Multidimensional OH local mode calculations for OH−(H2O)3—Importance of intermode anharmonicity†
Masato
Morita
and
Kaito
Takahashi
*
Institute of Atomic and Molecular Sciences, Academia Sinica, PO Box 23-166, Taipei, 10617, Taiwan R.O.C. E-mail: kt@gate.sinica.edu.tw
First published on 23rd October 2012
Abstract
We present theoretical calculations on the vibrational spectra between the energy range of 2000–4000 cm−1 for 4 isoenergetic conformers of gas phase OH−(H2O)3 and OH−(H2O)3·Ar clusters. The peak positions and associated intensities of the OH stretching vibrations were calculated using an extended local mode model on multi-dimensional ab initio potential energy surfaces and dipole moment functions obtained by MP2/6-311++G(3df,3pd). Furthermore, to simulate the experimental spectra directly, both the homogeneous and inhomogeneous line widths were determined theoretically. For the ionic hydrogen bonded OHs, which directly bind to the OH−, anharmonic coupling between the OHs on different water units is crucial for the reproduction of experimentally observed features. On the other hand, the coupling between free OH stretching vibrations is so small that the usual 1-dimensional local mode model provides a good and economical way to obtain the spectra. By comparing the theoretical spectra for 4 isoenergetic conformers, we found that the ionic hydrogen bonded OH stretching peaks can act as a descriptor for the subtle conformational differences in the first solvation shell. Furthermore, we showed that the coupling between the ionic hydrogen bonded OH and low frequency O⋯O stretching vibrations can cause fairly strong combination bands, and that the weakly bound argon, which was used as a messenger in the experiment by Robertson et al. [Science; 2003, 299, 1367], causes shifts on the peak positions for the ionic hydrogen bonded OHs. In addition, we quantified the effect of counterpoise correction on the simulated spectra for the ionic hydrogen bonded OHs.
I. Introduction
The molecular level mechanisms of the anomalously high mobility of a proton, H+, and a hydroxide ion, OH−, in aqueous solutions have been studied due to their fundamental importance toward acid–base reactions in chemistry and biology. Compared to the advances in the understating of proton transport,1 the mechanism of OH− transport is still controversial.2,3 The heart of the problem lies in the uncertainty of the first solvation shell coordination number of OH−, namely the number of water molecules pinned to the O atom in OH−.The experimental vibrational spectra of hydroxide ion water clusters, OH−(H2O)n (n = 1–5), by Robertson et al. in 2003 was an attempt to provide the answer from the gas phase cluster community.4 Due to the emergence of distinct peaks within the 3400–3600 cm−1 region, in the argon predissociation spectra, following the addition of the fourth water to the hydroxide anion, they concluded that three waters constitute the first solvation shell in gas phase OH−(H2O)n clusters. This conclusion was supported by theoretical simulations that showed that the second solvation shell water will result in peaks in the weak hydrogen bonded region of 3400–3600 cm−1.4,5
One may argue that the peak position of the OH− stretching vibration itself may provide more useful information on the environment around OH−. Indeed, as shown by Lin et al.,6 it is possible to extract the coordination number of water in the protonated water cluster through the peak position of its free OH stretching vibration. Thereby, it may be straightforward to investigate the vibrational peak of OH− as a marker of the environment around OH− for clusters and solution phase. Using the local mode model for OH− stretching vibration in the cluster cut out from snapshots of CPMD OH− (aq.) simulation, Hermansson et al. showed that solvent-induced frequency blue-shift for the peak position of OH− is related to the hydroxide hydration structure.7
However, as pointed out in our previous paper and shown in the experimental spectra by Robertson et al.,4,5 the intensity of the OH− stretching vibration in the OH−(H2O)n cluster is too weak to be observed except for small clusters, n = 1, 2. Moreover, the OH− peak is easily masked by the more intense free OH stretching peaks because the peak position of OH− generally emerges in the similar energy region as the free OHs. We note that although there is no free OH to be observed in OH− (aq.), the broad and intense peak by the weakly hydrogen bonded OH stretching vibration obscures the OH− peak as shown by Roberts et al.8
Therefore, in this paper, we suggest the observation of the OH stretching vibrations of the OH group directly bound to OH−, the ionic hydrogen bonded (IHB) OHs. Indeed they have large intensities, and their peak positions are largely red-shifted thus they are isolated from other OH stretching modes due to their large anharmonicity.4 Thereby detailed analysis and assignment of the IHB OH peaks should provide an understanding of the coordination number as well as subtle structural details of the first solvation shell water. In fact, Roberts et al. have used 2DIR spectroscopy method for the strongly red-shifted OH bonds to understand the dynamics of the first solvation shell water in OH− (aq.).8 Despite such an attractive characteristic of IHB OH, most of the previous studies on gas phase clusters have only focused on the peaks observed at around the free OH stretching energy region. One apparent reason is that, due to the strong hydrogen bonding, it is difficult to perform reliable calculation for the IHB OH stretching vibration with the usual normal mode analysis and empirical scaling prescriptions. In addition, the experimental fundamental IHB OH peaks observed in the strong hydrogen bonded region, less than 3000 cm−1, usually show up as broad peaks making comparison with theoretical stick spectra very hard. We note that having a large width also makes it hard to observe experimentally. These theoretical limitations have led to the difficulty in the assignment of the experimental spectra in this frequency range. Thus, it is important to develop calculation methods to treat such strong hydrogen bonded OH vibration.
For the accurate treatment of the strong hydrogen bonded OH stretching vibrations, it is crucial to perform variational calculations. However, it is a difficult task to obtain the full dimensional potential energy surface (PES) and the dipole moment function (DMF) for general clusters using accurate ab initio calculations. Thereby, in our previous paper,5 we developed the LU + LLC method as a general procedure for calculating the vibrational spectra for water and water–ion clusters, and applied it to several isomers of OH−(H2O)n (n = 3, 4) with PESs and the DMFs calculated by B3LYP/6-31+G(d,p). The LU + LLC model calculation is constructed from two procedures. First, the local unit (LU) calculation is performed based on the assumption that individual calculation of the water/OH− unit is valid by the adiabatic separation between the high frequency intra unit motions and the low frequency inter unit motions. This procedure has been successfully applied to the vibrational spectra calculations of other cluster systems by various authors.9–12 The goal of this LU was to solve the full three dimensional intramolecular vibration accurately to determine how the intra unit coupling between the stretching and bending vibrations, i.e. Fermi resonances, show up as observables in OH−(H2O)n spectra. Unfortunately, as shown in our previous paper, these couplings were minimal for strong hydrogen bonded OH stretching vibrations. Second, the inter unit vibrational mode coupling effects were included using the least order linear–linear coupling (LLC) terms for the interaction Hamiltonian. We note that this LLC term is equivalent to the harmonic coupling term. A similar kind of treatments of inter unit mode coupling calculations were performed for the water clusters by Kjaergaard et al.13 and Wang et al.11 Since the LU + LLC method includes the anharmonicity of the intra water high frequency vibrations in almost exact fashion, we obtained the overall feature of the spectra throughout a wide frequency range both for OH−(H2O)3 and OH−(H2O)4, and provided firm theoretical support for the previous assignment by Robertson et al.4 that the second solvation shell water starts at the fourth water. However, for OH−(H2O)3, our LU + LLC method could not describe the experimentally observed hump in the high energy region (2800 cm−1) of IHB OH stretching peak.
The main purpose of this paper is to remedy the above limitation of our previous approximations as well as to provide an economical yet reliable procedure to calculate the vibrational spectra for strongly hydrogen bonded systems. As postulated in our previous paper, possible reasons for the discrepancy between the LU + LLC model versus the experimental spectra are, (1) coupling of the high frequency intramolecular vibrations with the low frequency intermolecular vibration; (2) higher order couplings, namely anharmonic coupling, in the inter unit coupling; and (3) the effect of argon on the spectra. In this paper, we performed detailed analysis on these limitations. Using the obtained spectra, we discuss the possibility of using the IHB OH peak as a descriptor of subtle structural information for OH−(H2O)n clusters.
The remainder of the paper is organized in the following way. In Section II, we present the theoretical methods used for the present calculation. In Section III, we provide the results and discussions, and we conclude in Section IV.
II. Theoretical method
A. Search of conformers by geometry optimization
In the following, all electronic structure calculations were performed using Gaussian09 program.14 Since our interest includes the calculation of the argon complex OH−(H2O)3⋯Ar, we performed the geometry optimization for the singlet ground state of OH−(H2O)3 and OH−(H2O)3⋯Ar with MP2/6-311++G(3df,3pd) rather than the B3LYP/6-31+G(d,p) method used in our previous study. Schematic figures of 4 stable conformers of the bare cluster are given in Fig. 1; and electronic energies at the optimized geometries, zero point vibrational energy (ZPE) corrected energies and the Gibbs free energies at the temperature of 50 K are listed in Table 1. Schematic figures and energetic information of the stable structures of the argon bound conformers are given in Fig. 2 and Table 2, respectively. The Cartesian coordinates of the optimized geometries are given in the end of the ESI.†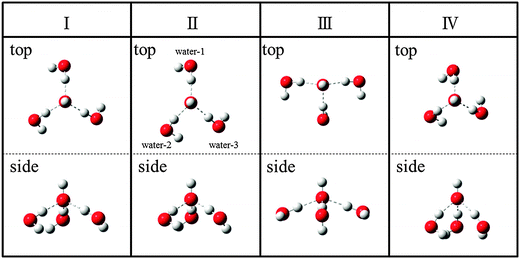 | ||
| Fig. 1 Schematic diagram of the 4 stable conformers, Conformer I to Conformer IV, of OH−(H2O)3 with MP2/6-311++G(3df,3pd). Upper and lower rows show top and side views, respectively. | ||
| Conformer | Electronic energy | Electronic energy + ZPE | Free energy (50 K) |
|---|---|---|---|
| Schematic figures of the optimized geometries are given in Fig. 1. | |||
| I | 0.026 | ![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) ![[. with combining low line]](https://www.rsc.org/images/entities/char_002e_0332.gif) |
|
| II | |
0.184 | 0.471 |
| III | 0.392 | 0.487 | 0.753 |
| IV | 0.020 | 0.849 | 1.381 |
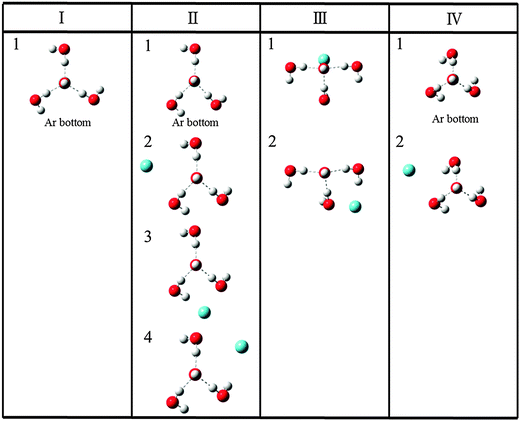 | ||
| Fig. 2 Schematic diagram of the stable structures of OH−(H2O)3·Ar for each conformer, Conformer I to Conformer IV, with MP2/6-311++G(3df,3pd). | ||
| Conformer | Electronic energy | Electronic energy + ZPE | Free energy (50 K) | |
|---|---|---|---|---|
| Schematic figures of the optimized geometries are given in Fig. 2. | ||||
| I | 1 | 0.004 | |
|
| II | 1 | |
0.190 | 0.191 |
| 2 | 0.164 | 0.405 | 0.329 | |
| 3 | 0.208 | 0.394 | 0.265 | |
| 4 | 0.205 | 0.402 | 0.292 | |
| III | 1 | 0.206 | 0.371 | 0.335 |
| 2 | 0.641 | 0.881 | 0.858 | |
| IV | 1 | 0.499 | 1.338 | 1.493 |
| 2 | 0.158 | 1.029 | 1.081 | |
B. Vibrational calculation with couplings
Since we are basically interested in the OH stretching peaks for the OH−(H2O)3, it is economical to resort to the local mode (LM) model, which has shown great success in the calculation of XH stretching spectra where X = O, C, N, and so on.15–20 The key in this success is the validity of the separation between the high frequency XH stretch and other low frequency vibrations. Additionally, our previous study5 using the LU model with B3LYP/6-31+G(d,p) has shown that IHB stretching vibrations are weakly coupled with the bending motion. However, considering OH⋯O systems, it is apparent that strong couplings can exist between the OH and O⋯O stretching vibrations. Furthermore, simple LM ignores the coupling between the XH bonds. Thus, for the symmetrically identical bonds, resultant vibrational peak positions are unphysically degenerate as recently studied by Wang et al. for water clusters.12 In this study, to avoid the above coupling treatment issue, we employed an extended local mode model using the multi-dimensional PES and DMF with MP2/6-311++G(3df,3pd). In the following, we use the term LM-nD to specify the extended local mode model, where n is the dimension of the employed vibrational degrees of freedom. We note that Kjaergaard et al. have developed the harmonic coupled anharmonic oscillator model, as an economical extension of the simple LM to account for the inter mode coupling at the harmonic level.21First, we report the details of the calculation for coupled OH and O⋯O stretching vibrations. Here we consider the coupling between the IHB OH and the low frequency vibration of O⋯O, in which one O atom is on the OH− unit and the other is the O on an IHB OH. Using the most stable Conformer I, we performed the 2 dimensional model, LM-2D, calculation to estimate its effect on the spectra. In the actual calculation, we considered both kinetic and potential energy coupling using the following Hamiltonian:22,23
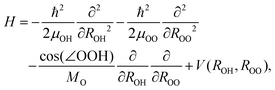 | (1) |
The vibrational problem was solved by diagonalizing the Hamiltonian matrix represented by the discrete variable representation (DVR) of the harmonic oscillator basis functions.24 We found that the result converges to within 1 cm−1 when we use 30 DVR basis for IHB OH and 60 DVR basis for O⋯O. Using the obtained eigenvalues, eigenvectors and DMF, the integrated absorption intensities (km mol−1) between the initial ground state, Ψ0, and the vibrational excited states, Ψf, were calculated by,25
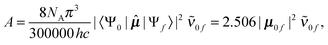 | (2) |
![[small mu, Greek, circumflex]](https://www.rsc.org/images/entities/b_i_char_e0b3.gif) is the DMF in Debye,
is the DMF in Debye, ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 0f is the transition energy in cm−1 and |μ0f| is the absolute value of the transition moment vector.
0f is the transition energy in cm−1 and |μ0f| is the absolute value of the transition moment vector.
Using the adiabatic separation between the fast OH stretching vibration and the slower O⋯O motion, many studies have utilized the mixed quantum classical model for the simulation of OH spectra in aqueous phase.7,8,26 In these models, the fast OH stretching vibration is treated quantum mechanically while the slower intermolecular motion is treated classically. Previously, Garden et al.27 have utilized a separation model for the water dimer where they first solved the fast OH stretching vibration then subsequently solved the slower inter molecular vibrational modes. Recently, Leforestier et al. also employed a method based on the adiabatic separation for water dimer peak position calculation using their flexible potential.28 To the best of our knowledge, there have not been any application of this adiabatic model to strong hydrogen bonded cases such as the present IHB, therefore, we also performed adiabatic separation treatment between the high frequency IHB OH and the low frequency O⋯O stretching vibrations to investigate the accuracy of such a simplified procedure. In the adiabatic separation calculation, we first obtain the potential energy curves of the IHB OH stretching vibrational states and transition dipole moments between these states by performing the usual LM, namely LM-1D, at each O⋯O distance. Then we performed another LM-1D calculation to obtain the vibrational states of O⋯O stretching vibration on the aforementioned potential energy curves. Using the resultant wave functions of O⋯O stretching vibrational states and the obtained transition dipole moment values, we calculated the integrated absorption coefficients. Since this adiabatic procedure ignores the contribution from the kinetic energy coupling term in eqn (1), we also performed LM-2D calculation without including the kinetic energy term for the comparison in equal footing. In the following, LM-2D(KC) and LM-2D will specify the calculations with and without the kinetic energy coupling term respectively.
Second, to determine the extent of coupling between the IHB OHs and between free OHs on different water units, we performed LM-3D calculations for 3 IHB OHs and 3 free OHs in OH−(H2O)3 using the following Hamiltonian:
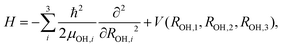 | (3) |
C. Simulation of the OH stretching vibrational spectra
To directly simulate the experimental vibrational spectra, it is necessary to consider the values of the linewidths. As described in our previous study,5,29 the usage of Voigt function30 is required to accurately reproduce the experimental spectrum. Since Voigt function is given as a convolution of the Lorentzian and Gaussian functions, it requires both the homogeneous (Lorentzian) and the inhomogeneous (Gaussian) linewidths.The homogeneous linewidth is determined from the intramolecular vibrational energy redistribution process which occurs after the excitation of the OH stretching vibration. In our previous paper,5 we reported on the fly direct dynamics method developed in ref. 31 for OH−(H2O)3 and OH−(H2O)4 to follow an ensemble of trajectories chosen to mimic the OH local mode excitation. As a result, we obtained the following power law relation between homogeneous linewidth Γ and peak position ω both in cm−1 unit,
| Γ = 0.011(ωref − ω)1.22, | (4) |
The inhomogeneous linewidth is rigorously obtained from the temperature dependent rotation population, and the direction of the transition moment with respect to the principal axes for inertia.32,33 However, as described in our previous paper,5 since the experimental spectra lack rotational line structure, our interest is in obtaining an estimate of the temperature dependent rotational envelope. For such purpose, we showed that the following scheme, initially employed by Uzer et al.,34 is effective.29 First, we obtain the most populated angular momentum J state, Jmax, at a given temperature, from the Boltzmann distribution by assuming symmetric top approximation. Then we approximate the width Δ due to temperature dependent inhomogeneous rotational broadening using the equation given by Uzer et al.34
| Δ = 4BJmax. | (5) |
III. Results and discussions
A. Stable conformers and their energetics
A schematic diagram of the 4 stable conformers of OH−(H2O)3 with MP2/6-311++G(3df,3pd) is given in Fig. 1. We note that these conformers have the same coordination number around the OH− and include only the first solvation shell waters. As mentioned in our previous paper,5 the isomers having a second solvation shell water is unstable by about 1 kcal mol−1 and should be ignored due to large energy differences. Furthermore, the absence of the peaks originating from the second solvation shell water in the experimental spectrum4 also supports this. Thereby, all the intense peaks observed in the experimental vibrational spectrum should be explained by considering the contributions from these 4 conformers.Conformer I has C3 symmetry where three O atoms in water monomers make an equilateral triangle with a O⋯O distance of 4.07 Å. This structure is essentially the same as the conformer considered in our previous paper using B3LYP/6-31+G(d,p),5 and has been reported by several authors.35–37 In Conformer II, one O⋯O distance between the water monomers, water-2 and water-3 in Fig. 1, is significantly shorter (3.33 Å) than the other two O⋯O distances (between water-1 and water-2 is 4.41 Å and water-1 and water-3 is 4.28 Å). Conformer III has T-shape as can be seen from the top view. Conformer IV also has C3 symmetry but the three water monomers are packed in a smaller space compared with Conformer I, and the O⋯O distance is 3.29 Å.
As shown in Table 1, Conformer II has the lowest relative electronic energies between these conformers. We note that Conformer II was not obtained in our previous paper with B3LYP/6-31+G(d,p) and we were not able to locate this conformer with the additional calculation using B3LYP/6-311++G(3df,3pd). On the other hand, the more sophisticated QCISD/6-311++G(3df,3pd) calculations confirmed the existence of this conformer.
As we can see, 4 conformers are isoenergetic, even for the most unstable Conformer III, the relative electronic energy difference from the most stable Conformer II is 0.3 kcal mol−1. Additionally, Conformers I and IV, both having C3 symmetry, have very similar electronic energies. However, when we consider the zero point vibrational energy corrected energy and the Gibbs free energy at 50 K, Conformer I becomes the most stable conformer, while Conformer IV becomes the most unstable conformer.
Similar to our previous discussion,5 although the Gibbs free energy of Conformer I is the most stable, the change in order of the stability between these conformers depends on the calculation levels and the properties used to gauge the stability, i.e. electronic energy, free energy and so on, and also depends on how we treat the basis set superposition error (BSSE). For example, if we focus on the electronic energy, Conformer IV is the lowest when one performs counterpoise correction at the geometry optimized without counterpoise correction, namely a posteriori procedure (Conformer I: +0.22 kcal mol−1, II: +0.19 kcal mol−1, III: +0.58 kcal mol−1). However, as shown in Table 1, Conformer IV has a very large zero-point energy due to its compact structure. In addition, the vibrational spectrum of Conformer IV is significantly different from the experimental spectrum observed by Robertson et al.4 as we will show later. Thereby, it is difficult to conclude the most dominant conformer under the experimental conditions from the energetic calculation. Indeed, we have to compare the theoretical vibrational spectrum for each conformer and the observed experimental spectrum to discuss the existence ratio of these conformers and to perform the complete assignment. Moreover, considering that the experiment4 was performed by the argon messenger method OH−(H2O)3·Ar + hν → OH−(H2O)3 + Ar, we must also consider the subtle argon binding effects to the energetics.
In most cases the argon messenger weakly bound to the cluster does not affect the spectra, but definitive proofs of the messenger effects on the observed spectra were reported previously.38–40 Schematic diagram of the stable structures obtained by attaching an argon to the 4 conformers is shown in Fig. 2. For the Conformer I, we obtained only one stable structure Conformer I-1, in which the argon atom attached to the bottom of Conformer I. Although we obtained a structure in which the argon atom is attached on top of the OH−, it had an imaginary frequency. On the other hand, Conformer II has 4 stable argon attached structures. As shown in the figure, other than the structure in which the argon atom attaches to the bottom, three other structures have argon attached to the sides. For both Conformers III and IV, we obtained two kinds of stable structures. We note that Conformer III-2 is degenerate with another symmetric structure in which the argon atom is attached to left-hand side. Similarly, Conformer IV-2 is triply degenerate.
Table 2 shows the energetics for the stable structures. For electronic energy, Conformer II-1 in which the argon atom attaches to the bottom side is the most stable structure. However, when we include the zero point energy correction or consider the Gibbs free energy, the most stable conformer is Conformer I-1. This tendency is similar to bare OH−(H2O)3 given in Table 1. However, compared to Conformer I, there exists many stable argon attached structures, for Conformers II, III and IV. In particular, relative energies between Conformer I-1 and the argon attached structures of Conformer II, namely from Conformer II-1 to Conformer II-4, are not so large. Thus, from simple Boltzmann estimation, the existence ratio of the Conformer II should be larger than that estimated from the bare clusters energetics in Table 1.
As mentioned above, there is no conclusive method to calculate the existence ratio between the isoenergetic isomers/conformers under the experimental conditions of messenger predissociation spectroscopy. Presently, we just consider the simple Boltzmann distribution obtained with the results of the free energy values at 50 K. First, for the case of bare conformers in Table 1, the calculated existence ratio for Conformer I is more than 99%. On the other hand, for the argon attached case in Table 2, the existence ratio for Conformer I-1 is 76%. As the result of the many possible structures for Conformer II, the existence ratio of the conformer is 23%. Conformer III is about 1% and Conformer IV is less than 0.1%. Thus, although Conformer I should be dominant, it is not certain whether other conformers, in particular Conformer II, are contributing to the experimentally observed spectrum. We will discuss this point in more detail in the Section C.
B. Coupling between IHB OH and O⋯O stretching vibrations
To discuss the effect of coupling on the vibrational spectrum, the two key vibrational states to be considered are the IHB OH stretch fundamental and the combination band between the fundamentals of IHB OH and O⋯O stretching vibrations. Table 3 shows the peak positions and intensities of IHB OH and the combination band between IHB OH and O⋯O stretching vibrations using the adiabatic separation treatment, LM-2D and LM-2D(KC). Additionally, the results for the IHB OH stretching vibration with the usual LM as well as the sum of the excitation energies of IHB OH and O⋯O fundamental stretching vibrations obtained by LM are also given. For a simple understanding of the results, we resort to the adiabatic model and plot the potential energy curves of a given IHB OH stretching vibrational quanta as a function of the O⋯O distance in Fig. 3. We note that the detailed list of the calculated peak positions and intensities including the other vibrational states are given in Table S1 and S2 of the ESI.†| OH fundamental | ||||
|---|---|---|---|---|
| LMa | Adiabaticb | LM-2Dc | LM-2D (KC)d | |
| a 1-Dimensinonal local mode model. b Adiabatic separation treatment between OH and O⋯O vibrations. c Local mode 2D model without the kinetic coupling term between OH and O⋯O vibrations. d Local mode 2D including the kinetic coupling term. e Sum of excitation energies for OH and O⋯O fundamentals calculated with LM. | ||||
| Excitation energy (cm−1) | 2423 | 2342 | 2344 | 2358 |
| Intensity (km mol−1) | 1997 | 1948 | 1900 | 1786 |
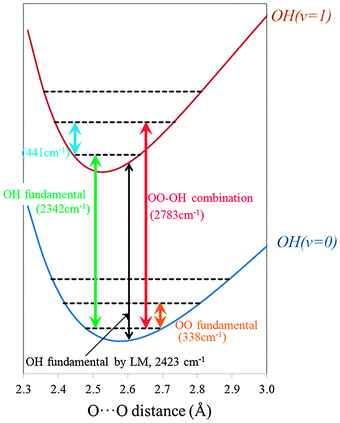 | ||
| Fig. 3 Schematic potential energy curves of the IHB OH stretching vibrational modes (OH) as a function of O⋯O distance (Conformer I). | ||
As shown in Table 3, for the peak position of IHB OH stretching vibration, the 2-dimensional models, namely the adiabatic separation, LM-2D and LM-2D(KC), give about 80 cm−1 red-shifted peak positions compared with the LM result of 2423 cm−1. Thus, we have to consider that such an amount of error is included when we ignore the coupling between OH and O⋯O stretching vibrations. The similarity in the peak positions of LM-2D and adiabatic separation treatment signifies that this simple treatment is valid even for the strongly hydrogen bonded OHs. In the following we will use the adiabatic model to explain the red-shift in comparison to the LM.
In LM, IHB OH is treated as a 1-dimensional problem at the equilibrium O⋯O distance of 2.61 Å. Thus, the IHB OH peak position, 2423 cm−1, corresponds to the difference of the potential energy curves between OH(v = 0) and OH(v = 1) at that point as shown by the black arrow in Fig. 3. On the other hand, with the adiabatic separation treatment, the peak position of the IHB OH fundamental is uniquely determined as the difference of the O⋯O vibrational ground states energies on OH(v = 0) and OH(v = 1), 2342 cm−1, as shown by green arrow in Fig. 3. If the curves of OH(v = 0) and OH(v = 1) had the same O⋯O minimum distance and curvature, the two values should match, however, as can be seen, the O⋯O minimum distance shifts greatly between OH(v = 0) versus OH(v = 1) and results in a red shift for the adiabatic 2D model. Although the peak position of IHB OH stretch fundamental does not largely depend on the kinetic coupling treatment in the 2-dimensional models, the inclusion of this coupling changes the intensity by more than 100 km mol−1. This shows the importance of the kinetic energy coupling term for accurate intensity calculation in this system.
For the combination band, once again the peak position dependence on the 2-dimensional models is very small, but in this case it is blue-shifted from the LM. Again, we resort to the adiabatic separation model to understand this change. As mentioned above, the peak position of the combination band by LM is obtained by simply summing up the LM fundamental frequencies, IHB OH (2423 cm−1) and O⋯O (319 cm−1). This fundamental peak position for O⋯O stretching vibration (319 cm−1) is similar to the result by the adiabatic separation treatment on OH(v = 0), 338 cm−1, shown by the orange arrow in Fig. 3. However, the peak position of the combination band in adiabatic separation treatment is given by the sum of the transitions energies of IHB OH fundamental (2342 cm−1), and of the O⋯O fundamental on the excited potential energy curve OH(v = 1), 441 cm−1, given by the light blue arrows. Thereby, the main reason for the blue-shift in the 2-dimensional models originates from the strengthening of the O⋯O bond as the IHB OH is vibrationally excited. As for the intensity, we can see a correlation between the decrease in the IHB OH transition and the increase in the combination band as the coupling treatment becomes more accurate, which is consistent with the intensity borrowing mechanism.
Going back to our motivation, we focus on the results of the most accurate LM-2D(KC) model calculation, and consider the impact of the combination band on the vibrational spectrum. As shown in Table 3, the difference of the peak positions between IHB OH and the combination band is 429 cm−1. From the view point of the peak position, the combination band exists in the energy range that corresponds to the hump seen in the experimental spectrum (∼2800 cm−1). However, the intensity of the combination band is less than 10% of the IHB OH, thus we conclude that the intensity of the combination band is too weak to cause significant features in the resultant vibrational spectrum around the energy region of IHB OHs peak. In the next section, we will consider the coupling between the IHB OHs in detail.
C. OH stretching vibrational spectra
| Conformer I | Conformer II | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Stretching mode | LM | LM-3D | Stretching mode | LM | LM-3D | ||||
| Frequency (cm−1) | Intensity (km mol−1) | Frequency (cm−1) | Intensity (km mol−1) | Frequency (cm−1) | Intensity (km mol−1) | Frequency (cm−1) | Intensity (km mol−1) | ||
| IHB OH | 2423 | 1997 | 2419 | 2361 | IHB OH | 2281 | 2013 | 2302 | 2580 |
| 2423 | 1997 | 2419 | 2361 | 2347 | 2178 | 2450 | 1657 | ||
| 2423 | 1997 | 2655 | 1075 | 2721 | 1543 | 2818 | 1309 | ||
| Free OH | 3773 | 6.8 | 3773 | 7.3 | Free OH | 3745 | 20.8 | 3745 | 21 |
| 3773 | 6.8 | 3773 | 6.6 | 3774 | 6.5 | 3774 | 6.3 | ||
| 3773 | 6.8 | 3773 | 6.6 | 3777 | 5.1 | 3777 | 5.1 | ||
| OH− | 3756 | 0.52 | — | — | OH− | 3755 | 0.76 | — | — |
| Conformer III | Conformer IV | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Stretching mode | LM | LM-3D | Stretching mode | LM | LM-3D | ||||
| Frequency (cm−1) | Intensity (km mol−1) | Frequency (cm−1) | Intensity (km mol−1) | Frequency (cm−1) | Intensity (km mol−1) | Frequency (cm−1) | Intensity (km mol−1) | ||
| IHB OH | 2271 | 1854 | 2314 | 1452 | IHB OH | 2682 | 1366 | 2666 | 976 |
| 2531 | 1904 | 2533 | 3286 | 2682 | 1366 | 2666 | 976 | ||
| 2531 | 1904 | 2718 | 715 | 2682 | 1366 | 2876 | 1970 | ||
| Free OH | 3765 | 8.7 | 3765 | 0.68 | Free OH | 3731 | 26 | 3727 | 16 |
| 3765 | 8.7 | 3765 | 17 | 3731 | 26 | 3732 | 32 | ||
| 3769 | 6.6 | 3769 | 6.3 | 3731 | 26 | 3732 | 32 | ||
| OH− | 3748 | 0.54 | — | — | OH− | 3773 | 8.2 | — | — |
Since Conformer II has no symmetry, there are no degenerate vibrational states for both IHB OHs and free OHs with the LM calculation. However, a large difference exists between the results of LM and LM-3D for IHB OHs. It shows the importance of the inter mode coupling between IHB OH stretching vibrations even if the conformer has no geometrical symmetry. On the other hand, the inter mode coupling effect between free OHs is once again small. The lowest energy state in free OH comes from the free OH of water-2 in Fig. 1. This can be considered as the result of a weak hydrogen bond between the free OH of water-2 and the O atom in water-3. In other words, the free OH of water-2 is not truly a “free OH” but a weakly hydrogen bonded OH. Therefore, this OH peak position is red-shifted and has larger intensity compared to the other two free OHs. Accordingly, compared with other two IHB OHs, the hydrogen bond of IHB OH in water-2 becomes weaker, and its peak position is blue-shifted and its intensity is smaller.
For the T-shape Conformer III, two IHB OHs are degenerate within the LM model, while the inclusion of mode coupling causes a 185 cm−1 spitting. The C3 symmetry Conformer IV has similar trend as Conformer I, but its much larger splitting is signifying the stronger coupling due to the more compact structure. Moreover, the energy splitting of free OHs for Conformer IV by LM-3D becomes 5 cm−1. All in all, comparing the LM and LM-3D results, inter mode coupling effects for IHB OHs are very large for all conformers, and ignoring the inter mode coupling results in a significant loss of accuracy.
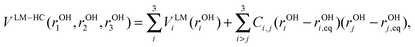 | (6) |
Fig. 4 shows the peak positions of IHB OHs and free OHs with LM, LM-HC and LM-3D for Conformer I. The splitting for the IHB OHs between LM-HC and LM-3D indicates that the anharmonicity in the inter mode coupling is crucial for this case. The splitting by harmonic coupling is only 76 cm−1 which is less than one-third of the splitting given by LM-3D. As described above, the importance of inter mode coupling strongly depends on the nature of OH bonds, and for the free OHs, the energy difference between LM, LM-HC and LM-3D is less than 1 cm−1. All in all, for strong hydrogen bonded OHs such as IHB, we have to pay attention to the anharmonicity of inter mode coupling as well as the anharmonicity of the OH stretching motion. As will be shown in the following, the large energy splitting of IHB OHs by the anharmonic inter mode coupling is pivotal to reproduce the experimental spectrum which we failed previously using the harmonic coupling (LLC) model. The detailed list of the peak positions with LM, LM-HC and LM-3D for all conformers is given in Table S3 and Fig. S1 of the ESI.†
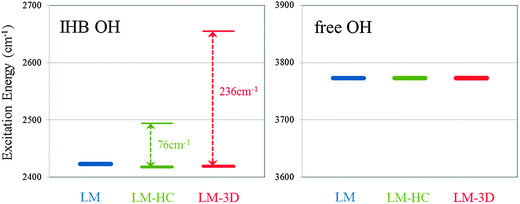 | ||
| Fig. 4 Excitation energies with the local mode (LM) model, the local mode plus harmonic inter mode coupling (LM-HC) model and the local mode 3D (LM-3D) model for Conformer I. Left-hand side is for the ionic hydrogen bonded (IHB) OH stretching vibration and right-hand side is for the free OH stretching vibration. | ||
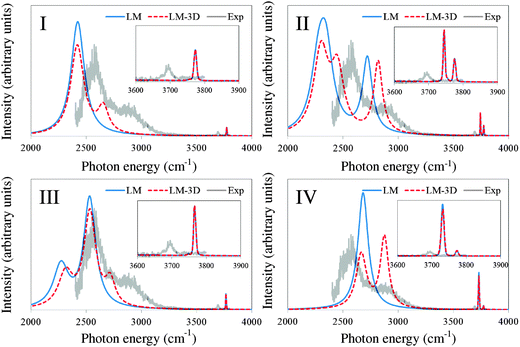 | ||
| Fig. 5 Simulated spectrum for each conformer of OH−(H2O)3, with the local mode (LM) model and the extended local mode (LM-3D) model. The homogeneous and inhomogeneous linewidths are calculated based on eqn (4) and (5) in the text. Experimental spectrum from ref. 4 is normalized in the figures so that the highest peak associated with IHB OHs matches between the experimental and the simulated LM-3D spectra. | ||
For Conformer I, with LM-3D, we clearly see two separate peaks coming from the IHB OHs. As mentioned above, these two peaks are due to the large intermode coupling between the three IHB OHs stretching vibrations. We note that LU + LLC calculation for this conformer in our previous paper did not predict such a clear separation due to the limitation of the harmonic intermolecular coupling approximation. We have also performed the LM-3D calculation using B3LYP/6-31+G(d,p) to confirm that this is a result due to the vibrational calculation not due to the difference in the quantum chemistry method (See Fig. S2 of ESI†). All in all, the overall structure of the experimental spectrum is reproduced by the simulated spectrum of Conformer I, which is consistent with the above result that the Conformer I is dominant from the view point of the energetic calculation with the Gibbs free energy at 50 K.
However, as mentioned above, Conformer II should be considered carefully because it has the most stable electronic energy, and a non-negligible existence ratio, 23%, from the simple Boltzmann distribution using the Gibbs free energies of argon attached structures. For Conformer II, three peaks of IHB OHs are observed in LM-3D. Thereby, subtle geometrical differences between Conformer I and Conformer II are reflected on the difference of IHB OH peaks. More importantly, a clear two peak structure in the free OH region emerges because one peak is red-shifted by the weak hydrogen bond mentioned above. Since only one peak is observed in the experimental spectrum, Conformer II should not be considered as the dominant conformer in the experiment.
The simulated spectrum for Conformer III has some similar characteristics as that of Conformer I, but the clear difference is that Conformer III has the smaller peak in the lower energy side to the main peak. Since the experimental spectrum did not observe this lower energy range, it is hard to confirm the existence of Conformer III, but our theoretical estimates show that the contribution from Conformer III is negligible, about 1%. Moreover, the energy difference between the main IHB OH peak and the higher energy sub peak is too small to explain the experimental spectrum shape. Thereby, we can conclude that Conformer III is possibly not dominant in the experiment. For Conformer IV, the relation of the intensities between two peaks from IHB OHs is apparently different from these experimental ones. Additionally, two peaks are seen in the free OH stretching region, which are assigned to the free OH stretching vibrations and the OH− stretching vibration.
All in all, our results show that IHB OH peaks reflect subtle structural difference between the conformers within the first solvation shell water. Thereby, by combining the information of IHB OHs and free OHs peaks, we can perform precise assignments for the OH−(H2O)n clusters. The similarity between the simulated spectrum for Conformer I to the experimental one indicates the possibility that only one kind of conformer is actually present in the experiment. Such high conformer selectivity in argon predissociation spectroscopy has been pointed out to be due to the efficient cooling/trapping by the attachment of the messenger.39,40 However, since there are no established mechanisms to confirm this, we will consider the remaining issues which can possibly affect the spectrum in the following section.
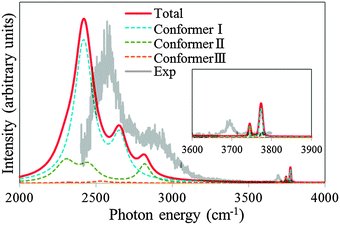 | ||
| Fig. 6 The theoretical spectrum obtained by summing up the contributions from the conformers (Total). The contributions are calculated with Boltzmann distribution at 50 K as described in the text. Each conformer contribution is described as Conformer I, Conformer II and Conformer III. The contribution from Conformer IV is negligible as described in the text. Experimental spectrum from ref. 4 is normalized in the figure so that the highest peak associated with IHB OHs matches between the experimental and the simulated (total) spectra. | ||
Second is the “direct effect” which is the perturbation induced by the argon to the peak positions and intensities. We investigate this “direct effect” on the spectrum of Conformer I. In Fig. 7, we compare the vibrational spectrum of the bare Conformer I (no argon in Fig. 7) and its argon attached counterpart Conformer I-1 (argon attached in Fig. 7). The “Direct effect” of the messenger has been previously reported for the free OH stretching vibration of the cationic water clusters.40 In such cases, the messenger binds directly to the free OH bond, in a O–H···Messenger structure, and causes a shift in the OH stretching peak position compared with other free OH peak positions. However, as shown in Fig. 2, such structure was not obtained for OH−(H2O)3·Ar. As a result, the apparent difference of the vibrational spectra between Conformer I and Conformer I-1 is not observed in the free OH region. On the other hand, for the IHB OH region, we can observe a small red-shift by the argon attachment. Thus, the subtle surrounding difference emerges more clearly for the IHB OH peak than the free OH peak. Finally, to investigate the effect of the counterpoise correction on the vibrational spectrum, we performed counterpoise optimization for Conformer I-1, then we calculated the vibrational spectrum using the counterpoise corrected potential energy surface (argon attached/CP in Fig. 7). Considering the counterpoise correction, blue-shift of the IHB OH is observed compared with non-corrected results, which shows the sensitivity of the IHB OH peak. However, we can conclude that it is possible to estimate the experimental spectrum without considering the “direct effect” of the argon messenger and counterpoise correction because the change in the overall shape of the spectrum is not drastic.
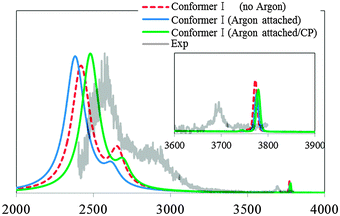 | ||
| Fig. 7 The theoretical spectra calculated with the extended local mode (LM-3D) model for Conformer I (no argon) in Fig. 1, and I-1 (argon attached) in Fig. 2. For I-1, the counterpoise corrected theoretical spectrum is also calculated (argon attached/CP). Experimental spectrum from ref. 4 is normalized in the figure so that the highest peak associated with IHB OHs matches between the experimental and the simulated spectrum (argon attached/CP). | ||
IV. Conclusions
We performed extended local mode simulation for the fundamental stretching vibrations of IHB OH and free OH and OH− for 4 isoenergetic conformers of OH−(H2O)3 and OH−(H2O)3·Ar. The main conclusions are (1) conformer I having C3 symmetry structure is probably dominant in the experiment by Robertson et al., and (2) the experimental spectrum is well reproduced by the LM-3D model for the IHB OHs and by the LM model for the free OHs stretching vibrations. Through this study, we also showed that we have to combine various calculations and compare various properties even for the determination of the most dominant conformer under the experimental conditions. For IHB OHs intermode anharmonic coupling is very important for the reproduction of the observed spectral feature, namely the hump observed in the high frequency region of IHB OH. Thereby failures in our previous LU + LLC model originated from the neglecting of the intermode anharmonic coupling. It has been well known that one must consider anharmonicities along the XH bond for hydrogen bonded systems, but the present results show that anharmonicities among XH bonds for the strong hydrogen bonded system should not be ignored, and must be carefully treated in the spectroscopy of the ion water cluster and aqueous ion solutions. Detailed analysis of the OH and O⋯O coupling for strong IHBs suggests that the adiabatic model is a very efficient method to calculate as well as understand spectral features.Acknowledgements
We thank Prof. Mark A. Johnson and Mr Arron Wolk for the numerical experimental data published in Science Magazine 2003, 299, 1367. KT thanks Academia Sinica Nano program and National Science Council (NSC98-2113-M-001-030-MY2 and NSC100-2113-M-001-004-MY2) of Taiwan for funding. We thank Academia Sinica High Performance Computer Center and National Center for High Performance Computing of Taiwan for computer time.References
- M. Eigen, Angew. Chem., Int. Ed. Engl., 1964, 3, 1 CrossRef CAS; G. Zundel, Adv. Chem. Phys., 2000, 111, 1 CrossRef; N. Agmon, Chem. Phys. Lett., 1995, 244, 456 CrossRef; D. Marx, M. E. Tuckerman, J. Hutter and M. Parinello, Nature, 1999, 397, 601 CrossRef; U. W. Schmitt and G. A. Voth, J. Chem. Phys., 1999, 111, 9361 CrossRef; D. Asthagiri, L. R. Pratt and J. D. Kress, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6704 CrossRef.
- N. Agmon, Chem. Phys. Lett., 2000, 319, 247 CrossRef CAS; D. Asthagiri, L. R. Pratt, J. D. Kress and M. A. Gomez, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7229 CrossRef; R. Ludwig, Angew. Chem., Int. Ed., 2003, 42, 258 CrossRef; M. E. Tuckerman, A. Chandra and D. Marx, Acc. Chem. Res., 2006, 39, 151 CrossRef.
- G. W. Brady, J. Chem. Phys., 1958, 28, 464 CrossRef CAS; R. Buchner, G. Hefter, P. M. May and P. Sipos, J. Phys. Chem. B, 1999, 103, 11186 CrossRef; A. Botti, F. Bruni, S. Imberti, M. A. Ricci and A. K. Soper, J. Chem. Phys., 2004, 120, 10154 CrossRef; I. A. Heisler, K. Mazur and S. R. Meech, J. Phys. Chem. Lett., 2011, 2, 1155 CrossRef.
- W. H. Robertson, E. G. Diken, E. A. Price, J.-W. Shin and M. A. Johnson, Science, 2003, 299, 1367 CrossRef CAS.
- M. Morita and K. Takahashi, Phys. Chem. Chem. Phys., 2012, 14, 2797 RSC.
- C.-K. Lin, C.-C. Wu, Y.-S. Wang, Y. T. Lee, H.-C. Chang, J.-L. Kuo and M. L. Klein, Phys. Chem. Chem. Phys., 2005, 7, 938 RSC.
- K. Hermansson, P. A. Bopp, D. Spångberg, L. Pejov, I. Bakó and P. D. Mitev, Chem. Phys. Lett., 2011, 514, 1 CrossRef CAS.
- S. T. Roberts, P. B. Petersen, K. Ramasesha, A. Tokmakoff, I. S. Ufimtsev and T. J. Martinez, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15154 CrossRef CAS.
- G. R. Low and H. G. Kjaergaard, J. Chem. Phys., 1999, 110, 9104 CrossRef CAS; D. P. Schofield and H. G. Kjaergaard, Phys. Chem. Chem. Phys., 2003, 5, 3100 RSC.
- T. Salmi, V. Hänninen, A. L. Garden, H. G. Kjaergaard, J. Tennyson and L. Halonen, J. Phys. Chem. A, 2008, 112, 6305 CrossRef CAS; T. Salmi, H. G. Kjaergaard and L. Halonen, J. Phys. Chem. A, 2009, 113, 9124 CrossRef; E. Sälli, T. Salmi and L. Halonen, J. Phys. Chem. A, 2011, 115, 11594 CrossRef.
- Y. Wang and J. M. Bowman, Chem. Phys. Lett., 2010, 491, 1 CrossRef CAS; Y. Wang and J. M. Bowman, J. Chem. Phys., 2011, 134, 154510 CrossRef; E. Kamarchik and J. M. Bowman, J. Phys. Chem. A, 2010, 114, 12945 CrossRef; E. Kamarchik, Y. Wang and J. M. Bowman, J. Chem. Phys., 2011, 134, 114311 CrossRef.
- Y. Wang and J. M. Bowman, J. Chem. Phys., 2012, 136, 144113 CrossRef.
- H. G. Kjaergaard, A. L. Garden, G. M. Chaban, R. B. Gerber, D. A. Matthews and J. F. Stanton, J. Phys. Chem. A, 2008, 112, 4324 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. L. Swofford, M. E. Long and A. C. Albrecht, J. Chem. Phys., 1976, 65, 179 CrossRef CAS.
- B. R. Henry, Acc. Chem. Res., 1977, 20, 207 CrossRef.
- M. S. Child and R. T. Lawton, Chem. Phys. Lett., 1982, 87, 217 CrossRef CAS; M. S. Child and L. Halonen, Adv. Chem. Phys., 1984, 57, 1 CrossRef.
- M. Lewerenz and M. Quack, Chem. Phys. Lett., 1986, 123, 197 CrossRef CAS; M. Quack, Annu. Rev. Phys. Chem., 1990, 41, 839 CrossRef.
- A. V. Fedorov and D. L. Snavely, Chem. Phys., 2000, 254, 169 CrossRef CAS.
- K. Takahashi, M. Sugawara and S. Yabushita, J. Phys. Chem., 2002, 106, 2676 CrossRef CAS; K. Takahashi, M. Sugawara and S. Yabushita, J. Phys. Chem. A, 2005, 109, 4242 CrossRef.
- H. G. Kjaergaard, H. Yu, J. S. Bernhard, R. H. Bryan and W. T. Allan, J. Chem. Phys., 1990, 93, 6239 CrossRef CAS.
- G. H. Gardenier, M. A. Johnson and A. B. McCoy, J. Phys. Chem. A, 2009, 113, 4772 CrossRef CAS.
- E. Kamarchik, Oleg Kostko, J. M. Bowman, M. Ahmed and A. I. Krylov, J. Chem. Phys., 2010, 132, 194311 CrossRef.
- J. C. Light, I. P. Hamilton and J. V. Lill, J. Chem. Phys., 1985, 82, 1400 CrossRef CAS.
- P. W. Atkins and R. S. Friedman, Molecular Quantum Mechanics, Oxford University, Oxford, 3rd edn, 1997 Search PubMed.
- B. M. Auer and J. L. Skinner, J. Chem. Phys., 2008, 128, 224511 CrossRef CAS; V. Buch, J. Phys. Chem. B, 2005, 109, 17771 CrossRef; H. Torii, J. Phys. Chem. A, 2006, 110, 9469 CrossRef.
- A. L. Garden, L. Halonen and H. G. Kjaergaard, J. Phys. Chem. A, 2008, 112, 7439 CrossRef CAS.
- C. Leforestier, K. Szalewicz and A. van der Avoird, J. Chem. Phys., 2012, 137, 014305 CrossRef.
- Y.-L. Cheng, H.-Y. Chen and K. Takahashi, J. Phys. Chem. A, 2011, 115, 5641 CrossRef CAS; H.-Y. Chen, Y.-L. Cheng and K. Takahashi, J. Phys. Chem. A, 2011, 115, 14315 CrossRef.
- M. R. Zaghloul, Mon. Not. R. Astron. Soc., 2007, 375, 1043 CrossRef CAS.
- K. Takahashi, Phys. Chem. Chem. Phys., 2010, 12, 13950 RSC.
- S. Ishiuchi, M. Fujii, T. W. Robinson, B. J. Miller and H. G. Kjaergaard, J. Phys. Chem. A, 2006, 110, 7345 CrossRef CAS.
- C. Murray, E. L. Derro, T. D. Sechler and M. I. Lester, Acc. Chem. Res., 2009, 42, 419 CrossRef CAS.
- T. Uzer, J. T. Hynes and W. P. Reinhard, J. Chem. Phys., 1986, 85, 5791 CrossRef CAS.
- H. M. Lee, P. Tarkeshwar and K. S. Kim, J. Chem. Phys., 2004, 121, 4657 CrossRef CAS; S. S. Xantheas, J. Am. Chem. Soc., 1995, 117, 10373 CrossRef.
- C. P. del Valle and J. J. Novoa, Chem. Phys. Lett., 1997, 269, 401 CrossRef.
- M. Masamura, Chem. Phys. Lett., 2001, 339, 279 CrossRef CAS; M. Masamura, J. Chem. Phys., 2002, 117, 5257 CrossRef.
- M. Okumura, L. I. Yeh, J. D. Myers and Y. T. Lee, J. Phys. Chem., 1990, 94, 3416 CrossRef CAS.
- K. Mizuse and A. Fujii, Phys. Chem. Chem. Phys., 2011, 13, 7098 RSC; K. Mizuse and A. Fujii, J. Phys. Chem. A, 2012, 116, 4868 CrossRef CAS.
- G. E. Douberly, R. S. Walters, J. Cui, K. D. Jordan and M. A. Duncan, J. Phys. Chem. A, 2010, 114, 4570 CrossRef CAS; P. D. Carnegie, B. Bandyopadhyay and M. A. Duncan, J. Phys. Chem. A, 2011, 115, 7602 CrossRef; R. Garza-Galindo, M. Castro and M. A. Duncan, J. Phys. Chem. A, 2012, 116, 1906 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of the peak positions and intensities by the LM-2D calculations for IHB OH and O⋯O stretching vibrations; tables of the vibrational excitation energies and intensities with LM, LM-HC and LM-3D; figures of the IHB OHs peak positions dependence on the inter mode coupling treatments; figures of the simulated vibrational spectra for the 4 conformers at the temperatures of 100 K and 150 K; table of vibrational excitation energies and intensities with the counterpoise correction.; energies of OH−(H2O)3·Ar with the counterpoise correction; lists of the optimized geometries for the conformers. See DOI: 10.1039/c2cp42501g |
| This journal is © the Owner Societies 2013 |