biochem. Biophys., 1995, 32, 442–446 CAS.
- A. Sengupta and P. Hazra, Chem. Phys. Lett., 2010, 501, 33–38 CrossRef CAS.
- A. Sengupta, R. V. Khade and P. Hazra, J. Phys. Chem. A, 2011, 115, 10398–10407 CrossRef CAS.
- A. Sengupta, W. D. Sasikala, A. Mukherjee and P. Hazra, ChemPhysChem, 2012, 13, 2142–2153 CrossRef CAS.
- A. Munoz de la Pena, T. Ndou, J. B. Zung and I. M. Warner, J. Phys. Chem., 1991, 95, 3330–3334 CrossRef CAS.
- A. Ueno, K. Takahashi and T. Osa, J. Chem. Soc., Chem. Commun., 1980, 921–922 RSC.
- S. Hamai, J. Phys. Chem., 1989, 93, 6527–6529 CrossRef CAS.
- R. S. Murphy, T. C. Barros, J. Barnes, B. Mayer, G. Marconi and C. Bohne, J. Phys. Chem. A, 1998, 103, 137–146 CrossRef.
- A. Nakamura and Y. Inoue, J. Am. Chem. Soc., 2002, 125, 966–972 CrossRef.
- A. Nakamura and Y. Inoue, J. Am. Chem. Soc., 2005, 127, 5338–5339 CrossRef CAS.
- N. Kandoth, S. Dutta Choudhury, J. Mohanty, A. C. Bhasikuttan and H. Pal, J. Phys. Chem. B, 2010, 114, 2617–2626 CrossRef CAS.
- C. Formoso, Biochem. Biophys. Res. Commun., 1973, 50, 999–1005 CrossRef CAS.
-
G. Fleming, Chemical applications of ultrafast spectroscopy, 1986, Medium: X; Size: p. 262 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
-
J. J. P. Stewart, MOPAC2009; Stewart Computational Chemistry, Colorado Springs, Colorado, 2008 Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS.
- M. O. Sinnokrot, E. F. Valeev and C. D. Sherrill, J. Am. Chem. Soc., 2002, 124, 10887–10893 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cp43282j |
|
| This journal is © the Owner Societies 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.