Temperature-controlled self-assembling structure with selective guest-recognition at the liquid–solid interface†
Yibao
Li
ab,
Chunhua
Liu
a,
Yunzhi
Xie
a,
Xiaokang
Li
a,
Xun
Li
a,
Xiaolin
Fan
*a,
Ke
Deng
b,
Qingdao
Zeng
*b and
Chen
Wang
b
aKey Laboratory of Organo-pharmaceutical Chemistry, Gannan Normal University, Ganzhou 341000, P. R. China. E-mail: Fanxl2008@gnnu.cn; Fax: +86 (0)797 8393536
bNational Center for Nanoscience and Technology (NCNST), Beijing 100190, P. R. China. E-mail: zengqd@nanoctr.cn; Fax: +86 (0)10 82545548
First published on 24th October 2012
Abstract
We investigate the influence of temperature on the self-assembly of 2,6,11-tricarboxydecyloxy-3,7,10-triundecyloxy triphenylene (asym-TTT) adsorbed on HOPG using scanning tunneling microscopy (STM) at the liquid/solid interface. We show that the packing structures of 2D self-assembled asym-TTT can be precisely tuned by adjusting the substrate temperature. The temperature change from 20 °C to 35 °C induces two phase transitions and increment in the packing density from 0.161 to 0.179 molecules per nm2. The density-functional theory (DFT) calculations reveal that their interaction energies are similar. In particular, the experimental findings further illustrate the preferential adsorption of guest molecules, such as phthalocyanine, only in the domain of more close packing structures.
Introduction
Nanoporous networks on solid surfaces have drawn much interest in the area of nanotechnology.1–3 Such two dimensional porous networks offer the possibility to immobilize functional guest molecules depending on the nature of porous networks, such as size, shape, and symmetry. A pressing issue in the design of the molecular nanoporous architectures in a controllable manner is inherent to the networks. In order to gain knowledge on controlling the nature of the network cavities, various approaches have been explored, including molecular structural designs and environmental factors.In general, self-assembly leads to the formation of nanopores through molecular structural designs via non-covalent interactions, such as van der Waals interaction,4–8 hydrogen bond,9–11 metal–ligand coordination,12–14etc. Hydrogen bonding is the most frequently encountered non-covalent force which determines the network structure at the liquid/solid interface because it is relatively strong and highly directional. Thus, two-dimensional (2D) hydrogen-bonded porous networks on surfaces have demonstrated unique potential of tailoring supramolecular assemblies, such as networks formed by 1,3,5-benzenetricarboxylic acid (trimesic acid, TMA),9 perylene tetra-carboxylic di-imide (PTCDI) and melamine.10 In addition, the feasible strategy for inclusion of molecular clusters may involve the improvement of network flexibility, such as introducing alkyl chains to the host molecules of the hydrogen bonded networks utilizing van der Waals interactions. With less selectivity and directionality as compared to hydrogen bonds, van der Waals interaction stabilized networks could bring in the broadened flexibility of functional molecular architectures complementary to the reported systems based on hydrogen bond interactions. This enhanced host network flexibility could facilitate the possible dimensional shrinking or expanding. For example, single or double phthalocyanine was entrapped into networks of 1,3,5-tris(10-carboxydecyloxy)benzene (TCDB) depending on the molar ratio between them.11
For the latter effect, several attempts to control the self-assembled structure by external stimuli have been recently reported. The changes in the self-assembled structures can be induced by light,15–18 temperature,19–26 concentration,27 the addition of appropriate guest molecules,28 and so on. An example of light-sensitive structures has been obtained with the binary system TCDB–4NN-macrocycle.18 Among all important parameters for self-assembly at the liquid–solid interface, temperature studies are probably less, and only a few examples are reported in the literature. Temperature-induced phase transitions were reported by Marie et al. for hexakis-(n-dodecyl)-peri-hexabenzocoronene (HBCC12).23 As the temperature increased from 20 °C to 50 °C, three irreversible phase transitions of HBCC12 self-assemblies were observed by STM at the liquid/solid interface. Although temperature is a vital parameter for any self-assembly because it directly affects both thermodynamics and kinetics, little is known about its influence on physisorbed monolayers at the liquid–solid interface.
Such reports have indicated that temperature allows tuning the 2D packing structure of a flexible host network. There is one particularly intriguing aspect to accommodate guest molecules. This effect may be utilized for the controlled recognition of molecular guests with conceivable medical and life-science applications. With this in mind, we designed the molecular building blocks with asymmetrical carboxyl groups, 2,6,11-tricarboxydecyloxy-3,7,10-triundecyloxytriphenylene (asym-TTT) (Fig. 1). The three carboxyl groups are connected by the alkyl chains with the triphenylene core to each other. The addition of the alkyl chains to the molecular cores is expected to enhance the flexibility of the network, providing an opportunity to further tune 2D packing structures. Recently, we have found that this molecule could construct a supramolecular network, which can facilitate the formation of the direction-oriented molecular arrays of zinc phthalocyanines.29 Herein we focus on structural transformation of asym-TTT monolayers at the 1-octanoic acid/graphite interface by altering the temperature. The experimental findings further demonstrate the preferential adsorption of guest molecules, such as phthalocyanines, in only one network.
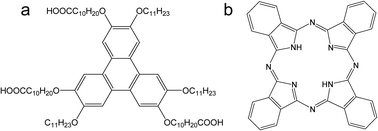 | ||
| Fig. 1 Chemical structures of asym-TTT (a) and phthalocyanine (Pc) (b). | ||
Experimental section
The chemical structures of asym-TTT and phthalocyanine (Pc) are shown in Fig. 1. The asym-TTT was synthesized according to the reported method.30 The phthalocyanine (Pc) was purchased from Aldrich and used without further purification. STM measurements were performed by using a Nanoscope IIId (Veeco Metrology, USA) with mechanically formed Pt/Ir (80/20) tips. The assemblies were formed by subsequent deposition of the components (about 10 μL) on a freshly cleaned HOPG (grade ZYB, Veeco Metrology, USA) surface. A sample holder was used to control the substrate temperature. The molar ratio was controlled by both the concentration and volume deposited. STM images were recorded after heating the samples up to a selected temperature for 20 min and also after cooling down the sample to room temperature. All images were recorded in constant current mode. The specific tunneling conditions are given in the corresponding figure captions. According to experimental results, solvent evaporation led to a maximum increase in asym-TTT concentration of about 12% during a measuring period, which is similar to the previous reported system.27We have performed theoretical calculations using DFT provided by DMol3 code.31 The Perdew and Wang parameterization32 of the local exchange-correlation energy are applied in local spin density approximation (LSDA) to describe exchange and correlation. We expand the all-electron spin-unrestricted Kohn–Sham wave functions in a local atomic orbital basis. For the large system, the numerical basis set is applied. All calculations are all-electron ones, and performed with the medium mesh. A self-consistent field procedure is done with a convergence criterion of 10−5 a.u. on the energy and electron density.
Results and discussions
At the solid–liquid interface, the relative surface coverage and stability of different phases can strongly depend on solution concentration.27 While previous studies were conducted with saturated solutions,29 further experiments were carried out with diluted solutions. When the concentration is diluted to 10−5 mol L−1, the asym-TTT can form two patterns with different packing densities. STM investigations on the 2D assembly of asym-TTT are presented in Fig. 2. Owing to the higher electronic density of states of the aromatic rings, the triphenylene core of asym-TTT appears in higher contrast than the alkyl chains in the image. The bright triangle is attributed to the triphenylene core, whereas the surrounding lines with reduced contrast to the alkyl substituents. There are two distinct domains, which can be observed in Fig. 2a. High-resolution STM images of Fig. 2b and c are presented for domain structures I and II, respectively. In domain I (Fig. 2b), it can be seen that every six molecules form a distorted hexagonal network. The alkyl chains of the six molecules are separated into two directions, in which there are two and four alkyl chains for the two directions, respectively. The angle between the two directions is about 120°. It can also be observed that there are twelve alkyl side chains in the center of the distorted hexagonal pore. According to the STM images, the corresponding molecular model is proposed in Fig. 2d. The stripes can be viewed as repeated dimers, in which the two triphenylene cores of the asym-TTT molecules are oriented in the same direction and the alkyl chains at both sides are aligned for maximizing the van der Waals interactions. The molecules of adjacent lamellae adopt tail-to-tail packing geometry, which can be ascribed to the hydrogen-bond formation between carboxyl groups from adjacent lamellae asym-TTT molecules. In addition, the measured unit cell parameters for domain I are a = 3.4 ± 0.1 nm, b = 7.3 ± 0.1 nm and α = 89 ± 1.0°, respectively.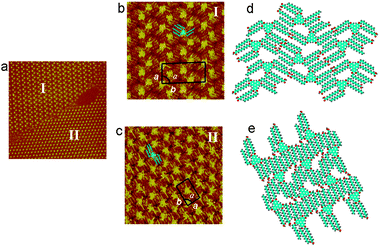 | ||
| Fig. 2 STM images of the assembly structure of asym-TTT at the 1-octanoic acid/graphite interface. (a) Large-scale (65.0 nm × 65.0 nm) STM image recorded with V = 531.4 mV, I = 518.8 pA. High-resolution STM images corresponding to domains I (b) and II (c). (b) Scan size: 16.1 nm × 16.1 nm, V = 531.4 mV, I = 425.4 pA. (c) Scan size: 18.6 nm × 18.6 nm, V = 638.9 mV, I = 394.9 pA. (d) Proposed packing molecular network structural model to domains I (d) and II (e). | ||
In domain II (Fig. 2c), asym-TTT forms a close-packed structure. The orientation of the triangle core could not be resolved clearly, which is very different from the orientation characteristics of the triangles in domain I. It should be noted that all alkyl side chains are adsorbed along one direction of the substrate. According to the STM image (Fig. 2c), the corresponding molecular model is proposed in Fig. 2e. The measured unit cell parameters are: a = 2.6 ± 0.1 nm, b = 4.3 ± 0.1 nm and α = 90 ± 1.0°, respectively.
In the molecular model of the asym-TTT supramolecular network, the unit cells of asym-TTT I and asym-TTT II include four and two molecules, respectively. According to the above STM analysis and results, the packing density of asym-TTT I or asym-TTT II is calculated to be 0.161 and 0.179 molecules per nm2, respectively. Their interaction energies may be similar, which can be concluded from packing density considerations. The DFT calculations for the interaction energies for two proposed structures indicate that the asym-TTT II (70.37 kJ mol−1 nm−2) is slightly more stable than asym-TTT I (65.57 kJ mol−1 nm−2). In the calculation, the effect is simplified. If solvent molecules are included, a more accurate result of the relative stabilities may be obtained. However, solvent is not observed in these two domains. According to above calculations, we think that the packing structures of 2D self-assembled asym-TTT might be tuned by altering the temperature.
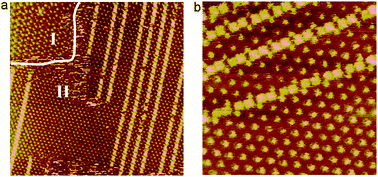
To investigate the temperature-controlled structural transformation, in situ STM experiments were performed to investigate the change of the asym-TTT supramolecular networks. The random lines in Fig. 3a–d highlight the domain boundary. Firstly, at room temperature (20 °C) asym-TTT molecules initially form an asym-TTT I structure, which fully covers the observed surface (Fig. 3a). Secondly, by heating to 25 °C, the asym-TTT II structure appears (Fig. 3b). Thirdly, upon continued heating to 30 °C in the same region, the area of the asym-TTT II structure increases (Fig. 3c). Finally, upon continued heating to 35 °C, the asym-TTT II structure fully covers the observed surface (Fig. 3d). From the in situ STM measurements, it could be noted that the packing structures of asym-TTT 2D self-assemblies can be tuned by adjusting the temperature. The increment in the temperature from 20 °C to 35 °C induces two structural transitions from asym-TTT I to asym-TTT II structure.
 | ||
| Fig. 3 Series of STM sequence images of a 162.0 × 162.0 nm region illustrating the structural transformation process of asym-TTT at different temperatures recorded with V = 935.3 mV; I = 223.3 pA. The condition of temperatures is inset in the images, and the random lines in Fig. 3b and c highlight the domain boundary. | ||
One fundamental question is whether this phase transition is under kinetic or thermodynamic control. In situ STM experiments were performed to investigate the structure of phase (I), as shown Fig. S1 (ESI†). Upon continued imaging in the same region under same imaging conditions, the structure of phase (I) can be recorded (Fig. S1a–c, ESI†). Once it is formed, it is not possible to form any novel structure by increasing the time. In addition, the assembling structures were observed after 20 min of thermal annealing. Longer annealing times or decreasing the sample temperature back to 20 °C does not change the arrangement. This indicates that the phase (II) is thermodynamically stable. Temperature-induced asym-TTT phase transitions are therefore irreversible.
Interestingly, upon the addition of Pc to the already formed supramolecular network of asym-TTT II at the solid/liquid interface, long molecular arrays of Pc are observed (Fig. 4a). It should be noted that growth of Pc molecular arrays could be observed only in domain II of asym-TTT and not in domain I (Fig. 4a). From the high resolution STM image (Fig. 4b), it can be clearly observed in detail. The adsorption behavior of Pc is similar to that of ZnPc, which has been reported.29 Since the domain II of asym-TTT is linked together by the weaker van der Waals interactions between the zig-zag molecular chains, the possible rearrangement of the inter-Z-chain interactions would lead to the inclusion of functional guest molecules. In addition, the angle between alkyl chains in domain II is about 180°, whereas that in domain I is 120°, which may lead to the preferential adsorption of Pc dimers in domain II of an asym-TTT supramolecular network. This controlled recognition of molecular guests may be conceivable in medical and life-science application.
 | ||
| Fig. 4 STM images showing asym-TTT II structure with selective recognition of guest molecules at the 1-octanoic acid/graphite interface. (a) Large-scale STM image (105.6 nm × 105.6 nm, V = 621.6 mV, I = 387.5 pA) indicates that 2D asym-TTT II can accommodate Pc molecules. The random lines in the image correspond to the boundary between domains I and II. (b) STM image of Pc–asym-TTT II demonstrates Pc is trapped within the cavity of the rearranged network (31.4 nm × 31.4 nm, V = 456.8 mV, I = 768.2 pA). | ||
Conclusion
In summary, the temperature is explored to control 2D supramolecular networks of asym-TTT molecules. It is observed that two packing structures of 2D self-assembled asym-TTT can be precisely tuned by adjusting the substrate temperature from 20 °C to 35 °C. In particular, the experimental findings further illustrate the preferential adsorption of guest molecules, such as phthalocyanine, only in one supramolecular network. These results also demonstrate that the temperature could provide promising approaches for modulating functional molecular networks and selectively recognizing guest molecules.Acknowledgements
This work was supported by the National Basic Research Program of China (No. 2011CB932303, 2013CB934200). Financial support from National Natural Science Foundation of China (No. 21073048, 51173031, 91127043, 2009BAI78B01, 2009BAI78B02), Key Laboratory of Optoelectronic Materials Chemistry and Physics, Chinese Academy of Sciences (2010KL0010), and the Youth Natural Science Foundation of Jiangxi Province are also gratefully acknowledged.Notes and references
- T. Kudernac, S. B. Lei, J. A. A. W. Elemans and S. De Feyter, Chem. Soc. Rev., 2009, 38, 402 RSC.
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671 CrossRef CAS.
- D. Bonifazi, S. Mohnani and A. Llanes-Pallas, Chem.–Eur. J., 2009, 15, 7004 CrossRef CAS.
- X. H. Qiu, C. Wang, Q. D. Zeng, B. Xu, S. X. Yin, H. Wang, S. L. Xu and C. L. Bai, J. Am. Chem. Soc., 2000, 122, 5550 CrossRef CAS.
- H. Spillmann, A. Kiebele, M. Stöhr, T. A. Jung, D. Bonifazi, F. Cheng and F. Diederich, Adv. Mater., 2006, 18, 275 CrossRef CAS.
- S. Lei, K. Tahara, X. Feng, S. Furukawa, F. C. De Schryver, K. Müllen, Y. Tobe and S. De Feyter, J. Am. Chem. Soc., 2008, 130, 7119 CrossRef CAS.
- S. Furukawa, K. Tahara, F. C. De Schryver, M. V. Auweraer, Y. Tobe and S. De Feyter, Angew. Chem., Int. Ed., 2007, 46, 2831 CrossRef CAS.
- D. Bléger, D. Kreher, F. Mathevet, A. J. Attias, G. Schull, A. Huard, L. Douillard, C. Fiorini-Debuischert and F. S. Charra, Angew. Chem., Int. Ed., 2007, 46, 7548 CrossRef.
- S. Griessl, M. Lackinger, M. Edelwirth, M. Hietschold and W. M. Heckl, Single Mol., 2002, 3, 25 CrossRef CAS.
- J. A. Theobald, N. S. Oxtoby, M. A. Philips, N. R. Champness and P. H. Beton, Nature, 2003, 424, 1029 CrossRef CAS.
- J. Lu, S. B. Lei, Q. D. Zeng, S. Z. Kang, C. Wang, L. J. Wan and C. L. Bai, J. Phys. Chem. B, 2004, 108, 5161 CrossRef CAS.
- S. Stepanow, M. Lingenfelder, A. Dmitriev, H. Spillmann, E. Delvigne, N. Lin, X. Deng, C. Cai, J. V. Barth and K. Kern, Nat. Mater., 2004, 3, 229 CrossRef CAS.
- M. A. Lingenfelder, H. Spillmann, A. Dmitriev, S. Stepanow, N. Lin, J. V. Barth and K. Kern, Chem.–Eur. J., 2004, 10, 1913 CrossRef CAS.
- S. Stepanow, N. Lin, D. Payer, U. Schilckum, F. Klappenberger, G. Zoppellaro, M. Ruben, H. Brune, J. V. Barth and K. Kern, Angew. Chem., Int. Ed., 2007, 46, 710 CrossRef CAS.
- R. Heinz, A. Stabel, J. P. Rabe, G. Wegner, F. C. De Schryver, D. Corens, W. Dehaen and C. Süling, Angew. Chem., Int. Ed., 1994, 33, 2080 CrossRef.
- P. Vanoppen, P. C. M. Grim, M. Rücker, S. De Feyter, G. Moessner, S. Valiyaveettil, K. Müllen and F. C. De Schryver, J. Phys. Chem., 1996, 100, 19636 CrossRef CAS.
- A. Miura, S. De Feyter, M. M. S. Abdel-Mottaleb, A. Gesquìere, P. C. M. Grim, G. Moessner, M. Sieffert, M. Klapper, K. Müllen and F. C. De Schryver, Langmuir, 2003, 19, 6474 CrossRef CAS.
- Y. T. Shen, L. Guan, X. Y. Zhu, Q. D. Zeng and C. Wang, J. Am. Chem. Soc., 2009, 131, 6174 CrossRef CAS.
- C. J. Li, Q. D. Zeng, Y. H. Liu, L. J. Wan, C. Wang, C. R. Wang and C. L. Bai, ChemPhysChem, 2003, 4, 857 CrossRef CAS.
- F. Klappenberger, A. Weber-Bargioni, W. Auwärter, M. Marschall, A. Schiffrin and J. V. Barth, J. Chem. Phys., 2008, 129, 214702–1 CrossRef.
- E. Treossi, A. Liscio, X. Feng, V. Palermo, K. Müllen and P. Samor, Small, 2009, 5, 112 CrossRef CAS.
- W. A. English and K. W. Hipps, J. Phys. Chem. C, 2008, 112, 2026 CAS.
- C. Marie, F. Silly, L. Tortech, K. Müllen and D. Fichou, ACS Nano, 2010, 4, 1288 CrossRef CAS.
- J. Adisoejoso, K. Tahara, S. B. Lei, P. Szabelski, W. Rżysko, K. Inukai, M. O. Blunt, Y. Tobe and S. De Feyter, ACS Nano, 2012, 4, 897 CrossRef.
- R. Gutzler, T. Sirtl, J. F. Dienstmaier, K. Mahata, W. M. Heckl, M. Schmittel and M. Lackinger, J. Am. Chem. Soc., 2010, 132, 5084 CrossRef CAS.
- J. M. MacLeod and F. Rosei, Aust. J. Chem., 2011, 64, 1297 CrossRef CAS.
- S. B. Lei, K. Tahara, F. C. De Schryver, M. V. Auweraer, Y. Tobe and S. De Feyter, Angew. Chem., Int. Ed., 2008, 47, 2964 CrossRef CAS.
- Y. B. Li, J. H. Wan, K. Deng, X. N. Han, S. B. Lei, Y. L. Yang, Q. Y. Zheng, Q. D. Zeng and C. Wang, J. Phys. Chem. C, 2011, 115, 6540 CAS.
- Y. B. Li, K. Deng, X. K. Wu, S. B. Lei, K. Q. Zhao, Y. L. Yang, Q. D. Zeng and C. Wang, J. Mater. Chem., 2010, 20, 9100 RSC.
- K. Q. Zhao, P. Hu, B. Q. Wang, W. H. Yu, H. M. Chen, X. L. Wang and Y. Shimizu, Chin. J. Chem., 2007, 25, 375 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244 CrossRef.
- A. Becke, J. Chem. Phys., 1988, 88, 2547 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cp43244g |
| This journal is © the Owner Societies 2013 |