 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly diastereoselective radical cyclisations of chiral sulfinimines†
Elise M. Rochettea,
William Lewisa,
Al G. Dossetterb and
Robert A. Stockman*a
aSchool of Chemistry, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: Robert.stockman@nottingham.ac.uk
bAstraZeneca, Alderley Park, Macclesfield, SK10 4TG, UK
First published on 22nd August 2013
Abstract
Chiral amines are formed by the highly diastereoselective intramolecular addition of alkyl and aryl radicals onto chiral mesityl sulfinimines.
Chiral amines are an important class of compounds, and find uses in the fields of drug discovery, materials synthesis and natural product synthesis. Of the many methods for the synthesis of chiral amines,1 the addition of nucleophiles to chiral sulfinimines is often the most reliable, simple and general method.2 Whilst there have been a large number of reports on the addition of anionic nucleophiles to both the Ellman tert-butanesulfinimines and Davis p-toluenesulfinimines,2 relatively little work has been disclosed on the use of radical addition to these motifs. Following on from a few reports throughout the past decade on their SmI2-mediated radical dimerisation or coupling with carbonyls3 and addition of cyclic ether-stabilised radicals to sulfinimines,4 recently García Ruano reported the first general study on intermolecular radical addition to Ellman-type sulfiminines.5 Their study looked at addition of alkyl radicals to (mainly) aryl tert-butanesulfinyl aldimines activated by complexation with the strong Lewis-acid boron trifluoride. They showed that radical addition gave the same type of 1,2 addition products as Grignard addition, with yields in the range of 32–98% and notably high diastereoselectivity (90 → 96% de), although a large excess (typically 10 eq.) of alkyl iodide radical precursor was used. Prompted by this disclosure, we herein detail our preliminary investigations into the intramolecular radical reactions of chiral mesitylsulfinimines.
Our work in this area was prompted by our on-going interest in the chemistry of sulfinimines,6 and by pioneering reports by Malacria7 and Clive8 on radical additions to imine derivatives. Clive found O-trityl oximes to be good substrates for radical cyclisation, whilst Malacria described a single example of radical addition to Davis-type p-toluenesulfinimines, which gave moderate diastereoselectivity in the presence of the Lewis Acid MAD (Scheme 1).
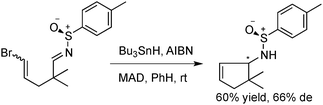 | ||
| Scheme 1 Malacria's report of radical cyclisation. Absolute configuration of diastereomers was not reported. | ||
We initially decided to investigate the intramolecular radical addition of an Ellman-type tert-butylsulfinimine, as our previous work on aziridine synthesis with carbenoid-type reagents9 had shown that these larger tert-butanesulfiminines gave higher stereoselectivity than the tolyl variants (Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Initiator | Propagator | Temp. (°C) | Yield 2 (%) | dr |
| All reactions carried out in benzene for 2 hours. | ||||||
| 1 | 1a | AIBN | Bu3SnH | 80 | — | — |
| 2 | 1a | AIBN | (Me3Si)3SiH | 80 | — | — |
| 3 | 1b | AIBN | Bu3SnH | 80 | — | — |
| 4 | 1c | AIBN | Bu3SnH | 80 | 50 | >98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 :2 |
| 5 | 1c | AIBN | (Me3Si)3SiH | 80 | — | — |
| 6 | 1c | Et3B/O2 | Bu3SnH | 40 | 61 | >98:2 |
| 7 | 1c | AIBN | 3 | 80 | 22 | >98:2 |
| 8 | 1c | Et3B/O2 | 3 | 40 | 33 | >98:2 |
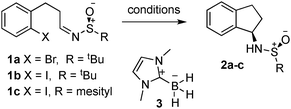
Thus we initially investigated the intramolecular cyclisation of compound 1a, which contained a bromide radical precursor, tethered to a tert-butanesulfinimine. After initial failure using AIBN with tributyltin hydride10 or tris(trimethylsilyl)silane, we then looked at using the aryl iodide radical precursor 1b. However, again we were not able to promote the radical cyclisation, although deiodination of the aryl iodide was observed. We thus surmised that a more electron rich sulfinimine might be more reactive towards radical addition: both Malacria and Clive's precedents used such electron rich imine derivatives. We therefore turned to mesitylsilfinimines. Initially introduced by Davis,11 we have found that they offer selectivity near that of the hindered tert-butanesulfinimines, but offer complementary reactivity in Grignard additions, giving high diastereoselectivities in an open-transition state addition.12 Furthermore, the mesityl group can be more easily removed, which can be useful for particularly sensitive substrates such as N-sulfinyl aziridines.10,11 We recently reported a convenient one-pot procedure for the synthesis of mesitylsulfinimines in >99% ee using a phenyl alanine-based template,13 and thus they are also readily available substrates. It is interesting to note that this is different to the course taken by García and co-workers, who also faced the issue of low reactivity.5 Working independently to us, they instead utilised activation of the tert-butanesulfinimines with boron trifluoride in order to increase their reactivity towards radical addition, as well as using an excess of radical precursor, in their intermolecular radical addition protocol. The stereoselctivity of our radical cyclisation follows the same sense of induction as García noted, as confirmed by the X-ray crystal structure of adduct 2c (Fig. 1).
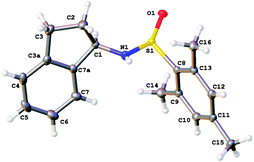 | ||
| Fig. 1 X-ray crystal structure14 of 2c. | ||
With mesitylsulfinimine 1c in hand, we then explored a range of radical initiators and propagators (Table 1). We were very happy to find that the mesitylsulfinimines were able to undergo radical cyclisation under a range of conditions, in each case giving the product 2c with complete diastereoselectivity, despite the elevated temperatures of these reactions. We were also pleased to find that tin-free conditions could be used, using the Malacria–Curran15 N-heterocyclic carbene borane complex 3 as a hydrogen atom donor with either triethyl borane/oxygen or AIBN as initiator, although attempts to use the more common and commercially available tris(trimethylsilyl)silane propagator were not fruitful.
Having found success with the mesitylsulfinyl group, we then turned our attention to investigating chain length, inclusion of heteroatoms in the chain, and the use of more substituted aryl groups, heteroaryl groups, and all-alkyl systems.
Initially, we looked at inclusion of an oxygen in the tether, and the length of the tether (Scheme 2). It was found that phenol-derivative 4a cyclised in similar yield to the all-carbon analogue explored previously. Increasing the chain length to form a six-membered ring (5b) also worked, albeit in slightly reduced yield. The six-ring 5b was found to be crystalline, and thus we were able to confirm the sense of induction in the cyclisation is consistent in the formation of the five-membered ring 2c and the six-membered ring 5b system (Fig. 2). We found the mesityl sulfinimine group of 5a was easily removed in near-quantitative yield by treatment with 4 M HCl (Scheme 3).
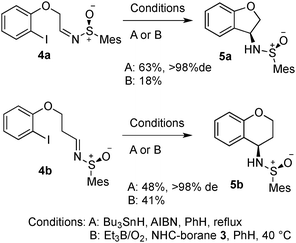 | ||
| Scheme 2 Investigation of tether length and heteroatom inclusion. | ||
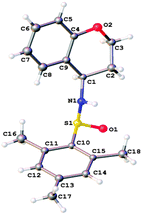 | ||
| Fig. 2 X-ray crystal structure14 of 5b. | ||
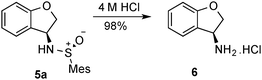 | ||
| Scheme 3 Removal of mesitylsulfinyl group. | ||
We then looked at aliphatic precursors. The simple N-tosyl tethered system 7a was prepared, but upon reaction it was not found to undergo radical cyclisation (Scheme 4). Rather, elimination of the terminal iodide to form alkene 8a was observed. Cyclohexane derivative 7b, which is not able to undergo an elimination, was successful in the cyclisation reaction, giving spirocycle 8b in a good 73% yield as a single diasteromer (X-ray structure, Fig. 3).
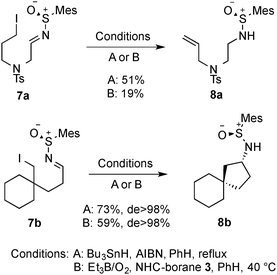 | ||
| Scheme 4 Investigation of aliphatic radical precursors. | ||
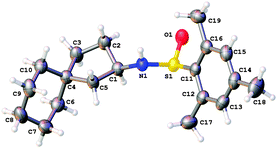 | ||
| Fig. 3 X-ray crystal structure14 of 8b. Structure shown is one of three independent molecules of compound 8b in the asymmetric unit. | ||
Finally we investigated two electron-rich aryl bromides as radical cyclisation precursors (9a and 9b, Scheme 5). In each case, we found that the reaction was able to proceed, albeit with modest yield. No other side-products or starting material was isolated from these reactions. Both products were formed as single diastereoisomers. In the case of thiophene adduct 10b, the absolute configuration was confirmed by X-ray crystallography (Fig. 4), and we assumed the absolute stereochemistry of 10a would be that shown by analogy with 10b.
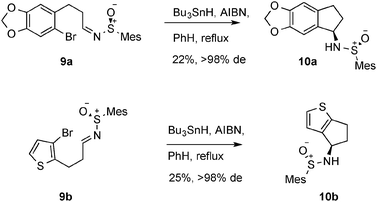 | ||
| Scheme 5 Cyclisation of electron-rich aryl bromides. | ||
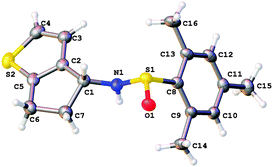 | ||
| Fig. 4 X-ray crystal structure14 of 10b. | ||
In conclusion, we have found that S-mesitylsulfinimines are able radical acceptors, and undergo radical cyclisations with exquisite control of diastereoselectivity. The product chiral sulfinamines are easily deprotected to give chiral amines which have fused aryl/aliphatic and spiro aliphatic ring systems, making them interesting as chiral building blocks for screening collection enhancement.16 Further studies are underway in our laboratories to fully explore the scope and limitations of this unusually diastereoselective radical cyclisation methodology, which will be reported in due course.
AstraZeneca (ER) and EPSRC (RAS, EP/E055346) are acknowledged for funding.
Notes and references
- Chiral Amine Synthesis: Developments and Applications, ed. T. C. Nugent, Wiley-VCH, Chichester, UK, 2010 Search PubMed.
- For a recent comprehensive review on sulfinimines in synthesis, see: M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600 CrossRef CAS PubMed.
- B. Wang and Y. Wang, Org. Lett., 2009, 11, 3410 CrossRef CAS PubMed; B. Wang and R.-H. Liu, Eur. J. Org. Chem., 2009, 2845 CrossRef; R. Wang, K. Fang, B.-F. Sun, M.-H. Xu and G.-Q. Lin, Synlett, 2009, 2301 Search PubMed; R.-C. Liu, K. Fang, B. Wang, B. F. Sun, M.-H. Xu and G.-Q. Lin, J. Org. Chem., 2008, 73, 3307 CrossRef PubMed; R.-C. Liu, J.-H. Wei, B.-G. Wei and G.-Q. Lin, Tetrahedron: Asymmetry, 2008, 19, 2731 CrossRef PubMed; Y.-W. Zhong, Y.-Z. Dong, K. Fang, K. Izumi, M.-H. Xu and G.-Q. Lin, J. Am. Chem. Soc., 2005, 127, 11956 CrossRef PubMed.
- T. Akindele, K. Yamada, T. Seijima, M. Maekawa, Y. Yamamoto, M. Nakano and K. Tomoika, Chem. Pharm. Bull., 2010, 58, 265 CrossRef CAS; T. Akindele, Y. Yamamoto, M. Maekawa, H. Umeki, K. Yamada and K. Tomoika, Org. Lett., 2006, 8, 5729 CrossRef PubMed.
- J. A. Fernández-Salas, M. C. Maestro, M. M. Rodríguez-Fernández, J. L. García-Ruano and I. Alonso, Org. Lett., 2013, 15, 1658 CrossRef PubMed.
- For our most recent work in the area of sulfinimine reactivity see: G. Procopiou, W. Lewis, G. Harbottle and R. A. Stockman, Org. Lett., 2013, 15, 2030 CrossRef CAS PubMed.
- E. Lacôte and M. Malacria, C. R. Acad. Sci., 1998, 191 Search PubMed.
- D. L. J. Clive, M. P. Pham and R. Subedi, J. Am. Chem. Soc., 2007, 129, 2713 CrossRef CAS PubMed.
- T. Moragas-Sola, I. Churcher and R. A. Stockman, Org. Biomol. Chem., 2011, 9, 5034 Search PubMed.
- For an excellent work-up procedure for tributyl tin hydride reactions, see: D. C. Harrowven, D. P. Curran, S. L. Kostiuk, I. L. Wallis-Guy, S. Whiting, K. J. Stenning, B. Tang, E. Packard and L. Nansona, Chem. Commun., 2010, 46, 6335 RSC.
- F. A. Davis, T. Ramachandar and Y. Wu, J. Org. Chem., 2003, 68, 6894 CrossRef CAS PubMed.
- C. Roe, T. Moragas-Sola, L. Sasraku-Neequye, H. Hobbs, I. Churcher, D. MacPherson and R. A. Stockman, Chem. Commun., 2011, 4791 Search PubMed.
- C. Roe, H. Hobbs and R. A. Stockman, Chem.–Eur. J., 2011, 17, 2704 CrossRef CAS PubMed; L. Sasraku-Neequaye, D. MacPherson and R. A. Stockman, Tetrahedron Lett., 2008, 49, 1129 CrossRef PubMed.
- Ellipsoids shown at 50% probability. Crystal-data notes can be found in the ESI†.
- A. Solovyev, S.-H. Ueng, J. Monot, L. Fensterbank, M. Malacria, E. lacôte and D. P. Curran, Org. Lett., 2010, 12, 2998 CrossRef CAS PubMed; S.-H. Ueng, L. Fensterbank, M. Malacria, E. lacôte and D. P. Curran, Org. Lett., 2010, 12, 3002 CrossRef PubMed.
- For interesting discussions on “Lead-Oriented-Synthesis” and three-dimensionality in screening compounds and drug discovery see: F. Lovering, MedChemComm, 2013, 4, 515 RSC; P. MacLellan and A. Nelson, Chem. Commun., 2013, 49, 2383 RSC; A. Nadin, C. Hattotuwagama and I. Churcher, Angew. Chem., Int. Ed., 2012, 51, 1114 CrossRef CAS PubMed; W. P. Walters, J. Green, J. R. Weiss and M. A. Murcko, J. Med. Chem., 2011, 54, 6405 CrossRef PubMed; F. Lovering, J. Bikker and C. Humbler, J. Med. Chem., 2009, 52, 6752 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details. CCDC 952387–952390. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc45452e |
| This journal is © The Royal Society of Chemistry 2013 |