A VUV photoionization study of the multichannel reaction of phenyl radicals with 1,3-butadiene under combustion relevant conditions†
Received
26th May 2012
, Accepted 2nd November 2012
First published on 5th November 2012
Abstract
We studied the reaction of phenyl radicals (C6H5) with 1,3-butadiene (H2CCHCHCH2) exploiting a high temperature chemical reactor under combustion-like conditions (300 Torr, 873 K). The reaction products were probed in a supersonic beam by utilizing VUV radiation from the Advanced Light Source and by recording the experimental PIE curves at mass-to-charge ratios of m/z = 130 (C10H10+), 116 (C9H8+), and 104 (C8H8+). Our data suggest that the atomic hydrogen (H), methyl (CH3), and vinyl (C2H3) losses are open with estimated branching ratios of about 86 ± 4%, 8 ± 2%, and 6 ± 2%, respectively. The isomer distributions were probed further by fitting the experimentally recorded PIE curves with a linear combination of the PIE curves of individual C10H10, C9H8, and C8H8 isomers. These fits indicate the formation of three C10H10 isomers (trans-1,3-butadienylbenzene, 1,4-dihydronaphthalene, 1-methylindene), three C9H8 isomers (indene, phenylallene, 1-phenyl-1-methylacetylene), and a C8H8 isomer (styrene). A comparison with results from recent crossed molecular beam studies of the 1,3-butadiene–phenyl radical reaction and electronic structure calculations suggests that trans-1,3-butadienylbenzene (130 amu), 1,4-dihydronaphthalene (130 amu), and styrene (104 amu) are reaction products formed as a consequence of a bimolecular reaction between the phenyl radical and 1,3-butadiene. 1-Methylindene (130 amu), indene (116 amu), phenylallene (116 amu), and 1-phenyl-1-methylacetylene (116 amu) are synthesized upon reaction of the phenyl radical with three C4H6 isomers: 1,2-butadiene (H2CCCH(CH3)), 1-butyne (HCCC2H5), and 2-butyne (CH3CCCH3); these C4H6 isomers can be formed from 1,3-butadiene via hydrogen atom assisted isomerization reactions or via thermal rearrangements of 1,3-butadiene involving hydrogen shifts in the high temperature chemical reactor.
1. Introduction
In the last few decades, the mechanisms of formation of polycyclic aromatic hydrocarbons (PAHs) have been the focus of extensive studies due to their importance in combustion,1,2 atmospheric,3 and interstellar chemistry.4,5 Combustion and interstellar chemistry models suggest that the phenyl radical in its ground electronic state (C6H5, X2A1) plays a crucial role in the formation of PAHs,6,7 especially via its reactions with unsaturated hydrocarbons such as (substituted) acetylenes, olefins, and aromatic molecules.8–14 Therefore, the reactions of phenyl radicals with unsaturated C3 and C4 hydrocarbons have been studied extensively both experimentally in crossed molecular beams, in high temperature reactors, and computationally. These systems probed the formation of C9Hx (x = 8, 10)15–21 and C10Hx (x = 6, 8, 10)22–27 molecules with indene and naphthalene cores, respectively. Recent crossed molecular beam studies provided compelling evidence that the reactions of the phenyl radical (C6H5) with methylacetylene (CH3CCH) and allene (H2CCCH2) lead to the formation of an aromatic indene molecule (C9Hx).18,19 Most importantly, bimolecular collisions of phenyl radicals with vinylacetylene (HCCC2H3) were found to form naphthalene (C10H8) under single collision conditions without an entrance barrier to reaction.23 Likewise, a recent study of the 1,3-butadiene (H2CCHCHCH2)–phenyl system via crossed molecular beams indicated the barrier-less formation of the 1,4-dihydronaphthalene molecule (C10H10) together with its trans-1,3-butadienylbenzene (C6H5HCCHCHCH2) isomer.27 These crossed beam studies were able to probe the outcome and hence reaction products of bimolecular reactions without the interference of wall effects, excluding stabilization of reaction intermediates by a third body, and without successive isomerization of the nascent reaction products and/or reactants.
However, in real combustion systems, the reaction mechanisms might be more complicated than those probed under single collision conditions. To account for this, we recently commissioned a high temperature chemical reactor, in which the products of phenyl radical reactions can be probed under combustion-relevant pressures and temperatures.28 This reactor has been applied very successfully to identify the aromatic indene molecule together with its acyclic isomers (phenylallene, 1-phenyl-1-methylacetylene, and 3-phenyl-1-methylacetylene) in the reactions of phenyl with methylacetylene and allene.18 Here, we expand these studies to probe the products of phenyl radicals reacting with 1,3-butadiene under combustion-like conditions (300 Torr, 873 K) in a high temperature chemical reactor; these findings are compared with the products formed under single collision conditions and predicted computationally.27
2. Methods
A resistively heated high temperature ‘chemical reactor’ interfaced with the molecular beams end station at the Chemical Dynamics Beamline (9.0.2) of the Advanced Light Source28 was utilized to study the reaction of the phenyl radical with 1,3-butadiene. Briefly, a continuous beam of phenyl radicals (C6H5) was generated in situ via a quantitative pyrolysis of the nitrosobenzene precursor (C6H5NO; Aldrich). The latter was seeded at levels of about 0.1% in an excess of neat 1,3-butadiene carrier gas (1,3-C4H6; Sigma), which was expanded at a pressure of 300 Torr through a 0.1 mm orifice into a resistively heated silicon carbide (SiC) tube at temperatures of about 873 K. The 1,3-butadiene molecules did not only act as a seeding gas, but also as a reactant with the pyrolytically generated phenyl radicals. The residence time of the reactant within the tube was estimated to be at least 26 ± 3 μs. This experimental setup guarantees a detection of the in situ generated products within the molecular beam: after passing a 2 mm skimmer located 10 mm downstream from the silicon carbide nozzle, the neutral molecular beam was crossed by quasi-continuous tunable VUV radiation from the ALS downstream at the extraction region of a Wiley–McLaren Reflectron Time-of-Flight (Re-TOF) mass spectrometer. The ions of the photoionized molecules were then extracted and collected using a microchannel plate detector in the Re-TOF mode utilizing a multi-channel scaler. A complete mass spectrum was obtained at intervals of 0.05 eV between 7.60 and 8.80 eV. The PIE curves of the products were obtained by plotting the integrated ion counts at desired mass-to-charge ratios, m/z, of 130 (C10H10+), 116 (C9H8+), and 104 (C8H8+), versus the photoionization energy between 7.60 and 8.80 eV. The signal was normalized to the photon flux. Based on known PIE curves of individual C10H10, C9H8, and C8H8 isomers, the recorded PIE curves were then fitted via a linear combination of known PIE curves of these isomers to extract the nature of the products formed.
Note that the PIE curves of individual C10H10 isomers (Fig. 1) do not exist in the literature, and, therefore, had to be recorded in separate experiments. Briefly, the PIE curves of five C10H10 isomers, 1,2-dihydronaphthalene (TCI, 98%), 1,4-dihydronaphthalene (TCI, 98%), 2-methylindene (TCI, 98%), 1-methylindene (TCI, 98%), and trans-1,3-butadienylbenzene (TCI, 98%), were collected in an identical setup as described above, but without heating the silicon carbide nozzle. For each molecule, a continuous beam of the C10H10 isomer was generated by passing helium (Airgas, 99.999%) carrier gas with a pressure of 300 Torr through a home-made stainless steel bubbler, which contained the individual C10H10 isomer at 293 K. This gas mixture was expanded into the source chamber before passing a 2 mm skimmer 10 mm downstream to reach the detector chamber. As for the reaction products, the ions of the photoionized molecules were extracted and collected using a microchannel plate detector in the Re-TOF mode. Individual PIE curves were obtained by plotting the integrated ion counts at a particular mass versus the photoionization energy between 7.60 eV and 8.80 eV in steps of 0.05 eV.
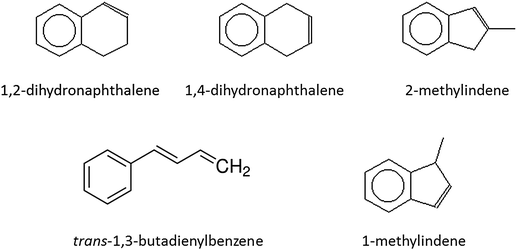 |
| | Fig. 1 Structures of distinct C10H10 isomers potentially formed in the reaction of phenyl radicals with 1,3-butadiene. | |
3. Results and discussion
Fig. 2 shows a characteristic mass spectrum recorded at a heater temperature of 873 K, where product peaks at m/z = 130, 116, and 104, which correspond to the molecular ions C10H10+, C9H8+, and C8H8+, respectively, can be discerned clearly. Considering the molecular mass of the reactant molecules, i.e. C6H5 (77 amu) and C4H6 (54 amu), the products are formed via the atomic hydrogen (reaction (1)), methyl radical (reaction (2)), and vinyl radical loss (reaction (3)) pathways.| | | C6H5 + C4H6 → C10H10 + H | (1) |
| | | C6H5 + C4H6 → C9H8 + CH3 | (2) |
| | | C6H5 + C4H6 → C8H8 + C2H3 | (3) |
Based on the raw data, peak intensities of the ion counts, and scaled photon absorption cross sections, branching ratios of 86 ± 4%, 8 ± 2%, and 6 ± 2% were estimated for reactions (1) to (3). Subsequently we attempt to fit the recorded PIEs with a linear combination of PIEs of the corresponding isomers to estimate their branching ratios. First, the PIE of m/z = 130 (C10H10+) (Fig. 3 (top)) could be fit with a linear combination of three isomers. These are trans-1,3-butadienylbenzene (44 ± 8%), 1-methylindene (34 ± 10%), and 1,4-dihydronaphthalene (22 ± 9%). We would like to stress that up to 9% of the 1,4-dihydronaphthalene product could be replaced by 2-methylindene without changing the fit. Therefore, the assignment of 2-methylindene at levels of 9 ± 9% is tentative; likewise, the potential contributions of the fifth isomer, 1,2-dihydronaphthalene, are marginal (1 ± 1%). Second, three C9H8 isomers were required to reproduce the PIE of m/z = 116 (C9H8+) with indene, phenylallene, and 1-phenyl-1-methylacetylene contributing 53 ± 14%, 27 ± 5%, and 20 ± 5%, respectively (Fig. 3 (center)). It was not necessary to include any contributions from the 3-phenyl-3-methylacetylene isomer. The fit of the PIE at 104 (C8H8+) is not trivial (Fig. 3 (bottom)). This graph could not be reproduced by assuming that the signal only originates from styrene (C6H5C2H3). A closer look at the mass spectra of the calibrated C10H10 isomers revealed that the molecular ions of 2-methylindene and 1,4-dihydronaphthalene fragmented to m/z = 104. After accounting for this, we could reproduce the PIE of m/z = 104 with two components: styrene (74 ± 12%) and 1,4-dihydronaphthalene (26 ± 4%). It is important to note that this fit is not unique, and up to 15% of the 2-methylindene isomer could be also included without changing the fit. This is due to the limited signal-to-noise ratio of this channel and also similar photoionization curves of 1,2- and 1,4-dihydronaphthalene.
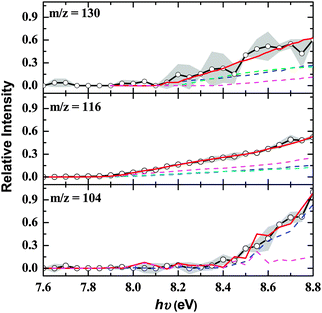 |
| | Fig. 3 Top: open circles present the PIE obtained at m/z = 130, whereas the red line is the simulation using PIEs of 1,4-dihydronaphthalene (blue), 1-methylindene (cyan), and 1,3-butadienyl-benzene (green). Center: open circles present the PIE obtained at m/z = 116, whereas the red line is the simulation using PIE curves of indene (blue), phenylallene (light blue) and 1-phenyl-1-propyne (magenta). Bottom: open circles present the PIE obtained at m/z = 104, whereas the red line presents the simulation using PIE curves of photoionization fragments of 1,4-dihydronaphathalene (purple) together with the PIE curve of styrene (blue). Experimental errors are defined by the grey curve. The individual PIE curves have been scaled according to their branching ratios. | |
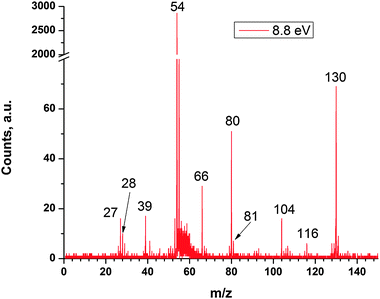 |
| | Fig. 2 Time of flight mass spectra recorded for an expansion of nitrosobenzene and 1,3-butadiene at a reactor temperature of 873 K and synchrotron photon energy of 8.8 eV. The ionization energy of the nitrogen monoxide radical formed in the pyrolysis of the nitrosobenzene precursor is 9.26 eV, i.e. above the photon energy of 8.8 eV utilized to obtain the mass spectrum. Hence there is no signal at m/z = 30 (NO+). m/z 104, 116 and 130 are singly ionized C8H8, C9H8 and C10H10, respectively, and are discussed in the text. Other ions at m/z 27, 28, 39, 54, 66, and 80 are attributed to singly ionized C2H3, C2H4, C3H3, C4H6, and C5H6, respectively. | |
A key question is, how are these reaction products formed in the chemical reactor? To explain these findings, we explore first the potential energy surface (PES) of the phenyl–1,3-butadiene reaction computed earlier at the G3(CC,MP2)//B3LYP/6-311G** level of theory (Fig. 4) and compare the data from the ‘chemical reactor’ with those obtained under single collision conditions via crossed molecular beam studies of the phenyl–1,3-butadiene system.27 According to the electronic structure calculations, the phenyl radical can add to the C1 or C2 atom of the 1,3-butadiene forming intermediates i1 and i2, respectively. Note that the addition to the C1 position is effectively barrier-less and involves the formation of a van der Waals complex and a submerged barrier in the entrance channel. The intermediate i1 can either undergo cis–trans isomerization to i3, isomerize to i2, or emit a hydrogen atom yielding trans-1,3-butadienylbenzene (p1). On the other hand, i2 decomposes via vinyl group loss to styrene (C6H5C2H3; C8H8). Isomer i3 can rearrange to a bicyclic structure i4, which then decomposes via hydrogen atom loss to form 1,4-dihydrobenzene (p2). Based on these considerations, the C10H10 isomers, trans-1,3-butadienylbenzene (p1) and 1,4-dihydronaphthalene (p2) together with styrene (C6H5C2H3; C8H8) can be formed as nascent reaction products in the bimolecular reaction of the phenyl radical with 1,3-butadiene. Hereafter, these products are classified as primary reaction products formed in the bimolecular reaction of the phenyl radical with 1,3-butadiene. Note that under single collision conditions at a collision energy of 55 kJ mol−1, crossed beam experiments established the formation of mainly 1,4-dihydronaphthalene and with smaller fractions, trans-1,3-butadienylbenzene. We would like to stress that under single collision conditions, the reaction of the phenyl radical with 1,3-butadiene can neither form the C10H10 isomer 1-methylindene nor any of the C9H8 isomers – indene, phenylallene, and 1-phenyl-1-methylacetylene. Therefore, additional reaction steps are likely involved in the formation of those molecules, which cannot be synthesized as a consequence of a single collision. These products are labeled as higher-order reaction products.
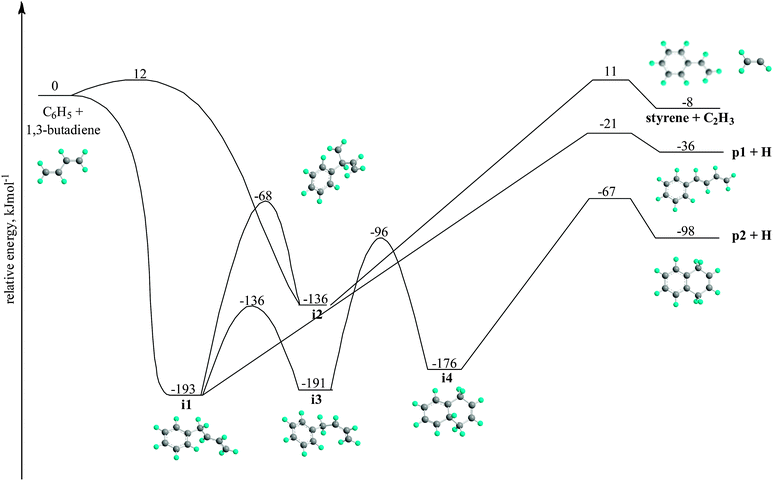 |
| | Fig. 4 Simplified C10H11 potential energy surface (PES) relevant to the reaction of phenyl radicals with 1,3-butadiene computed at the G3(MP2,CC)//B3LYP/6-311G** level of theory. Relative energies are given in kJ mol−1. | |
We now focus our attention on these reaction products and propose potential reaction pathways (Fig. 5). Previous electronic structure calculations suggested that the 1,3-butadiene molecule can undergo an atomic hydrogen induced isomerization to its less stable 1,2-butadiene, 1-butyne, and 2-butyne isomers.29 These processes are initiated by an initial addition of atomic hydrogen to 1,3-butadiene at the C1 and/or C2 carbon atom(s) via barriers not exceeding 12 kJ mol−1 (ref. 29) followed by isomerization and hydrogen loss. For instance, 1,3-butadiene and 1,2-butadiene have been monitored in combustion flames of hydrocarbon fuels,30 and mutual rearrangements between the C4H6 isomers were shown to be much faster than their decomposition at flame-relevant temperatures.31 In our chemical reactor, these hydrogen atoms can be the light byproducts of the reaction of the phenyl radical with 1,3-butadiene yielding the trans-1,3-butadienylbenzene and 1,4-dihydronaphthalene products (Fig. 4). Alternatively, all high energy C4H6 isomers can be formed easily from 1,3-butadiene through thermal rearrangements involving hydrogen shifts.32 These isomers – either formed via thermally induced hydrogen migrations or through hydrogen atom induced isomerization – can undergo secondary reactions with the phenyl radical.
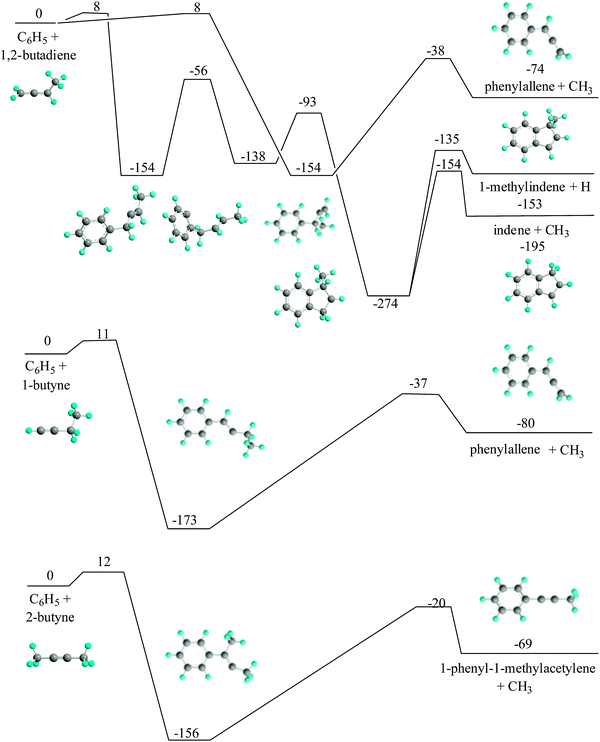 |
| | Fig. 5 Formation of secondary reaction products via the reactions of phenyl radicals with 1,2-butadiene, 1-butyne, and 2-butyne. Relative energies in kJ mol−1 are computed at the G3(MP2,CC)//B3LYP/6-311G** level of theory. | |
First, the phenyl radicals can react with 1,2-butadiene via addition to the C1 or C3 carbon atoms passing entrance barriers of about 8 kJ mol−1. The initial reaction intermediates either isomerize in two steps via hydrogen shifts followed by methyl and atomic hydrogen loss to form indene and 1-methylindene, respectively, or fragment to phenylallene plus the methyl radical.24,25 Second, the phenyl radical can add to the C1 carbon atom if it is 1-butyne via an entrance barrier of 11 kJ mol−1. The reaction intermediate can emit a methyl group forming phenylallene. Finally, the phenyl radical can add to the C1-atom of the 2-butyne isomer after passing a barrier of 12 kJ mol−1 thus yielding a doublet radical intermediate. The latter can undergo unimolecular decomposition and release a methyl group therefore forming the 1-phenyl-1-methylacetylene reaction product.26 Therefore, the initial additions of the phenyl radicals to the C4H6 isomers 1,2-butadiene, 1-butyne, and 2-butyne are correlated with entrance barriers of 12 kJ mol−1 at the most, which can be overcome easily in the chemical reactor. The initial reaction intermediate mainly decomposes via methyl group loss and partially – after isomerization – also via atomic hydrogen and methyl ejection pathways.
4. Summary
A high temperature chemical reactor was utilized to explore the reaction of phenyl radicals (C6H5) with 1,3-butadiene (H2CCHCHCH2) under combustion-like conditions (300 Torr, 873 K). The reaction products were monitored in a supersonic beam by utilizing VUV radiation from the Advanced Light Source and recording the PIE curves at mass-to-charge ratios of m/z = 130 (C10H10+), 116 (C9H8+), and 104 (C8H8+). Our data suggest that the atomic hydrogen (H), methyl (CH3), and vinyl (C2H3) losses are open with estimated branching ratios of about 86 ± 4%, 8 ± 2%, and 6 ± 2%, respectively. A detailed fit of the PIE curves and comparison with recent crossed beams and computational studies on the phenyl–1,3-butadiene reaction indicate that trans-1,3-butadienylbenzene (130 amu), 1,4-dihydronaphthalene (130 amu), and styrene (104 amu) can be rationalized as reaction products, which can also be formed under single collision conditions (Fig. 6). 1-Methylindene (130 amu), indene (116 amu), phenylallene (116 amu), and 1-phenyl-1-methylacetylene (116 amu) were identified as higher-order products synthesized upon reaction of the phenyl radical with the thermodynamically less stable C4H6 isomers 1,2-butadiene (H2CCCH(CH3)), 1-butyne (HCCC2H5), and 2-butyne (CH3CCCH3) as formed from 1,3-butadiene via hydrogen atom assisted isomerization or via thermal rearrangements involving hydrogen shifts. Our studies do not only verify the formation of three combustion-relevant polycyclic aromatic hydrocarbons (indene, 1-methylindene, and 1,4-dihydronaphthalene) in the chemical reactor, but also underline the necessity to compare experiments in the chemical reactor with those conducted under single collision conditions and with computed potential energy surfaces. This approach ensures differentiation between nascent and higher-order reaction products formed under combustion like conditions in the chemical reactor.
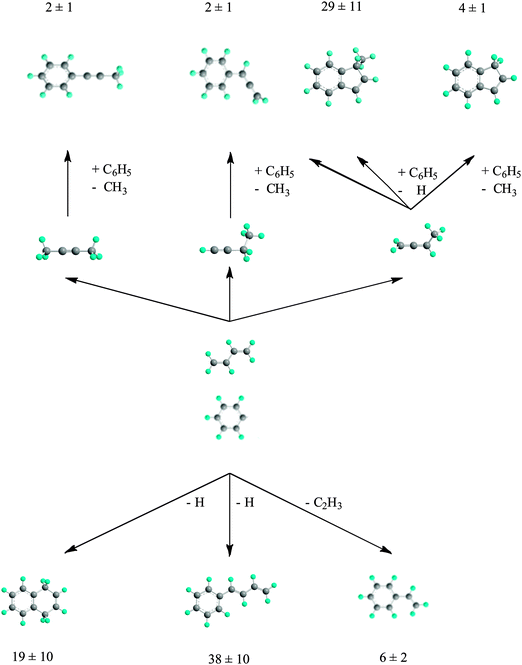 |
| | Fig. 6 Summary of primary (bottom) and secondary reaction products (upper section) probed in the present experiment together with the overall branching ratios. | |
Acknowledgements
This work was supported by the US Department of Energy, Basic Energy Sciences (DE-FG02-03ER15411 to the University of Hawaii and DE-FG02-04ER15570 to Florida International University). MA and AG are supported by the Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under Contract No. DE-AC02-05CH11231, through the Chemical Sciences Division. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. We thank Dr Fangtong Zhang (University of Hawaii) for his assistance with the experiments.
References
- H. Richter and J. B. Howard, Prog. Energy Combust. Sci., 2000, 26, 565–608 CrossRef CAS.
- W. W. Duley, Faraday Discuss., 2006, 133, 415–425 RSC.
- K. Hylland, J. Toxicol. Environ. Health, Part A, 2006, 69, 109–123 CrossRef CAS.
- M. Frenklach and E. D. Feigelson, Astrophys. J., 1989, 341, 372–384 CrossRef CAS.
- A. G. G. M. Tielens, Annu. Rev. Astron. Astrophys., 2008, 46, 289–337 CrossRef CAS.
- M. Hausmann and K. H. Homann, Ber. Bunsen-Ges., 1990, 94, 1308–1312 CAS.
- M. E. Law, P. R. Westmoreland, T. A. Cool, J. Wang, N. Hansen, C. A. Taatjes and T. Kasper, Proc. Combust. Inst., 2007, 31, 565–573 CrossRef.
- T. Yu and M. C. Lin, J. Phys. Chem., 1995, 99, 8599–8603 CrossRef CAS.
- T. Yu and M. C. Lin, Combust. Flame, 1995, 100, 169–176 CrossRef CAS.
- J. Park, G. J. Nam, I. V. Tokmakov and M. C. Lin, J. Phys. Chem. A, 2006, 110, 8729–8735 CrossRef CAS.
- I. V. Tokmakov, J. Park and M. C. Lin, ChemPhysChem, 2005, 6, 2075–2085 CrossRef CAS.
- J. Park, L. Wang and M. C. Lin, Int. J. Chem. Kinet., 2003, 36, 49–56 CrossRef.
- J. Park, S. Burova, A. S. Rodgers and M. C. Lin, Chem. Phys. Processes Combust., 1999, 308–311 CAS.
- H. Ismail, J. Park, B. M. Wong, W. H. Green Jr. and M. C. Lin, Proc. Combust. Inst., 2005, 30, 1049–1056 CrossRef.
- L. Vereecken, H. F. Bettinger and J. Peeters, Phys. Chem. Chem. Phys., 2002, 4, 2019 RSC.
- L. Vereecken and J. Peeters, Phys. Chem. Chem. Phys., 2003, 5, 2807 RSC.
- L. Vereecken, J. Peeters, H. F. Bettinger, R. I. Kaiser, P. v. R. Schleyer and H. F. Schaefer, III, J. Am. Chem. Soc., 2002, 124, 2781 CrossRef CAS.
- F. Zhang, R. I. Kaiser, V. V. Kislov, A. M. Mebel, A. Golan and M. Ahmed, J. Phys. Chem. Lett., 2011, 2, 1731–1735 CrossRef CAS.
- D. S. N. Parker, F. Zhang, R. I. Kaiser, V. V. Kislov and A. M. Mebel, Chem.–Asian J., 2011, 6, 3035–3042 CrossRef CAS.
- R. I. Kaiser, M. Goswami, F. Zhang, D. Parker, V. V. Kislov, A. M. Mebel, J. Aguilera-Iparraguirre and W. H. Green, Phys. Chem. Chem. Phys., 2012, 14, 720–729 RSC.
- V. V. Kislov, A. M. Mebel, J. Aguilera-Iparraguirre and W. H. Green, J. Phys. Chem. A, 2012, 116, 4176–4258 CrossRef CAS.
- D. S. N. Parker, F. Zhang, Y. S. Kim, R. I. Kaiser, A. Landera and A. M. Mebel, Phys. Chem. Chem. Phys., 2012, 14, 2997–3003 RSC.
- D. S. N. Parker, F. Zhang, R. I. Kaiser, A. Landera, V. V. Kislov, A. M. Mebel and A. G. G. M. Tielens, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 53–58 CrossRef CAS.
- X. Gu, F. Zhang, R. I. Kaiser, V. V. Kislov and A. M. Mebel, Chem. Phys. Lett., 2009, 474, 51–56 CrossRef CAS.
- V. V. Kislov and A. M. Mebel, J. Phys. Chem. A, 2010, 114, 7682–7692 CrossRef CAS.
- R. I. Kaiser, F. Zhang, X. Gu, V. V. Kislov and A. M. Mebel, Chem. Phys. Lett., 2009, 481, 46–53 CrossRef CAS.
- R. I. Kaiser, D. S. N. Parker, F. Zhang, A. Landera, V. V. Kislov and A. M. Mebel, J. Phys. Chem., 2012, 116, 4248–4258 CrossRef CAS.
- C. Nicolas, J. Shu, D. S. Peterka, M. Hochlaf, L. Poisson, S. R. Leone and M. Ahmed, J. Am. Chem. Soc., 2006, 128, 220–226 CrossRef CAS.
- J. L. Miller, J. Phys. Chem. A, 2004, 108, 2268–2277 CrossRef CAS.
- M. Musick, P. J. Van Tiggelen and J. Vandooren, Combust. Sci. Technol., 2000, 153, 247–253 CrossRef CAS.
- Y. Hidaka, T. Higashihara, N. Ninomiya, H. Masaoka, T. Nakamura and H. Kawano, Int. J. Chem. Kinet., 1996, 28, 137–142 CrossRef CAS; Y. Wang, W. Feng, M. Lei and R. Liu, Sci. China, Ser. B: Chem., 1998, 41, 60–65 CrossRef.
- S. D. Chambreau, J. Lemieux, L. Wang and J. Zhang, J. Phys. Chem. A, 2005, 109, 2190–2196 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cp42848b |
|
| This journal is © the Owner Societies 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.