Novel drug carriers: from grafted polymers to cross-linked vesicles†
Jiangtao
Xu
a,
Qiang
Fu
a,
Jing M.
Ren
a,
Gary
Bryant
b and
Greg G.
Qiao
*a
aDepartment of Chemical and Biomolecular Engineering, The University of Melbourne, Parkville, VIC 3010, Australia. E-mail: gregghq@unimelb.edu.au; Fax: +61 8344 4153; Tel: +61 8344 8665
bSchool of Applied Sciences, RMIT University, Melbourne, VIC 3000, Australia
First published on 23rd October 2012
Abstract
A simple and straightforward method of self-assembling grafted copolymers was developed to fabricate cross-linked polymer vesicles, which could conjugate anticancer drug cis-platinum and possess the capability of a high drug loading content, and a steady release rate.
Synthetic polymer nanoparticles have long been sought as nanocarriers for drug delivery because they are amenable to precise morphological control and surface modification.1 Polymer vesicles are particularly attractive materials since their sizes, structures and functions can be readily tailored to suit many desirable applications in nanomedicine through careful selection of the polymer structures and properties.2
Of particular interest is the preparation of polymer vesicles in aqueous solution from different topological copolymers. The intrinsic macromolecular parameters including block copolymer architecture, hydrophobic interaction, and hydrophobic/hydrophilic balance, determine the nature of the morphologies of these block copolymers. Among them, block copolymer architecture has been extensively investigated for simple linear–linear (coil–coil, coil–rod, rod–rod, and supplementary interaction),3 and linear-branched (or dendrimer) structures.4
Herein, we report on the construction of robust polymer vesicles self-assembled from linear-brush polyoxanorbornene-based diblock copolymers by pH triggering. The double bonds formed in the polymer backbone provide the capability of cross-linking vesicular structure by thiol–ene “click” chemistry with a biodegradable cross-linker. These cross-linked polymer vesicles can be utilized to conjugate anticancer drug cis-platinum with high loading content. To the best of our knowledge, this is the first example of a morphological investigation of grafted polynorbornene copolymers self-assembled in aqueous condition to form vesicles.
We designed linear-brush diblock copolymer 3 (Scheme 1) via ring-opening metathesis polymerization (ROMP) utilizing two monomers, oxanorbornenyl anhydride (ONBAn) and ω-oxanorbornenyl poly(ethylene glycol) (Mn = 2000) macromonomer (ONB-PEG2K, 2, ESI†). The anhydrides in the linear block would be modified to di-acid functionalities through hydrolysis, which would be responsive towards pH in aqueous solution and also provide binding sites for cis-platinum drug conjugation. The oxygen in the oxanorbornene monomer was expected to be more hydrophilic and hence increases the probability of biocompatibility for the resulting copolymers.5 PEG was employed to construct the brush block due to its prestige as the most important and widely used biocompatible polymer in pharmaceutical and biomedical applications. Brush and hyperbranched polymers generally display longer in vivo retention times compared to their linear polymer analogues.6
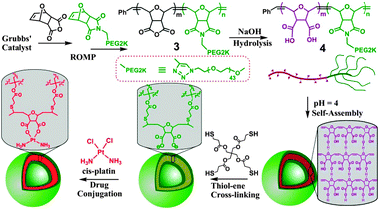 | ||
| Scheme 1 The chemistry of constructing cross-linked polymer vesicles and their drug conjugation with cis-platinum. | ||
ROMP initiated by ruthenium Grubbs' catalyst is a powerful tool to make well-defined polymers with low polydispersity, high conversion, and precisely controlled degrees of polymerization by easily adjusting the monomer to catalyst ratio. Particularly, it has been proved to be very efficient in making brush polymers.7 The linear-brush block copolymers, P(ONBAn)m-b-P(ONB-PEG2K)n, with two different block ratios were synthesized via sequential addition of (macro)monomers of ONBAn and ONB-PEG2K in the presence of pyridine modified 2nd generation Grubbs' catalyst. It gave polymer 3 (Mn (GPC) = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500, Mw/Mn = 1.14 for P(ONBAn)25-b-P(ONB-PEG2K)5 and Mn (GPC) = 19400, Mw/Mn = 1.16 for P(ONBAn)50-b-P(ONB-PEG2K)5).
500, Mw/Mn = 1.14 for P(ONBAn)25-b-P(ONB-PEG2K)5 and Mn (GPC) = 19400, Mw/Mn = 1.16 for P(ONBAn)50-b-P(ONB-PEG2K)5).
Hydrolysis was carried out in 0.1 M NaOH solution for 1 h. After dialysis against deionized water, the polymer solutions were freeze-dried to give viscous diblock copolymer 4 (with P(ONB-diacid)25-b-P(ONB-PEG2K)5 denoted as DP25 and P(ONB-diacid)50-b-P(ONB-PEG2K)5 denoted as DP50).
The self-assembly proceeded via dissolution of the diblock copolymer 4 at a concentration of 3 mg mL−1. The pH value of the solution was adjusted to ∼12 (measured by pH meter) by adding 2 M NaOH solution. Subsequently, 2 M HCl was added slowly into the solution to tune down the pH values. Dynamic light scattering (DLS) studies of these solutions at different pH values (Fig. 1A and Fig. S5, ESI†) for DP25 were performed. Fig. 1A showed that Dh did not change as the pH decreased from 12 to 5. It was around 5 nm, which means the polymers were dissolved molecularly. However, the size abruptly increased from 5 nm to 150 nm as pH decreased from 5 to 4, which indicated the formation of micellar aggregates.
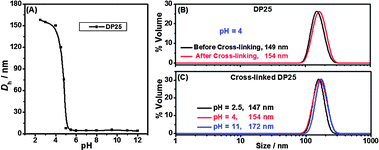 | ||
| Fig. 1 Hydrodynamic particle sizes (Dh) of self-assembled P(ONB-diacid)25-b-P(ONB-PEG2K)5 (DP25) (A) at varied pH before cross-linking, (B) at pH 4 before (black line) and after (red line) cross-linking, and (C) at different pH after cross-linking. | ||
It is well known that poly(carboxylic acid) chains undergo dramatic conformational changes, from an extended rod-like chain to a hypercoil chain as the pH value decreases. Such a change occurs gradually over a large pH range. Thus, the sharp transition at pH 4–5 in Fig. 1 cannot only be attributed to the simple conformational variation of poly(carboxylic acid) chains. In fact, the acid groups in poly(carboxylic acid) acting as proton-donating groups can form inter-polymer complexes (IPC) by H-bonding with proton-accepting ether groups in polymers like PEG and poly(vinyl ether) at low pH. This phenomenon had been extensively studied since it was first reported in 1959.8 Therefore, the sharp transition at pH 4–5 in this work could be attributed to IPC between poly(carboxylic acid) and polyoxanorbornene ether backbone or PEG brush (Scheme S2, ESI†). However, accompanying the hydrophobic intermolecular association of poly(carboxylic acid)s, it was proposed that the complexation between poly(carboxylic acid) and polyoxanorbornene ether backbone would be more favorable than that between poly(carboxylic acid) and PEG brush.
Taking into account the semi-rigidity of the polyoxanorbornene backbones, the molecular packing and IPC will tend to form bilayer structure, i.e. vesicles, at pH 4. The particle size (Dh = 150 nm) was far higher than the maximum theoretical value for spherical micelles (∼74 nm for the fully extended chain length of the constituent blocks, estimated by summing the average backbone length unit of C–C–O at 0.23 nm). Furthermore, light scattering (LS), transmission electron microscopy (TEM) and atomic force microscopy (AFM) measurements provided convincing evidence that the structures are vesicles. Cross-linking of the vesicles can provide a permanent vesicle for morphology observation.
Due to the olefin functionalities on polyoxanorbornene backbone, which can be applied for the thiol–ene click reaction9 with high tolerance in many different conditions and solvents,10 these vesicles were cross-linked by a tetra-functional thiol cross-linker, pentaerythritol tetrakis(3-mercaptopropionate) (PETMP), in the presence of photo initiator, 2,2-dimethoxy-2-phenyl acetophenone (DMPA). It is worthy to note that β-thioester bonds in PETMP were used as a labile linker because it can be easily hydrolyzed by esterase, which is abundant in cells.11 Therefore, the resultant cross-linked polymer vesicles are likely to be biodegradable if used as drug carriers. Although the grafted polymer segments cannot degrade, they can be cleared from blood circulation since their molecular weights are far below renal clearance threshold (40 K for linear PEG).12
The cross-linker and photo initiator were encapsulated into the hydrophobic part of polyacid aggregates due to their hydrophobic properties. The thiol–ene “click” reaction was performed under 365 nm UV light overnight. DLS measurements (Fig. 1B) confirmed that the particle size did not change (∼150 nm) before and after cross-linking.
DLS studies (Fig. 1C) of the cross-linked particles after dialysis showed the size increasing from 147 nm at pH 2.5 to 172 nm at pH 11 due to increased swelling. A control experiment was performed under identical reaction condition without PETMP: changing the pH from 2.5 to 7, no large-sized nanoparticles were observed by DLS, indicating the disassembly of uncross-linked polymer particles. The other grafted copolymer DP50 gave similar results in DLS (Fig. S6, ESI†) with particle size of ∼190 nm at low pH and ∼210 nm at high pH after cross-linking.
The combination of DLS and SLS measurement can provide detailed information of vesicles, including vesicle size, shell thickness and size distribution.13 Theoretical analysis of SLS measurements of the cross-linked polymer vesicles was conducted using established mathematical models.14 For the cross-linked polymer vesicle from diblock copolymer P(ONB-diacid)25-b-P(ONB-PEG2K)5 (denoted as CPV25), the best fit was achieved using a hollow sphere (vesicle) model (eqn (S1), red line in Fig. S7, ESI†) with a log-normal distribution of particle sizes. This model is in reasonable agreement with the SLS data, yielding a diameter of 132 nm, which matches well with the DLS data. Moreover, the shell thickness was calculated to be 27 nm. By comparison, no good fit could be obtained using the solid sphere (micelle) model (eqn (S3), green line in Fig. S7, ESI†). The other sample of cross-linked polymer vesicle from P(ONB-diacid)50-b-P(ONB-PEG2K)5 (denoted as CPV50) was fitted (Fig. S8, ESI†) using the vesicle model with a Schulz distribution, yielding a diameter of 188 nm and shell thickness of 37 nm. Again no good fit could be obtained using a micelle model.
Analysis of a dried sample of the CPV25 by TEM negatively stained with uranyl acetate showed polydisperse spherical micelles with average ∼155 nm particle size (Fig. 2A and Fig. S10, ESI†), which agrees with LS results. The high polydispersity originates from the formation mechanism of polymer vesicles. Generally, they are formed from a block copolymer to an assembled single bilayer membrane, with a hydrophobic middle layer and hydrophilic surfaces, and the resulting membrane folds over and rearranges by connecting its edges to enclose a space. No mechanism selects for the overall size, so the size distribution tends to be very polydisperse. It also induced the morphologies of fully enclosed and partially enclosed vesicles, which can be seen in TEM and AFM measurements.
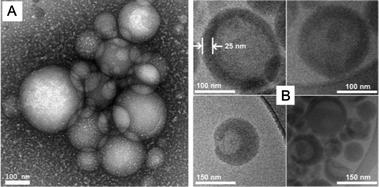 | ||
| Fig. 2 TEM images with negative staining (A) and cryo-TEM images with positive staining (B) for cross-linked polymer vesicle from P(ONB-diacid)25-b-P(ONB-PEG2K)5 (CPV25). | ||
Upon positive staining with loaded drug cis-platinum (selective staining for the di-acid block), definable bilayer structure with 25 nm shell thickness were observed from cryo-TEM (Fig. 2B and Fig. S11, ESI†), clearly indicating vesicular morphology. The shell thickness was a little smaller than that from LS results (27 nm) due to unstained thin PEG layers. Additionally, it indicated some bowl-shaped and partially enclosed vesicles (Fig. 2B bottom and Fig. S11C, ESI†).
AFM measurement (Fig. S12, ESI†) further confirmed the results by a diameter to height ratio of 7.5 to 1, a hollow property of the nanoparticles. The height showed doubled shell thickness of the polymer vesicles. With a single thickness of around 22 nm, which is slightly smaller than 25 nm from cryo-TEM measurements, as the AFM sample is in the dried state.
These novel cross-linked polymer vesicles are ideal nanocarriers for anti-cancer drug cis-platinum, because the diacid bilayer shell has the tendency to bind cis-platinum, forming metal complexes which can release drugs by hydrolytically cleaving carboxylic groups. Moreover, dicarboxylate–platinum complexes with a 6–8 membered ring structure can normally provide higher stability and loading contents to their conjugates.15 In this work, the di-acid structures have the potential to form stable platinum complex with 7-membered rings.
In a specific experiment, the basic solution (pH = 12) of CPV25 was mixed with freshly prepared cis-diaminediaqua platinum(II) complex solution from cis-dichlorodiamine–platinum(II) (CDDP) at given carboxylate/cis-platinum molar ratios (2/1), resulting in a light brown solution in an extremely short time (<5 min). It presented the rapid formation of polymer–platinum conjugates as well as high drug conjugation efficiency determined by TGA to be 88.4% (see Fig. S13 and its calculation in ESI†), which suggests that most carboxylate functional groups were involved in complex formation as 7-membered ring complexes. We believed that this stable complex promoted effective drug conjugation. Although the exact complex structure is not clear, it provides an extremely good drug loading and release profiles. The drug loading content was calculated to be 18.9% based on the conjugation efficiency, which is much higher than previously reported values (generally less than 10%16).
The in vitro release of the CDDP from a polymer vesicle solution was performed by a dialysis method.15 As shown in Fig. 3, the drug release profile followed a steady release rate, which meets the criterion that drug levels in the body remain constant while the drug is being administered. The release rate can be described by simple first-order kinetics. The release process may combine diffusion (out of polymer vesicles) and chemically controlled de-complexation processes.17 Similar results were given by Huynh et al.,15 with release constant (τ) at 47.84 hours.
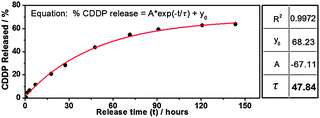 | ||
| Fig. 3 Release profile of CDDP from CPV25 in saline solution (0.9% NaCl, pH 7.4). The line was fitted according to equation % CDDP released = A exp(−t/τ) + y0. | ||
In summary, this work presents a straightforward method to prepare cross-linked polymer vesicles, which can conjugate cis-platinum drug at a high loading content and release it at a steady rate. The polymer vesicles were fabricated by self-assembly of linear-brush grafted copolymer forming inter-polymer complexes by H-bonding via pH control.
The authors wish to acknowledge the University of Melbourne's 2012 Early Career Researcher Grant (J.X.), Australian Research Council under the Future Fellowship (FT110100411, G.G.Q.), and valuable advice and suggestions from Dr Adrian Sulistio.
Notes and references
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2004, 55, 329 Search PubMed; M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771 CrossRef CAS.
- W. Meier, Chem. Soc. Rev., 2000, 29, 295 RSC; C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200 CrossRef CAS; D. M. Vriezema and A. M. Comellas, et al. , Chem. Rev., 2005, 105, 1445 CrossRef CAS; J. Du and R. K. O'Reilly, Soft Matter, 2009, 5, 3544 RSC.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967 CrossRef CAS; L. Zhang and A. Eisenberg, Science, 1995, 268, 1728 CrossRef CAS.
- J. del Barrio and L. Oriol, et al. , J. Am. Chem. Soc., 2010, 132, 3762 CrossRef CAS; W. Tao and Y. Liu, et al. , J. Am. Chem. Soc., 2012, 134, 762 CrossRef CAS; J. Xu, L. Tao, C. Boyer, A. B. Lowe and T. P. Davis, Macromolecules, 2011, 44, 299 CrossRef CAS.
- D. Le, V. Montembault, J.-C. Soutif, M. Rutnakornpituk and L. Fontaine, Macromolecule, 2010, 43, 5611 CrossRef CAS.
- M. E. Fox, F. C. Szoka and J. M. J. Frechet, Acc. Chem. Res., 2009, 42, 1141 CrossRef CAS; S. M. Grayson and W. T. Godbey, J. Drug Targeting, 2008, 16, 329 CrossRef CAS; M. Shokeen, E. D. Pressly and A. Hagooly, et al. , ACS Nano, 2011, 5, 738 CrossRef CAS.
- Y. Xia, B. Olsen, J. Kornfield and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 18525 CrossRef CAS; J. Johnson, Y. Lu, A. Burts, Y. Lim, M. Finn, J. Koberstein, N. Turro, D. Tirrell and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 559 CrossRef CAS.
- K. L. Smith, A. E. Winslow and D. E. Petersen, Ind. Eng. Chem., 1959, 51, 1361 CrossRef CAS; H. Dou and M. Jiang, et al. , Angew. Chem., Int. Ed., 2003, 42, 1516 CrossRef CAS.
- A. Wolfberger, B. Rupp, W. Kern, T. Griesser and C. Slugovc, Macromol. Rapid Commun., 2011, 32, 518 Search PubMed.
- L. A. Connal, C. R. Kinnane, A. N. Zelikin and F. Caruso, Chem. Mater., 2009, 21, 576 CrossRef CAS; M. Uygun, M. A. Tasdelen and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 103 CrossRef CAS; M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743 CrossRef CAS.
- Y. Shen and E. Jin, et al. , J. Am. Chem. Soc., 2010, 132, 4259 CrossRef CAS; H. Tang and C. J. Murphy, et al. , Nanomedicine, 2010, 5, 855 Search PubMed; E. Amir, P. Antoni and L. M. Campos, et al. , Chem. Commun., 2012, 48, 4833 RSC.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451 CrossRef CAS.
- J. H. van Zanten and H. G. Monbouquette, J. Colloid Interface Sci., 1994, 165, 512 CrossRef CAS; J. Pencer and F. R. Hallet, Langmuir, 2003, 19, 7488 CrossRef CAS.
- G. Bryant, S. Martin, A. Budi and W. van Megen, Langmuir, 2003, 19, 616 CrossRef CAS; H. J. Schöpe, O. Marnette, W. van Megen and G. Bryant, Langmuir, 2007, 23, 11534 CrossRef CAS; A. Sulistio, A. Blencowe, J. Wang, G. Bryant and G. G. Qiao, Macromol. Biosci., 2012, 12, 1220 Search PubMed.
- V. T. Huynh, G. Chen, P. de Souza and M. H. Stenzel, Biomacromolecules, 2011, 12, 1738 CrossRef; V. T. Huynh, J. Y. Quek, P. de Souza and M. H. Stenzel, Biomacromolecules, 2012, 13, 1010 CrossRef CAS.
- R. Tong and J. Cheng, Angew. Chem., Int. Ed., 2008, 47, 4830 CrossRef CAS.
- A. Patri, J. Kukowska-Latallo and J. Baker, Adv. Drug Delivery Rev., 2005, 57, 2203 CrossRef CAS; J. Siepmann and A. Gopferich, Adv. Drug Delivery Rev., 2001, 48, 229 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI): Experimental details and further characterization data are available. See DOI: 10.1039/c2cc37319j |
| This journal is © The Royal Society of Chemistry 2013 |