Molybdate templated assembly of Ln12Mo4-type clusters (Ln = Sm, Eu, Gd) containing a truncated tetrahedron core†
Yong
Zheng
a,
Qian-Chong
Zhang
a,
La-Sheng
Long
*a,
Rong-Bin
Huang
a,
Achim
Müller
b,
Jürgen
Schnack
c,
Lan-Sun
Zheng
a and
Zhiping
Zheng
a
aState Key Laboratory of Physical Chemistry of Solid Surface and Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: lslong@xmu.edu.cn; Fax: +86 592 218 3047
bFaculty of Chemistry, University of Bielefeld, P.O. Box 10 0131, D-33501 Bielefeld, Germany
cFaculty of Physics, University of Bielefeld, P.O. Box 10 0131, D-33501 Bielefeld, Germany
First published on 16th October 2012
Abstract
Three heterometallic cluster complexes {Ln12Mo4} featuring an Ln12 core of a distorted truncated tetrahedron were synthesized with the assistance of four MoO42− anions as ancillary ligands. Magnetic studies of the {Gd12Mo4} cluster revealed a large magnetocaloric effect due to the presence of the large number of weakly coupled Gd(III) ions.
Polynuclear lanthanide hydroxide complexes are of interdisciplinary interest.1–8 They are synthetically challenging as the oxophilicity of the lanthanide ions favors extensive interactions with hydroxo groups, leading frequently to intractable precipitates of lanthanide hydroxides. However, an increasing number of structurally pleasing polynuclear lanthanide hydroxide complexes with polyhedral cluster cores, though obtained serendipitously, have appeared in the literature and inspired the exploratory rational syntheses of these species.1–8 Such efforts have further been stimulated by their exciting applications as molecule-based magnetic materials,4 artificial nucleases for the hydrolytic cleavage of DNA and RNA,9 contrast agents for biomedical imaging,10 and fixation media for atmospheric CO2.7
The most successful synthetic approach to these compounds is the ligand-controlled hydrolysis of lanthanide ions in which preoccupation of the coordination sphere by certain ancillary ligands, such as α-amino acids5 and β-diketonates,11 limits the number of hydroxo ligands resulting from the deprotonation of the lanthanide-bound and activated aqua ligands, and thus preventing the precipitation of lanthanide hydroxides.
An interesting observation is the incorporation of small anionic species in many lanthanide hydroxide cluster complexes so-obtained. These include halides,5a–c carbonate,5d and oxalate,12 either present in the original reaction mixture or generated in situ due to ligand decomposition. Studies have established their indispensible role in templating the assembly of the cluster species. Their significance has also been demonstrated in the recent syntheses of a number of 3d–4f heterometallic clusters with giant magnetocaloric effects (MCEs) that are critical for the development of sub-Kelvin magnetic refrigeration technology.13
Due to the variation in composition, charge, and/or geometry, the use of different anions is expected to lead to clusters with distinct structures and interesting properties. Based on the templating role of the oxoanions mentioned above,5d,12 we set out to explore the use of simple oxometalates with the hope of producing novel lanthanide hydroxide clusters. The key objectives are to study the possible templating effects of these less commonly encountered anions, to obtain new heterometallic cluster species, and ultimately, to create new functional materials. In this communication, we report our results of utilizing molybdate anions (MoO42−) in the assembly of three novel lanthanide–Mo hydroxide clusters.
Compounds 1–3 of the general formula [Ln12Mo4O16(HL)6(μ3-OH)4(OOCCH3)12]·12MeOH·8H2O [Ln = Gd(1), Eu(2), Sm(3); H3L = (E)-2-(2,3-dihydroxypropylimino)methyl)-phenol] were synthesized under solvothermal conditions using a mixture containing the racemic Schiff-base ligand H3L (Fig. S1, ESI†), Ln(CH3CO2)3·3H2O, (n-Bu4N)4Mo8O26, and triethylamine and methanol. The formulation was established by single-crystal X-ray diffraction in conjunction with satisfactory elemental analyses (ESI†).
The crystal structure of 1 is discussed as the representative of the isomorphous compounds. The asymmetric unit contains one complete molecule (Fig. 1a), 12 methanol, and 8 water molecules. The Gd12 core, analogous to that of a previously reported cluster of nickel phosphonate,14 can be conveniently viewed as a distorted truncated tetrahedron (Fig. 1b) with each of its 12 vertices occupied by one Gd atom (the truncated tetrahedron is the simplest Archimedean polyhedron consisting of four triangles and four hexagons). Each of the triangular faces in 1 is capped by a triply bridging OH− group while each of the hexagonal faces is capped by a MoO42− anion (Fig. 2; average Mo = O distance 1.72 Å) with one of them (Mo4) showing a unique highly symmetrical coordination to the six Gd atoms within the related hexagon in a 6.2220 (Harris notation)15 bridging mode (Fig. S2a, ESI†) using three of its four O atoms; each of the remaining MoO42− (Mo1, Mo2, and Mo3) anions acts as a ligand using three of its four O atoms to coordinate to five Gd atoms in a 5.2210 bridging mode (Fig. 2 and Fig. S2b, ESI†).
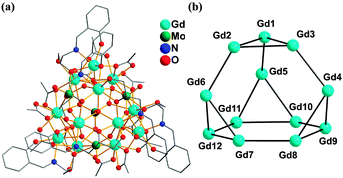 | ||
| Fig. 1 (a) Ball-and-stick plot of Gd12Mo4O16(HL2−)6(μ3-OH)4(CH3CO2)12; (b) the {Gd12} unit corresponds to a distorted truncated tetrahedron. | ||
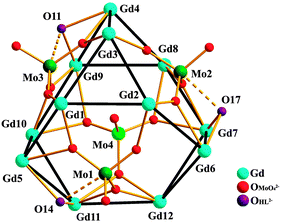 | ||
| Fig. 2 Demonstration of the {Gd12} unit capped by four MoO42− ions. The long Mo–O(HL) bond distances of 2.475(8)–2.548(11) Å are comparable to those of 2.482(11)–2.539(12) Å (dotted lines) are in a previously reported Mo complex but contain a different coordination type of the metal centers.16 | ||
The Gd atoms are further coordinated by six HL2− ligands in 2.2111 and 3.2211 bridging modes (Fig. S2c and S2d, ESI†) and by 12 acetate ligands in 2.21 and 3.21 modes. The Gd atoms are either octa- or nona-coordinated. Gd1, Gd2, and Gd3 are octacoordinated, featuring the coordination of the (N,O,O) moiety of one HL2− ligand, of two alkoxido O atoms from another HL2− ligand, of two molybdate O atoms, and of one μ3-OH group. Gd4, Gd5, and Gd6 are also octacoordinated by the (N,O,O) moiety of one HL2− ligand, furthermore by the phenolate O atom from another HL2− ligand, two acetate O atoms, one molybdate O atom, and one μ3-OH group. The remaining Gd atoms are all nonacoordinated with a tri-capped trigonal prismatic environment. Gd7, Gd9, and Gd11 are each coordinated by four acetate O, two alkoxido O, two molybdate O atoms, and one μ3-OH group, while the other three (Gd8, Gd10 and Gd12) are each coordinated by six acetate O, and two molybdate O atoms, and one μ3-OH group. The shortest Gd⋯Gd separation is 3.647(1) Å, comparable to the values found in similar μ3-OH-bridged Gd clusters.17 The shortest Gd⋯Mo distance is 3.610(2) Å, slightly shorter than 3.652 Å of a previously reported Gd(III)–Mo(VI) complex.18
The large number of magnetically interesting Gd(III) in 1 prompted us to investigate the magnetic properties of this unusual cluster compound. Its direct-current (DC) magnetic susceptibility was measured on a polycrystalline sample in the temperature range of 2.0 to 300 K under an applied magnetic field of 1000 Oe. As shown in Fig. S3 (ESI†), the χMT product at 300 K is 95.2 cm3 mol−1 K, in good agreement with the expected value of 94.5 cm3 mol−1 K for 12 non-interacting Gd(III) ions (8S7/2, g = 2.0).19 Upon lowering the temperature, the χMT value remains nearly constant above 75 K, below which it drops rapidly, to 50.6 cm3 mol−1 K at 2 K. The decrease of the χMT value at lower temperature, together with the easy saturation of magnetization at ca. 6 T (Fig. S4, ESI†), indicates weak yet dominant antiferromagnetic coupling between adjacent Gd(III) ions.
In order to estimate the magnitude of the antiferromagnetic exchange between the Gd(III) ions, a simulation of the magnetization was performed. The huge size of the Hilbert space of 12 spins s = 7/2 (68,719,476,736) precludes an exact quantum treatment even if symmetries are applied.20 Two approximations were adopted instead: (1) classical Monte-Carlo simulations and (2) the Finite Temperature Lanczos Method (FTLM).21 The former is expected to be rather accurate for the treatment of a large spin such as s = 7/2 and for thermal energies that are bigger than the exchange, while the latter is a very accurate quantum mechanical approximation for Hilbert space dimensions up to about 109. It was therefore applied to an analogous structure with single spins of 5/2. The Hilbert space dimension of 2.176.782.336 remains huge, but the magnetization can be obtained using the National Supercomputer at LRZ Garching. A single exchange constant J between nearest neighbor Gd spins was assumed, and a good agreement is reached with both methods for J = −0.04 K using a 2J-Heisenberg Hamiltonian. This number is somewhat smaller than in other Gd compounds,13e,22 but can be rationalized as some of the exchange pathways are long due to the involvement of a molybdenum atom.
The MCE of 1 is estimated from the magnetization data obtained at various fields and temperatures (Fig. S4, ESI†). Values of ΔSm are calculated by fitting the magnetization data with the equation ΔSm(T, ΔH) = ∫[∂M(T,H)/∂T]HdH (ΔSm: the change of magnetic entropy following a change of the applied magnetic field).23 As depicted in Fig. 3, the largest −ΔSm = 35.3 J kg−1 K−1 was obtained at 3 K for ΔH = 7 T, lower than the calculated value of 41.6 J kg−1 K−1 per mole for 12 uncorrelated Gd(III) spins (S = 7/2),23c possibly due to the presence of weaker intra-cluster antiferromagnetic interactions that contribute negatively to its MCE. Though not as large as the highest value of 47.7 J kg−1 K−1 recently reported for a Gd(III)-containing coordination polymer,24 the experimentally obtained −ΔSm value reported here is one of only a handful that are larger than 35.0 J kg−1 K−1.13b,d,24,25 That the observed −ΔSm value is smaller than that of the ferromagnetic coupled {Gd2} cluster25 but significantly larger than that of the antiferromagnetically coupled {Gd7} cluster26 suggests that the existence of the relatively large number of Gd(III) ions with weaker coupling is important in enhancing the field dependence of MCE due to an increase of the number of low-lying excited spin states.27
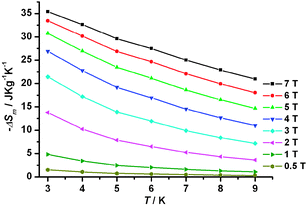 | ||
| Fig. 3 −ΔSm obtained from magnetization data of 1 at various fields and temperatures. | ||
How the material behaves in an adiabatic cooling process can be understood by evaluating the magnetic isentropes. In a perfect adiabatic demagnetization process, the system would follow its curve of constant entropy down to the respective temperature at B = 0. We suspect that sub-Kelvin temperatures should indeed be achievable, as can be deduced from Fig. S5 (ESI†).
In summary, three new heterometallic lanthanide–Mo cluster complexes have been prepared by lanthanide hydrolysis under solvothermal conditions and in the presence of molybdate anions. The four oxometalate ions serve as templates for the assembly of 12 lanthanide ions into a fascinating truncated tetrahedron. The unique templating role and reactivity of MoO42− and possibly other oxometalates merit further studies.28 For one of these clusters, the Gd-containing compound, magnetic studies revealed a large MCE, suggesting that the existence of a large number of high-spin Gd(III) ions and weak antiferromagnetic interactions29 are critical in enhancing the field dependence of the MCE. Thus, the present work not only offers a distinctly different approach to realizing structurally interesting heterometallic lanthanide-transition metal clusters, but also provides new insights into the design of materials with large MCEs for potential magnetic cooling applications.4c,29
We thank the 973 Project from MSTC (Grant No. 2012CB821704) and the NNSFC (Grant No. 20825103, 90922031, and 21021061) for the financial support. A.M. and J.S. thank the Deutsche Forschungsgemeinschaft for continuous support. Supercomputing time at the LRZ Garching is gratefully acknowledged.
Notes and references
- M. R. Bürgstein and P. W. Roesky, Angew. Chem., Int. Ed., 2000, 39, 549–551 CrossRef CAS.
- K. Manseki and S. Yanagida, Chem. Commun., 2007, 1242–1244 RSC.
- D. I. Alexandropoulos, S. Mukherjee, C. Papatriantafyllopoulou, C. P. Raptopoulou, V. Psycharis, V. Bekiari, G. Christou and T. C. Stamatatos, Inorg. Chem., 2011, 50, 11276–11278 Search PubMed.
- (a) J. K. Tang, I. Hewitt, N. T. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson, C. Benelli, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729–1733 CrossRef CAS; (b) L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC; (c) L. Bogani, C. Sangregorio, R. Sessoli and D. Gatteschi, Angew. Chem., Int. Ed., 2005, 44, 5817–5821 CrossRef.
- (a) Z. Zheng, Chem. Commun., 2001, 2521–2529 RSC; (b) R. Wang, Z. Zheng, T. Jin and R. J. Staples, Angew. Chem., Int. Ed., 1999, 38, 1813–1815 CrossRef CAS; (c) R. Wang, H. D. Selby, H. Liu, M. D. Carducci, T. Jin, Z. Zheng, J. W. Anthis and R. J. Staples, Inorg. Chem., 2002, 41, 278–286 CrossRef CAS; (d) X. J. Kong, Y. L. Wu, L. S. Long, L. S. Zheng and Z. Zheng, J. Am. Chem. Soc., 2009, 131, 6918–6919 CrossRef CAS; (e) B. Q. Ma, D. S. Zhang, S. Gao, T. Z. Jin, C. H. Yan and G. X. Xu, Angew. Chem., Int. Ed., 2000, 39, 3644–3646 CrossRef CAS.
- N. Mahé, O. Guillou, C. Daiguebonne, Y. Gérault, A. Caneschi, C. Sangregorio, J. Y. Chane-Ching, P. E. Car and T. Roisnel, Inorg. Chem., 2005, 44, 7743–7750 CrossRef CAS.
- P. C. Andrews, T. Beck, C. M. Forsyth, B. H. Fraser, P. C. Junk, M. Massi and P. W. Roesky, Dalton Trans., 2007, 5651–5654 RSC.
- O. A. Gerasko, E. A. Mainicheva, M. I. Naumova, M. Neumaier, M. M. Kappes, S. Lebedkin, D. Fenske and V. P. Fedin, Inorg. Chem., 2008, 47, 8869–8880 CrossRef CAS.
- M. Komiyama, N. Takeda and H. Shigekawa, Chem. Commun., 1999, 1443–1451 RSC.
- S. Aime, M. Botta, M. Fasano and E. Terreno, Chem. Soc. Rev., 1998, 27, 19–29 RSC.
- (a) V. Baskar and P. W. Roesky, Dalton Trans., 2006, 676–679 RSC; (b) J. C. Plakatouras, I. Baxter, M. B. Hursthouse, K. M. A. Malik, J. McALeese and S. R. Drake, Chem. Commun., 1994, 2455–2456 RSC.
- X. J. Kong, Y. P. Ren, L. S. Long, Z. Zheng, G. Nichol, R. B. Huang and L. S. Zheng, Inorg. Chem., 2008, 47, 2728–2739 CrossRef CAS.
- (a) G. Karotsis, S. Kennedy, S. J. Teat, C. M. Beavers, D. A. Fowler, J. J. Morales, M. Evangelisti, S. J. Dalgarno and E. K. Brechin, J. Am. Chem. Soc., 2010, 132, 12983–12990 CrossRef CAS; (b) J. B. Peng, Q. C. Zhang, X. J. Kong, Y. P. Ren, L. S. Long, R. B. Huang, L. S. Zheng and Z. Zheng, Angew. Chem., Int. Ed., 2011, 50, 10649–10652 CrossRef CAS; (c) Y. Z. Zheng, M. Evangelisti, F. Tuna and R. E. P. Winpenny, J. Am. Chem. Soc., 2012, 134, 1057–1065 CrossRef CAS; (d) J. B. Peng, Q. C. Zhang, X. J. Kong, Y. Z. Zheng, Y. P. Ren, L. S. Long, R. B. Huang, L. S. Zheng and Z. Zheng, J. Am. Chem. Soc., 2012, 134, 3314–3317 CrossRef CAS; (e) T. N. Hooper, J. Schnack, S. Piligkos, M. Evangelisti and E. K. Brechin, Angew. Chem., Int. Ed., 2012, 51, 4633–4636 CrossRef CAS.
- B. A. Breeze, M. Shanmugam, F. Tuna and R. E. P. Winpenny, Chem. Commun., 2007, 5185–5187 RSC.
- R. A. Coxall, S. G. Harris, D. K. Henderson, S. Parsons, P. A. Tasker and R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2000, 2349–2356 RSC.
- L. Ma, S. Liu and J. Zubieta, Polyhedron, 1989, 8, 1571–1573 CrossRef CAS.
- N. Kato, T. Mita, M. Kanai, B. Therrien, M. Kawano, K. Yamaguchi, H. Danjo, Y. Sei, A. Sato, S. Furusho and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 6768–6769 CrossRef CAS.
- C. D. Wu, C. Z. Lu, J. C. Liu, H. H. Zhuang and J. S. Huang, J. Chem. Soc., Dalton Trans., 2001, 3202–3204 RSC.
- C. Benelli and D. Gatteschi, Chem. Rev., 2002, 102, 2369–2387 CrossRef CAS.
- R. Schnalle and J. Schnack, Int. Rev. Phys. Chem., 2010, 29, 403–452 CrossRef CAS.
- (a) J. Jaklič and P. Prelovšek, Phys. Rev. B, 1994, 49, 5065–5068 CrossRef; (b) J. Schnack and O. Wendland, Eur. Phys. J. B, 2010, 78, 535–541 CrossRef CAS.
- S. Sanz, R. D. McIntosh, C. M. Beavers, S. J. Teat, M. Evangelisti, E. K. Brechin and S. J. Dalgarno, Chem. Commun., 2012, 48, 1449–1451 RSC.
- (a) M. Evangelisti, F. Luis, L. J. de Jongh and M. Affrote, J. Mater. Chem., 2006, 16, 2534–2549 RSC; (b) B. G. Shen, J. R. Sun, F. X. Hu, H. W. Zhang and Z. H. Cheng, Adv. Mater., 2009, 21, 4545–4564 CrossRef CAS; (c) M. Evangelisti and E. K. Brechin, Dalton Trans., 2010, 39, 4672–4676 RSC.
- F. S. Guo, J. D. Leng, J. L. Liu, Z. S. Meng and M. L. Tong, Inorg. Chem., 2012, 51, 405–413 CrossRef CAS.
- M. Evangelisti, O. Roubeau, E. Palacios, A. Camón, T. N. Hooper, E. K. Brechin and J. J. Alonso, Angew. Chem., Int. Ed., 2011, 50, 6606–6609 CrossRef CAS.
- J. W. Sharples, Y. Z. Zheng, F. Tuna, E. J. L. McInnes and D. Collison, Chem. Commun., 2011, 47, 7650–7652 RSC.
- S. K. Langley, N. F. Chilton, B. Moubaraki, T. Hooper, E. K. Brechin, M. Evangelisti and K. S. Murray, Chem. Sci., 2011, 2, 1166–1169 RSC.
- (a) A. Müller, E. Diemann, R. Jostes and H. Bögge, Angew. Chem., Int. Ed. Engl., 1981, 20, 934–955 CrossRef; (b) M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef; (c) H. Oshio, T. Kikuchi and T. Ito, Inorg. Chem., 1996, 35, 4938–4941 CrossRef CAS; (d) X.-Y. Yi, H. H. Y. Sung, I. D. Williams and W.-H. Leung, Chem. Commun., 2008, 3269–3271 RSC.
- R. Sessoli, Angew. Chem., Int. Ed., 2012, 51, 43–45 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses, crystallographic data and characterization details of 1–3, CCDC 861086–861088, Fig. S1–S5. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cc36530h |
| This journal is © The Royal Society of Chemistry 2013 |