Mesenchymal stem cell response to TGF-β1 in both 2D and 3D environments
Ryan S.
Stowers†
a,
Charles T.
Drinnan†
ab,
Eunna
Chung
a and
Laura J.
Suggs
*a
aLaboratory for Cardiovascular Tissue Engineering, Department of Biomedical Engineering, University of Texas at Austin, Austin, TX, USA. E-mail: laura.suggs@engr.utexas.edu; Fax: +512-471-0616; Tel: +512-232-1671
bDepartment of Bioengineering, Temple University, Philadelphia, PA, USA
First published on 21st May 2013
Abstract
Smooth muscle cells (SMC) are critical in stabilizing developing vascular networks, and transforming growth factor β1 (TGF-β1) has been shown to promote SMC differentiation from stem cells. Previously, our lab has developed a chemically modified fibrin-based hydrogel that induces endothelial cell (EC) phenotype and network formation from human mesenchymal stem cells (hMSCs) without exogenous cytokines. Additionally, we have shown that this hydrogel system is capable of releasing growth factors in a controlled manner. In the present work, the effects of TGF-β1 on hMSCs in both monolayer and fibrin-based gel culture systems were demonstrated. The objective was to enhance SMC properties through TGF-β1 signaling for vessel stability while maintaining EC gene expression and morphology. Proliferation was decreased with higher TGF-β1 concentration in both monolayer and 3D gel cultures. EC genes were predominantly downregulated in the presence of TGF-β1 in monolayer cultures, while SMC genes were generally upregulated. In fibrin-based gels, several SMC genes were significantly upregulated at high concentrations of TGF-β1. Even at elevated TGF-β1 concentrations, no significant differences were seen in EC genes for hMSCs in gels compared to controls. Network formation and growth occurred in PEGylated fibrin gels loaded with TGF-β1 and were not significantly different from gels without loaded growth factor. Additionally, production of smooth muscle α-actin (SMA) was significantly increased in gels loaded with TGF-β1. These results demonstrate a simultaneous response of hMSCs to both the 3D biomatrix and cytokine signaling cues.
Introduction
It has been well documented that clinical success in tissue engineering has been achieved with thin or avascular constructs.1–3 However, in order for seeded cells to remain viable and repopulate larger scaffolds, a patent microvascular network must be present.4,5 Researchers have demonstrated that cell viability is minimal when the distance between cells and a capillary source is greater than a few hundred microns.6 Thus, to successfully engineer larger constructs, robust neovascularization is required.Both adult and embryonic-derived endothelial cell sources may contribute towards the creation of new blood vessels and improve survival of engineered tissues.7 However, an insufficient number of endothelial cells (ECs) and the potential host immune response limit clinical applications. It is desirable for neovascularization to utilize an adult stem cell source that could be easily isolated and/or expanded rapidly in vitro. Mesenchymal stem cells (MSCs) derived from bone marrow,8–11 Wharton's jelly,12 amniotic membrane,13 umbilical cord blood,14 and amniotic fluid15 have the potential to differentiate towards EC lineages under various conditions. MSCs have also demonstrated a degree of immunoprivilege decreasing the likelihood of rejection.16 EC phenotype from bone marrow MSCs has been induced through growth factors and cytokines as well as mechanical stimulation such as shear force exposure.17–21 Recently, a number of groups, including ours, have demonstrated that 3D matrix culture can influence the differentiation of MSCs towards an EC phenotype.22–24 While the extent of terminal differentiation of bone marrow MSCs is uncertain,25 the preponderance of evidence suggests that the culture matrix can dramatically affect the EC phenotype of MSCs with potential clinical utility.
Fibrin gels are often employed as a matrix for in vitro vascularization; however, the degradation rate in vivo is rapid. Our lab has shown that derivatives of polyethylene glycol (PEG) can be covalently conjugated to fibrinogen before thrombin polymerization to significantly slow enzymatic degradation by hindering protease activity in addition to increased crosslinking.26 These PEGylated fibrin gels promote vasculogenesis in vitro from a population of MSCs, forming multicellular networks by day 3 that progressed over time.24 EC markers CD31, von Willebrand Factor (vWF), and vascular endothelial cadherin (VE-CAD) were upregulated in MSCs cultured in PEGylated fibrin without the addition of any soluble factors. Additional work by our group has demonstrated that the effect of PEGylation on network formation is quantifiable27 and that mesenchymal precursors eventually form fluid filled lumens as demonstrated by transmission electron microscopy.28 When implanted subcutaneously in rats, PEGylated fibrin gels were vascularized within 7 days.24,29 However, further analysis revealed the neovasculature was disorganized and immature, with weakly diffuse CD31 staining throughout the construct. Studies examining developing vasculature have demonstrated similar phenomena without the presence of mature smooth muscle cells (SMC) or pericytes.30 Thus, we hypothesized that enhancing the mural cell phenotype was necessary to stabilize the network formed in PEGylated fibrin gels.
Transforming growth factor-β1 (TGF-β1) is widely described to drive SMC differentiation from progenitors.31 In particular, studies investigating the effect of TGF-β1 on bone marrow MSCs in monolayer cultures have demonstrated upregulated expression of SMC genes including smooth muscle α-actin, calponin, and SM22α.32–36 Protein expression followed similar trends, and the induced cells could be expanded while maintaining the new phenotype.36 Further, SMC markers are upregulated in a dose-dependent manner with increasing TGF-β1 concentration and duration of exposure.34,37 However, the majority of these studies employed only monolayer culture systems, and additional evidence suggests that TGF-β1 signaling pathways can crosstalk with mechanical force-induced signaling in vascular cells.35 Thus, to fully understand the effects of TGF-β1 on MSCs, three-dimensional studies must be conducted to account for other microenvironmental signaling cues.
TGF-β1 has been used in 3D matrices to stimulate the differentiation of MSCs towards a SMC phenotype. Ross et al. demonstrated increased collagen synthesis and mechanical strength when multipotent adult progenitor cells (MAPCs) were cultured in fibrin hemispheres for 5 weeks in medium containing 2.5 ng ml−1 TGF-β1.36 Gong et al. seeded MSCs in a polyglycolic acid scaffold for 4 weeks and then exposed them for 6 weeks to pulsatile flow and TGF-β1. The presence of TGF-β1 and pulsatile flow conditions increased SMC marker expression, collagen production, and burst strength of the scaffolds.38 Additionally, MSCs have been seeded within porous collagen-glycosaminoglycan scaffolds and cultured with 2 ng ml−1 TGF-β1. The presence of TGF-β1 increased the scaffold contraction by 135%, indicating that MSCs have differentiated to contractile cells.34 In light of these studies, we sought to increase the stability of nascent networks observed in our fibrin-based gels by promoting SMC properties through gels loaded with TGF-β1.
While we hypothesized that TGF-β1 loaded in fibrin-based gels would induce SMC differentiation from hMSCs, the combined effect of TGF-β1 and biomatrix cues on EC phenotype and network morphology was unknown. Previously, we have loaded PEGylated fibrin gels with TGF-β1 via covalent conjugation and physical affinity for the matrix, resulting in controlled release of the cytokine for up to 12 days.39 Here, we sought to determine the effects of TGF-β1 on hMSC proliferation, morphology, gene expression and protein production when added either endogenously to monolayer culture or loaded within a 3D fibrin-based gel. We examined the gene and protein expression for both ECs and SMCs to determine whether the upregulation of a mural cell phenotype came at the expense of the endothelial phenotype and morphology.
Materials and methods
Cell culture
HMSCs (Lonza) were purchased commercially, maintained and expanded with DMEM containing 10% FBS, 1% Glutamax, and 1% penicillin/streptomycin (Life Technologies). Media was changed every 2–3 days and the cells were passaged according to the manufacturer's protocols. Passage 4–7 cells were used for all experiments. For monolayer experiments, hMSCs were plated into 6-well plates at 2000 cell per cm2 and exposed to exogenous addition of 0, 0.1, or 10 ng ml−1 TGF-β1.Fibrin-based gels
A porcine fibrinogen solution was prepared in Dulbecco's phosphate-buffered saline (DPBS) at a concentration of 80 mg ml−1 and pH 7.8. Recombinant human TGF-β1 (R&D Systems) at a concentration of 200 ng ml−1 or 800 ng ml−1 in 0.1% bovine serum albumin (BSA, Sigma) in PBS was added to the fibrinogen solution and incubated at 37 °C for 45 minutes. Controls utilized 0.1% BSA solution in DPBS in lieu of growth factor. Following TGF-β1 loading, homobifunctional succinimidyl glutarate polyethylene glycol (PEG-(SG)2) (3400 Da, NOF America) was added to the fibrinogen solution at a molar ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. For nonPEGylated gel conditions, DPBS was added in lieu of the PEG derivative. The reaction was allowed to proceed at room temperature for 3 minutes. Cells were added to the solution at 200000 per ml in DMEM at 1/4 of the final gel volume. To initiate crosslinking and gelation, growth factor loaded PEGylated fibrinogen was exposed to 25 U ml−1 human thrombin (Sigma) in a 40 mM CaCl2 solution. Gelation time is dependent on thrombin concentration and at this range the typical gelation time is 0.5–2 minutes. The final concentrations were 10 mg ml−1 fibrinogen, 1 mg ml−1 PEG-(SG)2, 25 or 100 ng ml−1 TGF-β1, 50000 hMSCs per ml and 12.5 U ml−1 thrombin (Fig. 1A).
:1. For nonPEGylated gel conditions, DPBS was added in lieu of the PEG derivative. The reaction was allowed to proceed at room temperature for 3 minutes. Cells were added to the solution at 200000 per ml in DMEM at 1/4 of the final gel volume. To initiate crosslinking and gelation, growth factor loaded PEGylated fibrinogen was exposed to 25 U ml−1 human thrombin (Sigma) in a 40 mM CaCl2 solution. Gelation time is dependent on thrombin concentration and at this range the typical gelation time is 0.5–2 minutes. The final concentrations were 10 mg ml−1 fibrinogen, 1 mg ml−1 PEG-(SG)2, 25 or 100 ng ml−1 TGF-β1, 50000 hMSCs per ml and 12.5 U ml−1 thrombin (Fig. 1A).
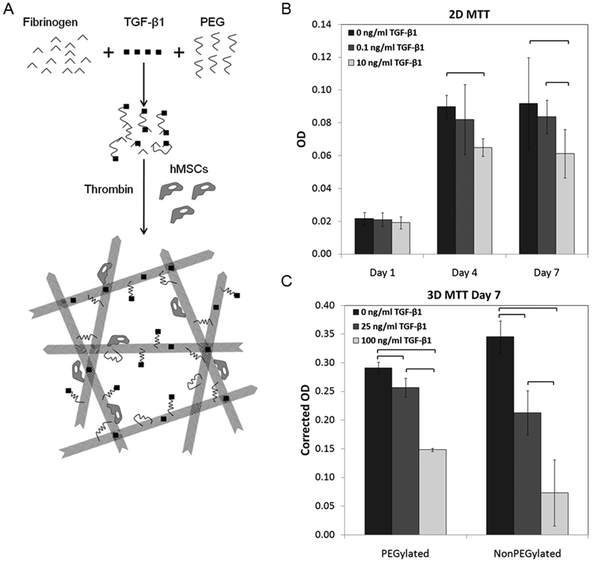 | ||
| Fig. 1 Schematic of mechanisms of fibrin PEGylation, TGF-β1 loading, and thrombin-mediated crosslinking in the presence of hMSCs (A). MTT assay for hMSCs cultured in monolayers (B) and in gels (C) exposed to various concentrations of TGF-β1. Brackets indicate significance of p < 0.05. | ||
MTT assay
The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was adapted from the commercial protocol. MTT reagent (Sigma) was prepared at 5 mg ml−1 in serum free media and added to the media of the cells at 10% of the volume (100 μl per 1 ml of media). For monolayers of hMSCs, MTT reagent was incubated for 3 hours at 37 °C. For hMSCs in gels, MTT reagent was exposed for 6 hours at 37 °C.40 Following incubation, formazan crystals were dissolved in 0.1 N HCl in isopropanol. Formazan crystals formed in monolayers of hMSCs were dissolved within 30 min while crystals in the gels were dissolved overnight. Color formation was quantified with a universal microplate reader ELX800 (BIO-TEK Instruments) at a wavelength of 560 nm. Corrected absorbance was calculated by subtracting the absorbance of media exposed to MTT reagent and gels without hMSCs from experimental group values.Quantitative real-time polymerase chain reaction
RNA was isolated from hMSCs in monolayer culture using Qiagen RNeasy Minikit according to the manufacturer's instructions. RNA extraction from hMSCs in gels was performed by following the Trizol-chloroform method.41Isolated RNA was quantified using a Nanodrop spectrophotometer (Fisher Scientific). RNA samples were reverse transcribed using a kit without RNase inhibitors (Life Technologies) according to the manufacturer's instructions. Blanks consisting of buffer and reagents were used in parallel as controls. Standard hMSCs taken from the maintenance stock were used for comparison.
TaqMan® gene expression assays (Life Technologies) were utilized to quantify mRNA production. CDNA from consistent amounts of starting RNA were mixed with TaqMan® reagents (Life Technologies). Genes and assay IDs used in this study can be found in Table 1. β-Actin was used as an endogenous control gene. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed utilizing an Applied Biosystems 7900HT with the following reaction conditions: 95 °C for 10 min and 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The reporter dye was 6-FAMTM. Relative quantification (RQ) was determined by comparing gene expression to β-actin and standard hMSCs using the 2-ΔΔCT method.42
| Gene name | Symbol | Assay ID | Target protein |
|---|---|---|---|
| Actin, alpha 2, smooth muscle, aorta | ACTA2 | Hs00426835_g1 | ACTA2 (SMA) |
| Desmin | DES | Hs00157258_m1 | DES |
| Calponin 1, basic smooth muscle | CNN1 | Hs00154543_m1 | CNN1 |
| Angiopoietin 1 | ANGPTI | Hs00181613_m1 | ANGPT1 |
| Angiopoietin 2 | ANGPT2 | Hs00169867_m1 | ANGPT2 |
| Platelet derived growth factor receptor, beta polypeptide | PDGFRB | Hs00182163_m1 | PDGFRB |
| Platelet/endothelial cell adhesion molecule | PECAM1 | Hs00169777_m1 | PECAM1 (CD31) |
| Vascular endothelial growth factor A | VEGFA | Hs00900054_m1 | VEGFA |
| Cadherin 5, type 2 (vascular endothelium) | CDH5 | Hs00174344_m1 | CDH5 |
| von Willebrand factor | VWF | Hs00169795_m1 | vWF |
| Alanyl (membrane) aminopeptidase | ANPEP | Hs00174265_m1 | ANPEP |
Light microscopy
Light micrographs were captured using a Leica DMI 3000 B microscope with attached camera. Images were taken once daily to monitor cell morphology and network formation and growth.Immunocytochemical analysis
For protein analysis, MSCs were cultured in 4 well chamber slides. After 7 days, the cells were fixed with 4% paraformaldehyde and rinsed in DPBS. The cells were then permeabilized with 0.25% Triton-X-100 and washed. The slides were blocked with 10% goat serum (Jackson Labs). After blocking, the primary antibody (anti-SMA, ab7817, Abcam) in 1% goat serum was applied and incubated overnight at 4 °C. Samples were then incubated in a 1:100 dilution of Cy-5 conjugated goat anti-mouse IgG secondary antibody (ab6563, Abcam) for 1 hour at room temperature. The slides were rinsed in DPBS and counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) in mounting medium (Santa Cruz Biotechnology). Images were captured on a Zeiss Axiovert microscope equipped with a Zeiss Axiocam.
For confocal microscopy, gels were prepared and cultured in 4 well chamber slides or permeable membrane-bottomed inserts (8.0 μm pores, transparent PET membrane, BD Biosciences) under the conditions described above. For calcein-AM staining, the gels were washed with serum-free media and incubated with 8 μM calcein-AM (Invitrogen) in PBS for 1 hour at 37 °C. The samples were then fixed for 10 min with 4% paraformaldehyde and imaged. For immunofluorescent staining, the gels were washed with DPBS and fixed for 15 minutes with 10% neutral-buffered formalin (PSL Equipment, Inc.). After blocking with 10% goat serum (Life Technologies) for 1 hour at room temperature, the gels were incubated with primary antibody (anti-SMA, ab7817, Abcam) in 1% goat serum overnight at 4 °C followed by incubation with the secondary antibody Alexa 488 goat anti-mouse IgG (H+L) (A11001, Life Technologies) at room temperature for 1 hour. A Leica SP2 AOBS confocal microscope was used to obtain images of all gels.
Western blot analysis
SMA and vWF expression in hMSCs were investigated by western blot analysis. Cells from 3D samples were lysed in RIPA buffer (Santa Cruz Biotechnology). Lysed samples were centrifuged at 14000g for 20 minutes. Isolated proteins in the supernatant were quantified using the BCA Protein Assay Kit (Pierce) to derive the concentration of total protein in each sample. Proteins were mixed 1:1 with Laemmli sample buffer (Bio-Rad Laboratories)/β-mercaptoethanol (19:1 volume ratio) followed by heating at 95 °C for 5 min. For electrophoresis, the heated protein (20 μg per lane) was loaded in a 10% Mini-PROTEAN® TGX™ Precast Gel (Bio-Rad Laboratories). Proteins transferred onto the PVDF membranes were immunoblotted using mouse and rabbit primary antibodies for SMA (ab7817, Abcam) and vWF (ab6994, Abcam), respectively, and matched secondary antibodies diluted in the blocking buffer composed of 5% non-fat dry milk solution in Tween-20/Tris buffered saline (TBS). For mouse and rabbit primary antibodies, goat anti-mouse (sc-2005, Santa Cruz) and anti-rabbit (ab6721, Abcam) IgG conjugated with horse radish peroxidase were used as secondary antibodies. For a loading control, the band of β-actin was detected using a rabbit polyclonal anti-β-actin antibody (ab75186, Abcam). The chemiluminescent signal of each band was visualized by FluorChem Q (ProteinSimple) and quantified using AlphaView software.
Statistical analysis
ANOVA was performed for each experiment with post hoc Tukey's test when applicable using MATLAB (MathWorks). Groups were considered significantly different for p-values less than 0.05. Error bars represent standard error of the mean of at least three independent samples per group.Results
Cell proliferation
The addition of TGF-β1 inhibited proliferation in monolayer cultures of hMSCs (Fig. 1B). There were significantly fewer cells at day 4 in the 10 ng ml−1 TGF-β1 compared to controls without additional TGF-β1. By day 7, a significant decrease was observed in the 10 ng ml−1 group compared to both 0 and 0.1 ng ml−1 TGF-β1 groups.When hMSCs were cultured in gels, significant differences were demonstrated between all groups in both PEGylated and nonPEGylated fibrin gels at day 7 (Fig. 1C). Proliferation steadily decreased with increasing TGF-β1 concentration. It should be noted that the optical properties of the two gel types are dissimilar, and while an attempt was made to correct for absorption by the gels, comparisons should only be made within gel groups and not between PEGylated and nonPEGylated fibrin gels.
TGF-β1 affects gene expression of 2D HMSC cultures
HMSCs were cultured until day 7 with 0, 0.1 (low), or 10 ng ml−1 (high) concentrations of TGF-β1. Reverse transcription, qRT-PCR was then performed to examine the expression of SMC or EC genes. After 7 days, the highest concentration of TGF-β1 downregulated the EC marker, VE-CAD, compared to both low and no growth factor addition (Fig. 2A). Interestingly, VEGF was significantly upregulated compared to hMSC controls in the presence of low TGF-β1, but downregulated by the higher TGF-β1 concentration compared to the low concentration.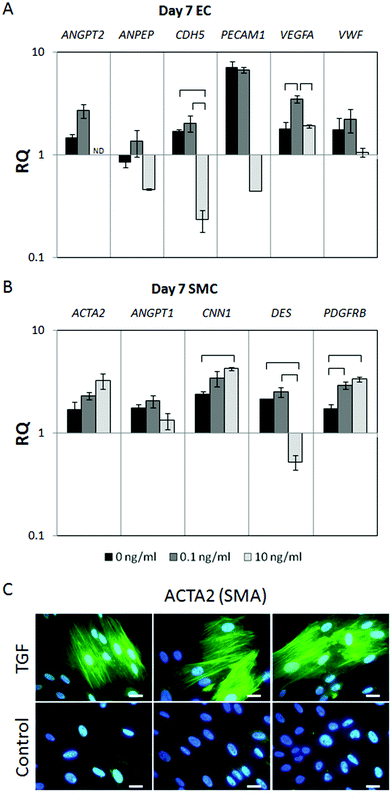 | ||
| Fig. 2 Gene expression of hMSCs in monolayer culture exposed to various levels of TGF-β1 after 7 days. Endothelial (A) and smooth muscle markers (B) were tested. Brackets indicate significance of p < 0.05. Immunostaining of hMSCs in monolayer culture treated with 10 ng ml−1 TGF-β1 or untreated control. SMA (green) was highly expressed and found in prominent stress fibers in cultures with TGF-β1, compared to low expression levels and diffuse cytoplasmic staining in controls (3 representative replicates shown in C). Nuclei were counter-stained with DAPI (blue). Scale bar represents 20 μm. | ||
In the presence of TGF-β1, significant upregulation of several SMC markers was observed (Fig. 2B). CAL was upregulated in the high TGF-β1 concentration group and PDGF-RB was upregulated with both TGF-β1 groups. Additionally, DES was significantly downregulated at the highest concentration of TGF-β1 compared to both hMSCs cultured with low and no TGF-β1.
Immunocytochemical analysis of 2D HMSCs in the presence of TGF-β1
HMSCs were cultured in chamber slides either with or without an additional 10 ng ml−1 TGF-β1 in the culture medium, and stained for SMA. Monolayers cultured with TGF-β1 showed a high degree of SMA production, with most cells staining positive after 7 days (Fig. 2C). SMA was localized in prominent stress fiber bundles. In the control group, SMA was produced at low levels and was diffusely spread throughout the cytoplasm. Additionally, very few stress fibers were evident.HMSC morphology in PEGylated fibrin gels
Light micrographs confirmed that hMSCs form branched, multicellular structures by day 3 and grew into extensive networks by day 7 in PEGylated fibrin gels (Fig. 3A), as previously demonstrated.24 Networks were more extensive in PEGylated fibrin gels compared to nonPEGylated fibrin. Though the opacity of the nonPEGylated fibrin gels limited visibility under phase contrast, this was clearly demonstrated through calcein-AM staining (Fig. 3B). The addition of 100 ng ml−1 TGF-β1 to PEGylated fibrin gels did not inhibit network formation or growth. By day 5, extensive networks were observed both in the presence and absence of TGF-β1. Additionally, network extension was stable over a range of TGF-β1 concentrations and was not diminished at the highest concentration used in these studies.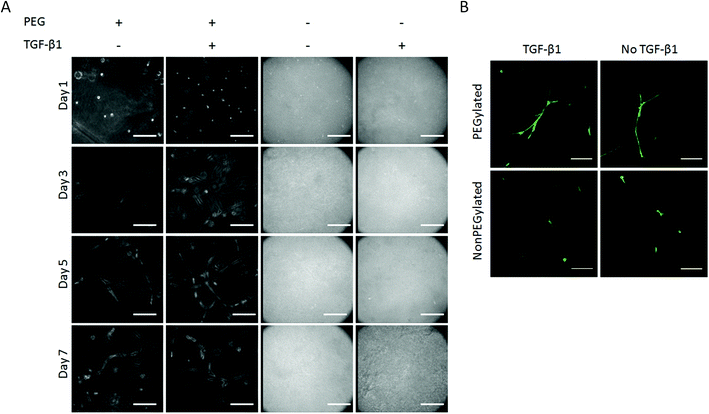 | ||
| Fig. 3 Light micrograph images of hMSCs cultured in various gel conditions. Network formation and growth is evident in PEGylated fibrin gels with and without 100 ng ml−1 TGF-β1. Scale bar represents 200 μm. Fluorescent images of calcein-AM stained hMSCs within gels at day 7 (B). HMSCs demonstrated network formation in PEGylated gels and were not inhibited by 100 ng ml−1 TGF-β1. Scale bar represents 150 μm. | ||
TGF-β1 affects gene expression of HMSCs in gels
Gene expression of hMSCs was assessed at day 7 for gels loaded with 25 ng ml−1 of TGF-β1. While changes in gene expression are typically assessed within hours of inductive signalling, our previous studies have shown marked upregulation of gene expression at day 7 in 3D gel cultures.28 Additionally, TGF-β1 is continuously released in PEGylated fibrin gels for up to 12 days, presumably leading to differences in gene expression after one week. Three markers for both ECs (CD31, VE-CAD, and VEGF) and SMCs (SMA, CAL, and DES) were selected based on the gene expression results of hMSCs cultured in monolayers, and those of known importance from prior studies.24,38 The addition of TGF-β1 to the gels did not significantly inhibit expression of EC markers (Fig. 4A). A significant increase in VEGF expression was found between PEGylated and nonPEGyated fibrin with TGF-β1, and trends are consistent with our prior report that PEGylation increases EC gene expression.24 While significance was not demonstrated in any SMC genes, trends in several groups indicated upregulation in the presence of TGF-β1 (Fig. 4B). In order to establish the validity of the trends seen in SMC markers, further tests were conducted with a higher concentration of TGF-β1, 100 ng ml−1 (Fig. 4C and D). In several cases, SMC markers were significantly upregulated compared to unexposed controls. SMA was upregulated in nonPEGylated fibrin gels with TGF-β1 compared to the same gel without the growth factor. Additionally, CAL was upregulated in both PEGylated and nonPEGylated fibrin gels when loaded with TGF-β1 over unloaded controls. EC markers showed significant upregulation of VEGF and vWF in PEGylated fibrin with TGF-β1 groups, as well as the trends elsewhere suggesting increased EC expression in PEGylated fibrin gels. Notably, at the higher concentration of TGF-β1, EC markers were not significantly altered from controls. HMSCs of a different passage, and thus growth characteristics, were used for analysis of the higher TGF-β1 concentration, which may account for differences in relative quotient (RQ).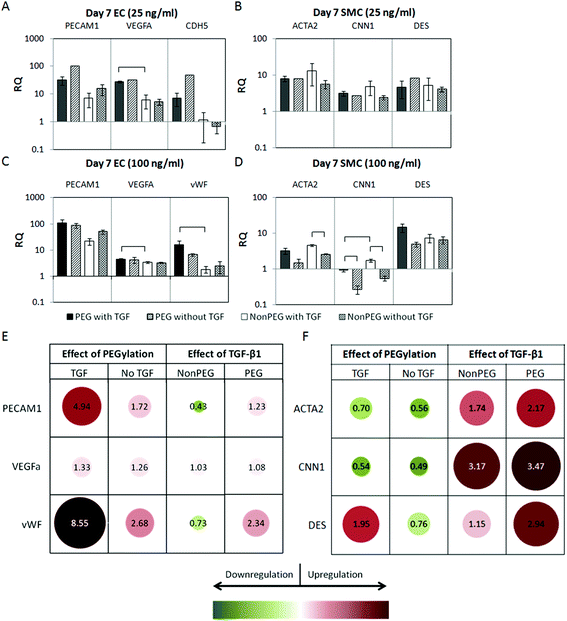 | ||
| Fig. 4 QRT-PCR of hMSCs in gels after 7 days. EC markers with 25 ng ml−1 TGF-β1 loaded (A). SMC markers with 25 ng ml−1 TGF-β1 loaded (B). EC markers with 100 ng ml−1 TGF-β1 loaded (C). SMC markers with 100 ng ml−1 of TGF-β1 loaded (D). Brackets indicate significance of p < 0.05. Relative gene expression between groups based on 100 ng ml−1 TGF-β1 qRT-PCR data (E, F). Values represent the ratio of RQ for the groups listed within a given gene. A value of 1 indicates equal gene expression between the two groups. Note that the color and size scaling differ from E to F and should only be compared within each matrix. | ||
To more clearly demonstrate the impact of TGF-β1 addition or PEGylation of fibrin gels, ratios of expression levels were calculated. A value of one for this metric indicates equal RQ between the two groups. Values greater than 1 represent relative upregulation and values less than 1 indicate relative downregulation due to the condition. Overall, the addition of TGF-β1 results in moderate upregulation of SMC genes, while PEGylation tends to slightly downregulate SMC genes (Fig. 4E). Expression levels of EC genes did not vary significantly with respect to TGF-β1 loading; however PEGylation was seen to promote EC marker expression (Fig. 4F).
Protein analysis of HMSCs in gels
Protein expression of hMSCs at day 7 was assessed by western blotting. PEGylation induced a marked upregulation of vWF expression both with and without TGF-β1 (Fig. 5A). The inclusion of TGF-β1 did not significantly alter the production of vWF in either PEGylated or nonPEGylated gels.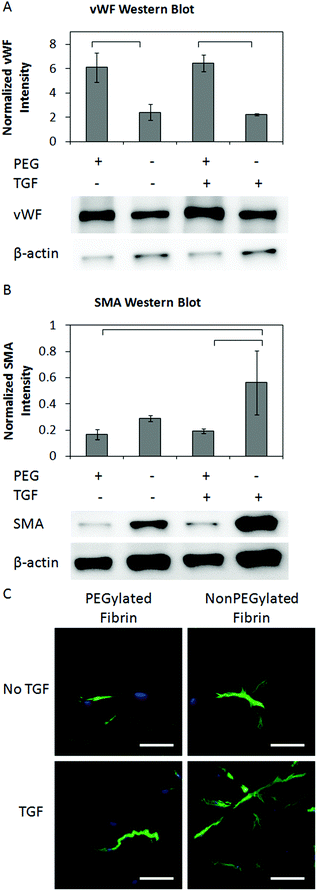 | ||
| Fig. 5 Western blot images and quantification of for vWF normalized to β-actin expression (A). Representative images of the blot are shown below the corresponding lane. Western blot images and quantification of for SMA normalized to β-actin expression (B). Representative images of the blot are shown below the corresponding lane. Brackets indicate significance of p < 0.05. Confocal Z-projections of hMSCs in gels stained for SMA (C). Scale bar represents 100 μm. | ||
SMA expression was significantly upregulated in nonPEGylated fibrin gels compared to PEGylated fibrin gels when both were loaded with TGF-β1 (Fig. 5B). TGF-β1 loaded gels demonstrated higher SMA expression compared to unloaded controls, especially in the nonPEGylated gels, though the difference was not statistically significant.
The morphologic distribution of SMA in cellular networks was examined with confocal microscopy. In PEGylated gels without TGF-β1, SMA was sparsely observed. HMSCs in nonPEGylated gels without TGF-β1 demonstrated moderate expression, but extensive networks were not seen, as mentioned previously (Fig. 5C). The addition of TGF-β1 to the nonPEGylated gels resulted in robust expression with considerably more extensive networks compared to controls without TGF-β1. A fraction of cells within the PEGylated fibrin gels with TGF-β1 displayed very high expression of SMA with extensive network formation. However, in many networks, the expression of SMA was low or undetectable.
Discussion
Proliferation assays performed on monolayer cultures of hMSCs demonstrated decreased proliferation in response to increasing TGF-β1 concentration, which is consistent with literature.38 Proliferation in 3D fibrin-based gels decreased in response to loaded TGF-β1 in a dose dependent manner. These results are consistent with a cell population that is transitioning from a proliferative phenotype to one that is synthesizing proteins. Furthermore, this gives evidence that matrix-bound TGF-β1 maintains bioactivity toward hMSCs up to 7 days and does not require supplementation in the media.In a previous report, we have demonstrated that TGF-β1 is loaded within PEGylated fibrin gels both by covalent conjugation and physical affinity for the matrix.39 While the half-life of TGF-β1 is less than 2 hours in aqueous environments, we achieved release of bioactive TGF-β1 up to 7 days in PEGylated fibrin matrices as employed in the current study. While we had initially examined a gel loading of 25 ng ml−1, the lack of significant differences in gene and protein expression among groups led us to consider a higher loading condition. Based on the loading and release kinetics measured in the prior study, we would expect approximately 25 ng released over this time period from a PEGylated fibrin gel loaded with 100 ng ml−1. We therefore report here both a high (100 ng ml−1) and low (25 ng ml−1) loading condition, and were able to demonstrate significant differences in gene and protein expression with the high loading condition.
A biphasic relationship was observed with respect to hMSC gene expression in 2D culture in response to varying levels of TGF-β1 in culture media. VEGF was significantly upregulated at the low concentration compared to either no TGF-β1 or the high concentration. A similar trend was demonstrated in the EC markers VE-CAD and vWF, angiogenic regulator ANPEP, and in SMC markers DES and ANGPT1; however differences were not statistically significant. This result is consistent with literature reports regarding the biphasic effects of TGF-β1 concentration levels on gene expression, protein production, and migration, both in similar angiogenic studies and other developmental process such as osteogenesis.43,44
Immunostaining revealed high levels of SMA organized into cytoskeletal filaments in monolayer cultures with TGF-β1. This is in accordance with previously published reports.36,45 In contrast, control cultures without TGF-β1 showed low levels of SMA dispersed throughout the cytoplasm. We hypothesized that loading TGF-β1 in 3D fibrin-based gels would upregulate SMC genes and protein synthesis, which could lead to more stable vascular networks. Indeed, our results suggest that SMC genes are promoted upon exposure to released TGF-β1 in 3D fibrin-based matrices. Significant differences were seen in the key markers, SMA and CAL, at the high loading concentration. Additionally, TGF-β1 caused an increase in production of SMA in nonPEGylated groups compared to controls shown both in the immunostained images as well as quantified by western blotting. Other groups have demonstrated enhanced SMA production in hMSCs in 3D matrices cultured with low levels of TGF-β1 supplemented to culture media (2 ng ml−1). This effect from a much lower growth factor concentration may be due to the repeated administration in growth media compared to what is released from our matrix.37
In 3D cultures, we demonstrate an enhancement by PEGylation in key endothelial cell genes including VEGF and vWF. Western blotting for vWF revealed that protein expression was increased in the PEGylated fibrin gels. Immunostaining for vWF was problematic as it is a secreted protein that remained associated with the fibrin matrix. The resulting images could not be clarified due to the high background signal. No significant reductions were seen in either gene or protein expression with the introduction of TGF-β1. The morphology of MSC networks in PEGylated fibrin was also not affected by TGF-β1 and was markedly distinct from the cells in nonPEGylated fibrin.
The effect of TGF-β1 on angiogenic phenomena is complex and not entirely understood. TGF-β1 is a pleiotropic cytokine with an indirect effect on the angiogenic cascade via upregulation of VEGF and bFGF production.46,47 Mallet et al. demonstrated an increase in EC differentiation from ESCs independent of VEGF, but an inhibition of tube formation in experiments with 0.1–10 ng ml−1 TGF-β1 added to culture medium.48 Further, TGF-β1 has been shown to inhibit EC proliferation, invasion, and tube formation in collagen gels at concentrations as low as 0.1 ng ml−1.48 These reports and others demonstrate the need to consider the contextual presentation (i.e. concentration, duration, matrix, and cell type) of TGF-β1 when comparing results.
In the current study, we demonstrated TGF-β1 does promote SMC-like properties while maintaining network morphology and EC gene and protein expression levels in vitro. We did not examine whether the phenotypic changes represented two distinct cell populations, although we did not see anything to suggest that a second cell type was developing in a perivascular position relative to the first. It is possible that the time point examined here was too early to see that type of development or it may be that what we are demonstrating here is the plasticity of MSCs that can express multiple phenotypic characteristics. Future in vivo assays will be performed to determine if vessel stability is enhanced as a result of TGF-β1 loaded within fibrin-based matrices.
Conclusions
This study demonstrates the effect of TGF-β1 on hMSC populations in monolayer and fibrin-based gel cultures. Decreased proliferation was observed in response to TGF-β1 concentration in 2D cultures. Gene expression of smooth muscle markers was largely upregulated, while endothelial cell markers were downregulated. In fibrin-based gel cultures, TGF-β1 also decreased proliferation. Several SMC genes were upregulated in response to growth factor addition, while EC genes did not differ significantly. Further, microscopic analysis showed persistent network formation, and positive staining for SMA with high intensity in PEGylated fibrin gels with TGF-β1. This evidence suggests that hMSCs respond simultaneously to distinct cytokine and biomatrix induction cues.Acknowledgements
L. J. Suggs acknowledges funding from an American Heart Association Beginning Grant-in-Aid as well as funding from the National Science Foundation (CBET-0853996 ARRA). The authors would like to thank fellow lab members for helpful discussion and the Microscopy and Imaging Facility of the Institute for Cellular and Molecular Biology at The University of Texas at Austin.Notes and references
- A. Atala, S. B. Bauer, S. Soker, J. J. Yoo and A. B. Retik, Lancet, 2006, 367, 1241–1246 CrossRef.
- P. Macchiarini, P. Jungebluth, T. Go, M. A. Asnaghi, L. E. Rees, T. A. Cogan, A. Dodson, J. Martorell, S. Bellini, P. P. Parnigotto, S. C. Dickinson, A. P. Hollander, S. Mantero, M. T. Conconi and M. A. Birchall, Lancet, 2008, 372, 2023–2030 CrossRef.
- F. Oberpenning, J. Meng, J. J. Yoo and A. Atala, Nat. Biotechnol., 1999, 17, 149–155 CrossRef CAS.
- R. K. Jain, P. Au, J. Tam, D. G. Duda and D. Fukumura, Nat. Biotechnol., 2005, 23, 821–823 CrossRef CAS.
- J. J. Moon and J. L. West, Curr. Top. Med. Chem., 2008, 8, 300–310 CrossRef CAS.
- J. Rouwkema, N. C. Rivron and C. A. van Blitterswijk, Trends Biotechnol., 2008, 26, 434–441 CrossRef CAS.
- S. Levenberg, J. Rouwkema, M. Macdonald, E. S. Garfein, D. S. Kohane, D. C. Darland, R. Marini, C. A. van Blitterswijk, R. C. Mulligan, P. A. D'Amore and R. Langer, Nat. Biotechnol., 2005, 23, 879–884 CrossRef CAS.
- M. Jazayeri, A. Allameh, M. Soleimani, S. H. Jazayeri, A. Piryaei and S. Kazemnejad, Cell Biol. Int., 2008, 32, 1183–1192 CrossRef CAS.
- J. W. Liu, S. Dunoyer-Geindre, V. Serre-Beinier, G. Mai, J. F. Lambert, R. J. Fish, G. Pernod, L. Buehler, H. Bounameaux and E. K. Kruithof, J. Thromb. Haemost., 2007, 5, 826–834 CrossRef CAS.
- J. Oswald, S. Boxberger, B. Jorgensen, S. Feldmann, G. Ehninger, M. Bornhauser and C. Werner, Stem Cells, 2004, 22, 377–384 CrossRef.
- C. Zhen-Zhou, J. Xiao-Dan, L. Gui-Tao, S. Jiang-Hua, L. Ling-Hui, D. Mou-Xuan and X. Ru-Xiang, Cytotherapy, 2008, 10, 611–624 CrossRef CAS.
- M. Y. Chen, P. C. Lie, Z. L. Li and X. Wei, Exp. Hematol., 2009, 37, 629–640 CrossRef CAS.
- F. Alviano, V. Fossati, C. Marchionni, M. Arpinati, L. Bonsi, M. Franchina, G. Lanzoni, S. Cantoni, C. Cavallini, F. Bianchi, P. L. Tazzari, G. Pasquinelli, L. Foroni, C. Ventura, A. Grossi and G. P. Bagnara, BMC Dev. Biol., 2007, 7, 11 CrossRef.
- E. J. Gang, J. A. Jeong, S. Han, Q. Yan, C. J. Jeon and H. Kim, Cytotherapy, 2006, 8, 215–227 CrossRef CAS.
- O. M. Benavides, J. J. Petsche, K. J. Moise Jr., A. Johnson and J. G. Jacot, Tissue Eng. Part A, 2012, 18, 1123–1131 CrossRef CAS.
- J. M. Kanczler, P. J. Ginty, L. White, N. M. Clarke, S. M. Howdle, K. M. Shakesheff and R. O. Oreffo, Biomaterials, 2010, 31, 1242–1250 CrossRef CAS.
- K. Bai, Y. Huang, X. Jia, Y. Fan and W. Wang, J. Biomech., 2010, 43, 1176–1181 CrossRef.
- G. P. Duffy, S. D'Arcy, T. Ahsan, R. M. Nerem, T. O'Brien and F. Barry, Tissue Eng. Part A, 2010, 16, 2755–2768 CrossRef CAS.
- H. Lin, A. Shabbir, M. Molnar, J. Yang, S. Marion, J. M. Canty Jr. and T. Lee, J. Cell Physiol., 2008, 216, 458–468 CrossRef CAS.
- J. Xu, X. Liu, J. Chen, A. Zacharek, X. Cui, S. Savant-Bhonsale, Z. Liu and M. Chopp, Am. J. Physiol. Cell Physiol., 2009, 296, C535–C543 CrossRef CAS.
- P. Zhang, J. Baxter, K. Vinod, T. N. Tulenko and P. J. Di Muzio, Stem Cells Dev., 2009, 18, 1299–1308 CrossRef CAS.
- H. Colley, S. L. McArthur, A. Stolzing and A. Scutt, Biomed. Mater., 2012, 7, 045015 CrossRef.
- K. Wingate, W. Bonani, Y. Tan, S. J. Bryant and W. Tan, Acta Biomater., 2012, 8, 1440–1449 CrossRef CAS.
- G. Zhang, C. T. Drinnan, L. R. Geuss and L. J. Suggs, Acta Biomater., 2010, 6, 3395–3403 CrossRef CAS.
- S. J. Zhang, H. Zhang, M. Hou, Z. Zheng, J. Zhou, W. Su, Y. Wei and S. Hu, Stem Cells Dev., 2007, 16, 683–690 CrossRef CAS.
- H. Liu, S. F. Collins and L. J. Suggs, Biomaterials, 2006, 27, 6004–6014 CrossRef CAS.
- J. A. Rytlewski, L. R. Geuss, C. I. Anyaeji, E. W. Lewis and L. J. Suggs, Tissue Eng. Part C, Methods, 2012, 18, 507–516 CrossRef.
- S. Natesan, G. Zhang, D. G. Baer, T. J. Walters, R. J. Christy and L. J. Suggs, Tissue Eng. Part A, 2011, 17, 941–953 CrossRef CAS.
- G. Zhang, X. H. Wang, Z. L. Wang, J. Y. Zhang and L. Suggs, Tissue Eng., 2006, 12, 9–19 CrossRef CAS.
- M. C. Dickson, J. S. Martin, F. M. Cousins, A. B. Kulkarni, S. Karlsson and R. J. Akhurst, Development, 1995, 121, 1845–1854 CAS.
- K. K. Hirschi, S. A. Rohovsky and P. A. D'Amore, J. Cell Biol., 1998, 141, 805–814 CrossRef CAS.
- B. Kinner, J. M. Zaleskas and M. Spector, Exp. Cell Res., 2002, 278, 72–83 CrossRef CAS.
- Y. Narita, A. Yamawaki, H. Kagami, M. Ueda and Y. Ueda, Cell Tissue Res., 2008, 333, 449–459 CrossRef CAS.
- J. S. Park, J. S. Chu, A. D. Tsou, R. Diop, Z. Tang, A. Wang and S. Li, Biomaterials, 2011, 32, 3921–3930 CrossRef CAS.
- D. Wang, J. S. Park, J. S. Chu, A. Krakowski, K. Luo, D. J. Chen and S. Li, J. Biol. Chem., 2004, 279, 43725–43734 CrossRef CAS.
- J. J. Ross, Z. Hong, B. Willenbring, L. Zeng, B. Isenberg, E. H. Lee, M. Reyes, S. A. Keirstead, E. K. Weir, R. T. Tranquillo and C. M. Verfaillie, J. Clin. Invest., 2006, 116, 3139–3149 CAS.
- B. Kinner, J. M. Zaleskas and M. Spector, Exp. Cell Res., 2002, 278, 72–83 CrossRef CAS.
- Z. Gong and L. E. Niklason, FASEB J., 2008, 22, 1635–1648 CrossRef CAS.
- C. T. Drinnan, G. Zhang, M. A. Alexander, A. S. Pulido and L. J. Suggs, J. Controlled Release, 2010, 147, 180–186 CrossRef CAS.
- G. Zhang, X. Wang, Z. Wang, J. Zhang and L. Suggs, Tissue Eng., 2006, 12, 9–19 CrossRef CAS.
- P. Chomczynski and N. Sacchi, Anal. Biochem., 1987, 162, 156–159 CrossRef CAS.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS.
- D. J. J. de Gorter, M. van Dinther, O. Korchynskyi and P. ten Dijke, J. Bone Miner. Res., 2011, 26, 1178–1187 CrossRef CAS.
- M. S. Pepper, J. D. Vassalli, L. Orci and R. Montesano, Exp. Cell Res., 1993, 204, 356–363 CrossRef CAS.
- M. Seruya, A. Shah, D. Pedrotty, T. du Laney, R. Melgiri, J. A. McKee, H. E. Young and L. E. Niklason, Cell Transplant., 2004, 13, 93–101 Search PubMed.
- E. Brogi, T. Wu, A. Namiki and J. M. Isner, Circulation, 1994, 90, 649–652 CrossRef CAS.
- J. M. Isner and A. Takayuki, Front. Biosci., 1998, 3, e49–e69 CAS.
- C. Mallet, D. Vittet, J. J. Feige and S. Bailly, Stem Cells, 2006, 24, 2420–2427 CrossRef CAS.
Footnote |
| † Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |