Catalytic activity of Co–Nx/C electrocatalysts for oxygen reduction reaction: a density functional theory study
Received 29th July 2012, Accepted 18th October 2012
First published on 19th October 2012
Abstract
First-principles DFT computations are performed to explain the origin and the mechanism of oxygen reduction reaction (ORR) on Co–Nx (x = 2, 4) based self-assembled carbon supported electrocatalysts in alkaline and acidic media. The results show that the formation of graphitic Co–N4 defect is energetically more favorable than the formation of graphitic Co–N2 defect. Furthermore graphitic Co–N4 defects are predicted to be stable at all potentials (U = 0–1.23 V) in the present study while Co–N2 defects are predicted to be unstable at high potentials. Therefore the Co–N4 defect is predicted to be the dominant in-plane graphitic defect in Co–Nx/C electrocatalysts. O2 chemisorbs to Co–N4 and Co–N2 defects indicating that both defect motifs are active for the reduction of O2 to peroxide. However, the weak interaction between peroxide and Co–N4 defect shows that this defect does not promote complete ORR and a second site for the reduction of peroxide is required, supporting a 2 × 2e− dual site ORR mechanism independent of pH of the electrolyte. In contrast, the much stronger interaction between peroxide and Co–N2 defect supports a 2 × 2e− single site ORR mechanism in alkaline and acidic media.
Introduction
Energy production is expected to be one of the most significant challenges in the 21st century. This is mainly due to the decreasing natural energy resources, population growth and the goal to reduction of carbon emissions. Fuel cell technology may provide at least a partial solution to the increasing societal demand of non-stationary energy. Currently, platinum group metals (PGMs) are the best performing cathode catalysts for oxygen reduction reaction (ORR) in fuel cells.1,2 However, PGMs are rare and comparatively expensive. Thus, it is important to develop more cost-effective non-PGM based fuel cell catalysts that reduce or eliminate PGMs from the catalyst design.Nitrogen containing carbon nanostructures and carbon supported self-assembled materials with and without transition metal have attracted significant interest as non-PGM ORR electrocatalysts.3–28 Recent experimental studies have shown excellent ORR activity of TM–Nx/C (TM = Fe, Co) electrocatalysts.29–31 However the chemical nature of the active site remains unclear. Several previous studies have attributed the ORR activity of nitrogen containing carbon supported catalysts to the nitrogen moieties in the sample.11,15–17,19,20 However, often transition metals (TMs) such as Fe, Co and Ni are present during catalyst synthesis22–27 and it is difficult to ensure that the TM's have been completely removed during acid washing from these catalysts. Other catalyst synthesis methods incorporate TM directly in the catalyst design, such as pyrolysis of CoTMPP/CoPPy.21,32,33 Available XPS studies of these catalysts support the presence of various nitrogen (N) moieties (pyridinic, pyrrolic and graphitic), transition metal–nitrogen (TM–Nx, x = 2, 4) centers and transition metal oxides, TMxOy.32,33 Experimental studies on Co-based pyrolyzed porphyrins ORR electrocatalysts in acidic medium show that Co–N4 centers promote a complete 4e− reduction of O2 to H2O and Co–N2 centers are responsible for the formation of H2O2 intermediate.21 Later RRDE measurements on pyrolyzed Co-polypyrrole and Co-porphyrins derived electrocatalysts show that Co–N4 centers are responsible for the reduction of O2 to peroxide and peroxide is further reduced on CoxOy/Co particles in both acidic and alkaline media supporting a dual site 2 × 2e− ORR mechanism.32,33 However, the location of the Co–N2 type site is ambiguous since it can be either graphitic or located at the edges of the carbon support. Previous computations have found weak binding of Co to graphite edge N atoms (which form a Co–N2 edge defect) suggesting that Co will easily dissolve from the support.34 This indicates that in the presence of Co the ORR activity is likely dominated by graphitic Co–Nx defects. A recent combined experimental and computational study on molecular Co-phthalocyanine carbon supported electrocatalysts (Co–N4 active site) in alkaline medium has reported that peroxide is the main ORR product.6 But, it remains unclear if these conclusions can be transferred directly to pyrolyzed Co–Nx/C electrocatalysts where the active sites are embedded in a carbon matrix.
Several previous computational studies investigated adsorption energies of O2, peroxide, O, OH and OOH to proposed TM–Nx sites to explain the ORR activity and the mechanism.3,6,10,18 However, binding energies alone do not provide a rationale for the experimental observation that the catalyst performs differently in acidic and alkaline electrolytes.35 Furthermore it remains unclear what the elementary rate limiting ORR reaction steps are and how they depend on pH and external potential.
In the present study, first-principles density functional theory (DFT) calculations are performed to study the chemistry of ORR active sites and the ORR mechanism of Co–Nx/C (x = 2, 4) electrocatalysts in alkaline and acidic media in the absence and presence of an external potential. The ORR activity and the mechanism of Co–Nx/C electrocatalysts are explored through the interaction of Co–Nx (x = 2, 4) sites embedded in a carbon matrix with O2 and H2O2.
Computational methods
The spin-polarized density functional theory (DFT)36,37 computations were performed using the plane wave basis set Vienna ab initio Simulation Package (VASP) code.38,39 The generalized gradient approximation (GGA) as parametrized by Perdew, Burke and Ernzerhof40 was used to describe the electronic exchange and correlation effects within the PAW formalism.41,42 The carbon support in the catalyst was modeled as a 4 × 4 graphene orthorhombic supercell with lattice parameters a = 9.842 Å, b = 8.524 Å (containing 32 atoms) and with periodic boundary conditions. Our computed in-plane C–C bond length of 1.42 Å in graphene sheet agrees well with previous computations.43,44 The artificial interactions between the sheet and its periodic images were minimized by separating slab and its images through a vacuum layer of 14 Å thickness. For geometry optimization, a plane wave energy cutoff of Ecut = 800 eV and a 4 × 4 × 1 Monkhorst–Pack grid45 were sufficient to obtain energy convergence better than 1 meV per atom. All atoms were allowed to relax during geometry optimization. Energies of atomic Co, and O and molecules N2, O2, OH, OOH and H2O2 were optimized in an orthorhombic 12 Å × 13 Å × 14 Å unit cell. The O–O bond lengths in our optimized O2 and H2O2 are 1.22 Å and 1.48 Å, respectively, which agree well with experimental O–O bond lengths of 1.21 Å and 1.48 Å in O2 and H2O2 molecules.46 Graphitic Co–N2 and Co–N4 defects were created by decorating graphene double vacancy (DV) with two and four nitrogen atoms, respectively, and then incorporating Co at the center of DV in a single step. The formation energy (ΔE) of Co–Nx defects was calculated by using the expression,| | | ΔE = Egraphene+(Co–Nx) + yμC − (Egraphene + xμN + ECo) | (1) |
Here, Egraphene+(Co–Nx) and Egraphene are the energies for optimized graphene with Co–Nx defects and pristine graphene sheet, respectively. μC is the chemical potential of carbon defined as the total energy per carbon atom for defect free graphene,47–49μN is the chemical potential of nitrogen defined as half of the total energy of an N2 molecule49–51 and ECo is the total energy of Co2+ calculated using the expression,where E(Co) is the total energy of an isolated Co atom in the gas phase and U is the external potential. x is the number of nitrogen atoms added and y is the number of carbon atoms removed from the graphene sheet during defect formation.Adsorbates were relaxed from different initial geometries in order to avoid trapping in a local minimum of the potential energy surface (PES). The binding energies that are reported below refer to the most stable binding sites. The binding/adsorption energy (BE) of molecules on defect motifs is calculated as:
| | | BE= Edefect+molecule − (Edefect + Emolecule) | (3) |
where
Edefect+molecule is the energy of the molecule adsorbed defective graphene configuration,
Edefect is the energy of defective graphene configuration and
Emolecule is the energy of an isolated molecule. The presence of a solvent (water) can affect the energetics of the ORR intermediates.
52 This is mainly due to the formation of hydrogen bonds between water in the liquid state with adsorbed intermediates. Thus, adsorbates tend to be stabilized in the presence of an electrolyte. In the present study, the binding energies of adsorbates are corrected by solvation energies taken from
ref. 52. Throughout this work we follow the convention that higher (more positive) formation energy (binding energy) means that the presence of a defect in the equilibrium (weaker interaction) is less likely.
The change in free energy (ΔG) of a reaction step is given by the expression in ref. 53 and 54,
| | | ΔG = ΔE + ΔZPE − TΔS + ΔGU + ΔGpH + ΔGfield | (4) |
Here, Δ
E is the reaction energy obtained from DFT calculations, ZPE is the zero point energy and
S is entropy. Δ
GU = −
eU where
U is potential at the electrode,
e is charge transferred and Δ
GpH =
kBT × ln 10 × pH where
kB is the Boltzmann constant and
T = 300 K. We assume pH = 0 for acidic medium and pH = 14 for alkaline medium, following previous work.
19,53 The free energy correction due to an electrochemical double layer was neglected as in previous study.
54 The free energy of O
2 was obtained from the reaction O
2 + 2H
2 → 2H
2O for which a free energy change is 4.92 eV.
19,54 The entropies and vibrational frequencies of molecules in gas phase were taken from NIST database. The free energy of H
2O was calculated in the gas phase at 298 K with a pressure of 0.035 bar, following
ref. 19 and 54. The vibrational frequencies of adsorbed species *O, *O
2, *OH and *OOH were taken from
ref. 3 and 54.
Results and discussion
The formation of graphitic Co–Nx defects (x = 2, 4 Fig. 1; formation energies −0.90 eV and −3.56 eV, respectively, at U = 0) in a carbon matrix is energetically favorable in agreement with the previous computations55,56 and XPS measurements.21,32,33 However, these formation energies are strictly only valid under open-circuit conditions (U = 0 V) and change in response to an applied potential (U). Following eqn (2) the formation energy remains only negative (favorable) in the relevant potential range (0 to 1.23 V) if the formation energy under open-circuit conditions is lower than −2.46 eV. Effectively, the reactant side is stabilized with the net effect that the formation energy becomes more positive (less favorable). Interestingly, the two defects show different behavior. The formation energy of Co–N4 remains negative in this potential range. In contrast, the formation energy of Co–N2 changes sign at U = 0.45 V, indicating that this defect is likely unstable at potentials that are relevant for fuel cells. Thus, graphitic Co–N4 defects are expected to dominate in catalysts that are synthesized at low pyrolysis temperatures and that the abundance of graphitic Co–N2 defects may increase with increasing pyrolysis temperature. This predicted temperature dependence of the defect chemistry is supported by XPS measurements on pyrolyzed Co–Nx/C electrocatalysts which show an increased concentration of Co–N2 defect motifs with increasing pyrolysis temperature.21 Previously, Co–N2 defects located on the edges of the carbon support have been predicted to be ORR active.34 However, the same study also showed that the Co binds to N2 edge sites with a BE ∼−1.5 eV and suggested that Co bound to N2 edge sites is not stable against dissolution. This view is supported by eqn (2) that shows this binding energy would become positive at U ∼ 0.75 V. In contrast, our computed Co BEs in graphitic Co–Nx defects (Co–N2: −7.92 eV and Co–N4: −7.54 eV) are significantly more favorable. In addition, we find that Co |BEs| are higher than the experimental Co bulk cohesive energy of 4.39 eV57 which further suggests that Co in Co–Nx defects is stable against clustering and maintains dispersed.55 Thus, graphitic Co–Nx defects and especially Co–N4 defect motifs may not only contribute to the overall ORR activity but also increase the durability and lifetime of the catalyst as compared to the edge defects.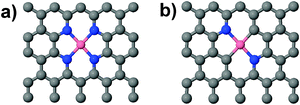 |
| | Fig. 1 (a) In-plane/graphitic Co–N4 and (b) Co–N2 defects embedded in graphene modeled as a carbon support for a Co based non-PGM catalyst. Grey: C, blue: N and pink: Co atoms. | |
The ORR activity of graphitic Co–Nx (x = 2, 4) defect motifs is explored through their interactions with O2 (Table 1). The chemisorption of O2 to Co–Nx indicates that both Co–N2 and Co–N4 defect motifs promote at least the reduction of O2 to peroxide. However, the O2/Co–Nx binding geometries are different: O2 binds in an end-on configuration on Co–N4 (Fig. 2a and b) and in a side-on configuration (parallel to the graphene sheet) on Co–N2 (Fig. 2c and d). The optimized geometry of O2 on the Co–N4 defect in the present study is similar to the previously reported geometry of O2 on Co-phthalocyanine molecular catalyst.6 Our computed O2/Co–N4 BE = −0.67 eV (Table 1) excluding solvation effects for comparison is slightly higher than the BE = −0.4 eV that was reported for Co-phthalocyanine.6 This indicates that TM–Nx catalytic centers in molecular fragments may show similar character as that of embedded TM–Nx defects in an extended carbon support at least for O2 adsorption. Furthermore, the binding geometry of O2 on the Co–N4 defect is similar to the binding geometry of O2 on Fe–N4 centers on carbon nanotubes10 which suggest that the effect of curvature may not be significant at least for the first step in ORR. The interaction of O2 with Co–N4 and Co–N2 results in a ∼7% and ∼9% increase of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond length, respectively (Table 1). The side-on adsorption of O2 on the Co–N2 defect allows for a larger overlap of Co-3d orbitals with O2-2p orbitals as compared to the end-on adsorption, where interactions are mediated by the doubly oxygen lone electron pair. Thus more electrons are transferred to the antibonding 2π* orbital of O2 in side-on adsorption which weakens the OO bond to the extent resulting in a ∼2% larger OO bond length. Furthermore, the geometry of O2 binding on catalytic sites may select the reaction pathway.58 The end-on adsorption geometry of O2 which corresponds to a weaker binding and a smaller OO bond elongation facilitates 2e− transfer to form the peroxide intermediate. On the other hand, the breaking of the OO bond is easier for a side-on binding configuration and ORR is likely a net 4e− reaction.
O bond length, respectively (Table 1). The side-on adsorption of O2 on the Co–N2 defect allows for a larger overlap of Co-3d orbitals with O2-2p orbitals as compared to the end-on adsorption, where interactions are mediated by the doubly oxygen lone electron pair. Thus more electrons are transferred to the antibonding 2π* orbital of O2 in side-on adsorption which weakens the OO bond to the extent resulting in a ∼2% larger OO bond length. Furthermore, the geometry of O2 binding on catalytic sites may select the reaction pathway.58 The end-on adsorption geometry of O2 which corresponds to a weaker binding and a smaller OO bond elongation facilitates 2e− transfer to form the peroxide intermediate. On the other hand, the breaking of the OO bond is easier for a side-on binding configuration and ORR is likely a net 4e− reaction.
Table 1 Binding energies (BEs in eV) of molecules without and with water solvation energies, the shortest distance between Co in Co–Nx defects and O of adsorbates (dCo–O in Å), and the optimized O–O distance (dO–O in Å) of adsorbates
| Defect | Molecules | BE (without solvation) | Solvation energya | BE (with solvation) | dCo–O | dO–O |
|---|
| Ref. 52. |
|---|
| Co–N4 | O | −3.18 | −0.70 | −3.88 | 1.71 | — |
| O2 | −0.67 | −0.41 | −1.08 | 1.93 | 1.30 |
| OH | −2.44 | −0.38 | −2.82 | 1.88 | — |
| OOH | −1.02 | −0.47 | −1.49 | 1.87 | 1.47 |
| H2O2 | −0.06 | — | — | 3.36 | 1.48 |
| Co–N2 | O | −4.15 | −0.70 | −4.85 | 1.65 | — |
| O2 | −1.16 | −0.41 | −1.57 | 1.92 | 1.33 |
| OH | −2.71 | −0.38 | −3.09 | 1.83 | — |
| OOH | −1.75 | −0.47 | −2.22 | 1.77 | 1.47 |
| H2O2 | −1.90 | — | — | 1.93 | 2.51 |
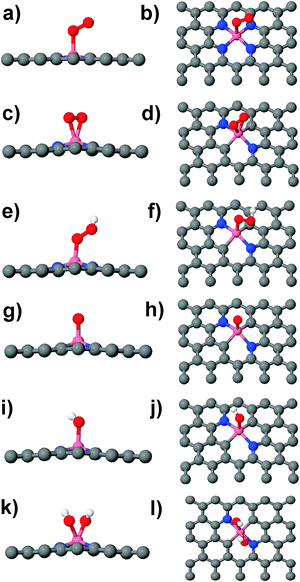 |
| | Fig. 2 O2 adsorption on Co–N4: (a) side view, (b) top view; O2 adsorption on Co–N2: (c) side view, (d) top view; OOH adsorption on Co–N2: (e) side view, (f) top view; O adsorption on Co–N2: (g) side view, (h) top view, OH adsorption on Co–N2: (i) side view, (j) top view and H2O2 decomposition on Co–N2: (k) side view and (l) top view. Grey: C, blue: N, pink: Co, red: O and white: H atoms. | |
The complete 4e− ORR in alkaline medium proceeds by the reactions:19
| | | O2 + 2H2O + 4e− → *O2 + 2H2O + 4e− | (5) |
| | | *O2 + 2H2O + 4e− → *OOH + OH− + H2O + 3e− | (6) |
| | | *OOH + OH− + H2O + 3e− → *O + 2(OH−) + H2O + 2e− | (7) |
| | | *O + 2(OH−) + H2O + 2e− → *OH + 3(OH−) + e− | (8) |
| | | *OH + 3(OH−) + e− → 4(OH−) | (9) |
Alternatively, a peroxide intermediate (OOH
−) can form instead of the product in
reaction (7) and desorbs from the catalytic site as a stable intermediate.
19Similarly, the complete 4e− ORR in acidic medium occurs via the reactions:54
| | | O2 + 4(H+ + e−) → *O2 + 4(H+ + e−) | (10) |
| | | *O2 + 4(H+ + e−) → *OOH + 3(H+ + e−) | (11) |
| | | *OOH + 3(H+ + e−) → *O + H2O + 2(H+ + e−) | (12) |
| | | *O + H2O + 2(H+ + e−) → *OH + H2O + (H+ + e−) | (13) |
| | | *OH + H2O + (H+ + e−) → 2H2O | (14) |
Alternatively, H
2O
2 can form instead of the product in
eqn (12) and desorb from the catalytic site as a stable intermediate similar to that in alkaline medium. Here (*) represents adsorbed species on graphitic Co–N
x defects.
The interaction of peroxide with Co–Nx defects determines the ORR pathway in this class of electrocatalyst. More precisely, the breaking of the O–O bond in the peroxide accounts for the complete ORR with a net 4e− transfer.6,20 Conversely, the lack of O–O bond breaking suggests that peroxide is a stable intermediate. In alkaline medium, the desorption of *OOH + e− as OOH− results in a stable peroxide intermediate.19 Our computation shows that OOH binds to Co–N4 defects (Table 1). However, the interaction of OOH−, a closed shell molecule is expected to be weaker as compared to the open-shell OOH molecule. Thus OOH− will desorb more easily from a Co–N4 catalytic site and a second site is needed for its further reduction. This suggests that Co–N4 mediated ORR in alkaline medium is a 2 × 2e− dual site mechanism consistent with experimental observation.32 Similarly, the weak interaction of H2O2 with Co–N4 (Table 1) shows that this defect only promotes the first step of ORR. In detail, the O–O distance (dO–O = 1.48 Å) in adsorbed H2O2 remains unchanged as compared to the gas phase value. The computations were initiated with the molecular adsorbate H2O2 positioned such that the closest Co–O distance was 1.9 Å, close to the sum of the covalent radii of Co and O, 1.99 Å.59 During the relaxation this closest Co–O distance increases to 3.36 Å and the peroxide O–O distance approached our DFT optimized gas phase value of 1.48 Å, corroborating the weak interaction between peroxide and graphitic Co–N4 defects. Thus, in the presence of graphitic Co–N4 ORR in an acidic electrolyte is predicted to occur via a dual-site 2 × 2e− mechanism. In contrast, the computations show a much stronger interaction of peroxide with graphitic Co–N2 defect motifs. Here, H2O2 is predicted to form a Co(OH)2 complex (Fig. 2k and l) as supported by the large O–O distance (2.51 Å) after geometry optimization which is ∼70% larger than the corresponding distance in molecular H2O2. This Co(OH)2 complex reacts with 2(H+ + e−) to form 2H2O which restores the catalytic site and complete ORR is predicted to occur on a single site.
Therefore, our results show that a second catalytic site in acidic and alkaline media is needed for the reduction of peroxide in the presence of graphitic Co–N4 defects. This finding for extended surfaces is consistent with previous work on the molecular Co-phthalocyanine electrocatalyst that was deposited on glassy carbon and only promoted the reduction of O2 to peroxide.6 These results are also consistent with available RRDE measurements on Co–Nx/C electrocatalysts derived from pyrolyzed Co-porphyrins and Co-polypyrrole.32,33 Co–N2 defect motifs are predicted to promote a complete single site 2 × 2e− ORR without the formation of a peroxide intermediate. This observation is analogues to the experimental finding of ORR activity of Fe–N2 sites in Fe–Nx/C ORR electrocatalysts that promote complete ORR without peroxide formation.28
To explore the presence of likely rate limiting (uphill) processes in alkaline and acidic media and their dependence on an external potential (U) we computed free-energy diagrams for ORR, following ref. 54. The ORR free energy diagrams are computed for the Co–N4 defect with H2O2 as a stable end product and for Co–N2 defects which are predicted to promote complete ORR. Reactions (5–9) are used to compute the free energy diagram in alkaline medium while reactions (10–14) are used to compute the corresponding diagram in acidic medium.
The computed free energy diagram (Fig. 3) for the Co–N4 defect in acidic medium shows that OOH formation remains uphill for all U > 0 V. But for U > 0.4 V, reduction of OOH to H2O2 is also uphill which indicates that the first step in ORR on the Co–N4 defect is facilitated at low U. In contrast, the free energy diagram for Co–N2 catalytic site for complete O2 reduction in alkaline medium consists of only downhill processes at high potential (Fig. 4). In alkaline medium *OOH formation (reaction 6) and OH removal (reaction 9) are uphill processes for U up to 0.4 V while *OOH formation is the only uphill process for 0.4 V < U < 0.5 V. At a higher potential, U ≥ 0.5 V, all elementary reaction steps of ORR are predicted to be downhill. In contrast, an increasing number of uphill processes are found with increasing potential in acidic medium (Fig. 5): all processes are downhill for potentials up to ∼0.3 V. For U > 0.4 V, formation of *OOH (reaction 11) and the formation of the final H2O form *OH (reaction 14) are also uphill. In summary, the free-energy diagrams for graphitic Co–N2 defects show that O2 reduction at high potential is facilitated in alkaline medium similar to the previous observation in Ni–Nx/C electrocatalysts.60 These findings provide an explanation for the previously poorly understood observation that the ORR activity of Co–Nx/C electrocatalysts is higher in high pH electrolytes.35 Interestingly, this observation may explain an initially high ORR activity that degrades in proportion to the disappearance of the Co–N2 defects which we predict to be unstable at potentials higher than 0.45 V. Thus, ORR activity among the two considered graphitic defect types can be attributed to Co–N4 and the ORR occurs via a dual site mechanism consistent with experiment.32,33 However, the ORR performance of Co–Nx/C electrocatalyst is expected to be lower than that of Pt-based electrocatalysts due to the low density of catalytically active sites as compared to that on Pt and Pt-alloy catalysts. In addition, the ORR intermediates OH and OOH adsorb more strongly on Co–Nx defects than that on the Pt(111) surface.61 Thus, the reduction of these intermediates may not be as facile as that on Pt surfaces.
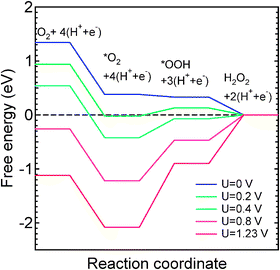 |
| | Fig. 3 Free-energy diagram for the reduction of O2 to H2O2 on the Co–N4 defect in acidic medium. | |
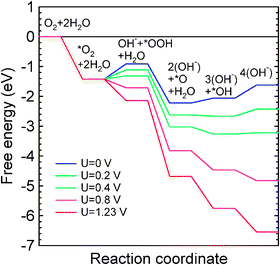 |
| | Fig. 4 Free-energy diagram for complete O2 reduction on the Co–N2 defect in alkaline medium. | |
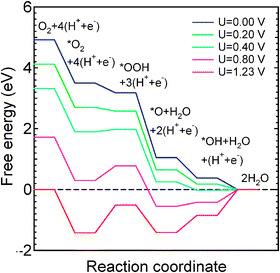 |
| | Fig. 5 Free-energy diagram for complete O2 reduction on the Co–N2 defect in acidic medium. | |
Conclusions
The computations predict that graphitic Co–N4 defect motifs are energetically favorable as compared to Co–N2 defect motifs. Moreover, graphitic Co–N4 defects are predicted to be stable for potentials in the range U = 0–1.23 V while the stability of Co–N2 defects is limited to potentials below 0.45 V. Thus, Co–N4 is expected to be the dominant catalytically active graphitic defect motif in Co–Nx/C electrocatalysts. O2 reacts with both types of graphitic Co–Nx (x = 2, 4) defects and the presence of either or both defects will enhance at least the reduction of O2 to peroxide. However, Co–N4 does not interact strongly with peroxide. Thus, a second site for the reduction of peroxide is required, supporting a 2 × 2e− dual site ORR pathway in both alkaline and acid solutions. In contrast, the much stronger interaction of Co–N2 with peroxide supports a 2 × 2e− single site ORR independent of pH value. However, the results show that even though this defect may be present after pyrolysis, it is not stable at high potentials. Thus, ORR activity is predicted to decrease with the disappearance of Co–N2 defects when the catalyst is exposed to potentials higher than 0.45 V. Subsequent catalytic activity of the Co–Nx/C electrocatalyst will be determined by the abundance of the stable Co–N4 defects. Thus, ORR on graphitic Co–Nx/C electrocatalysts is predicted to occur via a 2 × 2e− dual site mechanism.Acknowledgements
This work was supported in part by the DOE-EPSCoR Implementation Program: Materials for Energy Conversion. S. Kattel and B. Kiefer gratefully acknowledge computing resources provided by the New Mexico Computing Applications Center (NMCAC) and by the National Science Foundation through TeraGrid resources provided under grant number DMR TG-110093.References
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Norskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS.
- F. Calle-Vallejo, J. I. Martinez and J. Rossmeisl, Phys. Chem. Chem. Phys., 2011, 13, 15639–15643 RSC.
- F. Charreteur, F. Jaouen, S. Ruggeri and J. P. Dodelet, Electrochim. Acta, 2008, 53, 2925–2938 CrossRef CAS.
- F. Charreteur, S. Ruggeri, F. Jaouen and J. P. Dodelet, Electrochim. Acta, 2008, 53, 6881–6889 CrossRef CAS.
- R. R. Chen, H. X. Li, D. Chu and G. F. Wang, J. Phys. Chem. C, 2009, 113, 20689–20697 CAS.
- E. B. Easton, A. Bonakdarpour and J. R. Dahn, Electrochem. Solid-State Lett., 2006, 9, A463–A467 CrossRef CAS.
- E. B. Easton, R. Z. Yang, A. Bonakdarpour and J. R. Dahn, Electrochem. Solid-State Lett., 2007, 10, B6–B10 CrossRef CAS.
- T. Ikeda, M. Boero, S. F. Huang, K. Terakura, M. Oshima and J. Ozaki, J. Phys. Chem. C, 2008, 112, 14706–14709 CAS.
- D. H. Lee, W. J. Lee, W. J. Lee, S. O. Kim and Y. H. Kim, Phys. Rev. Lett., 2011, 106, 175502 CrossRef.
- K. R. Lee, K. U. Lee, J. W. Lee, B. T. Ahn and S. I. Woo, Electrochem. Commun., 2010, 12, 1052–1055 CrossRef CAS.
- M. Lefevre and J. P. Dodelet, Electrochim. Acta, 2003, 48, 2749–2760 CrossRef CAS.
- M. Lefevre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. B, 2000, 104, 11238–11247 CrossRef CAS.
- M. Lefevre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. B, 2002, 106, 8705–8713 CrossRef CAS.
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS.
- C. V. Rao, C. R. Cabrera and Y. Ishikawa, J. Phys. Chem. Lett., 2010, 1, 2622–2627 CrossRef CAS.
- R. A. Sidik, A. B. Anderson, N. P. Subramanian, S. P. Kumaraguru and B. N. Popov, J. Phys. Chem. B, 2006, 110, 1787–1793 CrossRef CAS.
- A. Titov, P. Zapol, P. Kral, D. J. Liu, H. Iddir, K. Baishya and L. A. Curtiss, J. Phys. Chem. C, 2009, 113, 21629–21634 CAS.
- L. Yu, X. L. Pan, X. M. Cao, P. Hu and X. H. Bao, J. Catal., 2011, 282, 183–190 CrossRef CAS.
- L. P. Zhang and Z. H. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CAS.
- J. M. Ziegelbauer, T. S. Olson, S. Pylypenko, F. Alamgir, C. Jaye, P. Atanassov and S. Mukerjee, J. Phys. Chem. C, 2008, 112, 8839–8849 CAS.
- P. H. Matter and U. S. Ozkan, Catal. Lett., 2006, 109, 115–123 CrossRef CAS.
- P. H. Matter, E. Wang, M. Arias, E. J. Biddinger and U. S. Ozkan, J. Phys. Chem. B, 2006, 110, 18374–18384 CrossRef CAS.
- P. H. Matter, E. Wang, M. Arias, E. J. Biddinger and U. S. Ozkan, J. Mol. Catal. A: Chem., 2007, 264, 73–81 CrossRef CAS.
- P. H. Matter, E. Wang, J. M. M. Millet and U. S. Ozkan, J. Phys. Chem. C, 2007, 111, 1444–1450 CAS.
- P. H. Matter, E. Wang and U. S. Ozkan, J. Catal., 2006, 243, 395–403 CrossRef CAS.
- P. H. Matter, L. Zhang and U. S. Ozkan, J. Catal., 2006, 239, 83–96 CrossRef CAS.
- C. Medard, M. Lefevre, J. P. Dodelet, F. Jaouen and G. Lindbergh, Electrochim. Acta, 2006, 51, 3202–3213 CrossRef CAS.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS.
- M. Lefevre, E. Proietti, F. Jaouen and J. P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS.
- T. S. Olson, S. Pylypenko, P. Atanassov, K. Asazawa, K. Yamada and H. Tanaka, J. Phys. Chem. C, 2010, 114, 5049–5059 CAS.
- T. S. Olson, S. Pylypenko, J. E. Fulghum and P. Atanassov, J. Electrochem. Soc., 2010, 157, B54–B63 CrossRef CAS.
- E. Vayner and A. B. Anderson, J. Phys. Chem. C, 2007, 111, 9330–9336 CAS.
- R. Z. Yang, K. Stevens and J. R. Dahn, J. Electrochem. Soc., 2008, 155, B79–B91 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev. Sect. B, 1964, 136, B864 Search PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133 CrossRef.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- D. E. Jiang, B. G. Sumpter and S. Dai, J. Chem. Phys., 2007, 126, 134701 CrossRef.
- C. Rajesh, C. Majumder, H. Mizuseki and Y. Kawazoe, J. Chem. Phys., 2009, 130, 124911 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- N. N. Greenwood and A. Earnshaw, Chemistry of the elements, Pergamon Press, Oxford [Oxfordshire], New York, 1st edn, 1984 Search PubMed.
- R. Faccio, L. Fernandez-Werner, H. Pardo, C. Goyenola, O. N. Ventura and A. W. Mombru, J. Phys. Chem. C, 2010, 114, 18961–18971 CAS.
- A. G. Garcia, S. E. Baltazar, A. H. R. Castro, J. F. P. Robles and A. Rubio, J. Comput. Theor. Nanosci., 2008, 5, 2221–2229 CrossRef CAS.
- Y. Shang, J. X. Zhao, H. Wu, Q. H. Cai, X. G. Wang and X. Z. Wang, Theor. Chem. Acc., 2010, 127, 727–733 CrossRef CAS.
- E. Finazzi, C. Di Valentin, A. Selloni and G. Pacchioni, J. Phys. Chem. C, 2007, 111, 9275–9282 CAS.
- K. S. Yang, Y. Dai, B. B. Huang and S. H. Han, J. Phys. Chem. B, 2006, 110, 24011–24014 CrossRef CAS.
- Y. Sha, T. H. Yu, Y. Liu, B. V. Merinov and W. A. Goddard, J. Phys. Chem. Lett., 2010, 1, 856–861 CrossRef CAS.
- S. Zuluaga and S. Stolbov, J. Chem. Phys., 2011, 135, 134702 CrossRef.
- J. K. Norskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef CAS.
- W. I. Choi, S. H. Jhi, K. Kim and Y. H. Kim, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085441 CrossRef.
- S. Kattel, P. Atanassov and B. Kiefer, J. Phys. Chem. C, 2012, 116, 8161–8166 CAS.
- P. H. T. Philipsen and E. J. Baerends, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 5326–5333 CrossRef CAS.
- Z. Shi, H. S. Liu, K. Lee, E. Dy, J. Chlistunoff, M. Blair, P. Zelenay, J. J. Zhang and Z. S. Liu, J. Phys. Chem. C, 2011, 115, 16672–16680 CAS.
- J. E. Huheey, Inorganic chemistry: principles of structure and reactivity, Harper & Row, New York, SI units edn, 1975 Search PubMed.
- S. Kattel, P. Atanassov and B. Kiefer, J. Phys. Chem. C, 2012, 116, 17378–17383 CAS.
- Z. Y. Duan and G. F. Wang, Phys. Chem. Chem. Phys., 2011, 13, 20178–20187 RSC.
|
| This journal is © the Owner Societies 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.