Cell phenotypic changes of mouse connective tissue fibroblasts (L-929) to poly(ethylene glycol)-based gels†
Christine
Strehmel
a,
Zhenfang
Zhang
a,
Nadine
Strehmel
*b and
Marga C.
Lensen
*a
aTechnische Universität Berlin, Department of Chemistry, Nanostrukturierte Biomaterialien, Straße des 17. Juni 124, 10623 Berlin, Germany. E-mail: Lensen@Chem.TU-Berlin.de
bLeibniz Institute of Plant Biochemistry, Department of Stress and Developmental Biology, Weinberg 3, 06120 Halle (Saale), Germany. E-mail: Nadine.Strehmel@ipb-halle.de
First published on 16th May 2013
Abstract
Cellular responses to various gels fabricated by photoinitiated crosslinking using acrylated linear and multi-arm poly(ethylene glycol) (PEG)-based and poly(propylene glycol)-b-poly(ethylene glycol) precursors were investigated. While no protein adsorption and cell adhesion were observed on the hydrophilic PEG-based gels, protein adsorption and cell adhesion did occur on the more hydrophobic gel generated from the block copolymer precursor. Murine fibroblast viability on the poly(ethylene glycol)-based gels was studied in the course of 72 h and the results indicated no cytotoxicity. In a systematic study, extra- and intracellular metabolites of the murine fibroblasts cultured on these PEG-based gels were examined by GC-MS. Distinct intra- and extracellular changes in primary metabolism, namely amino acid metabolism, glycolysis and fatty acid metabolism, were observed. Cells cultured on the polymeric gels induced more intense intracellular changes in the metabolite profile by means of higher metabolite intensities with time in comparison to cells cultured on the reference substrate (tissue culture polystyrene). In contrast, extracellular changes of metabolite intensities were comparable.
Introduction
Synthetic polymers, such as poly(ethylene glycol) (PEG), poly(2-hydroxyethyl methacrylate) (PHEMA) and poly(vinyl alcohol) (PVA), have been frequently used in medicine and pharmacology for fabrication of various devices and for tissue engineering applications due to their versatile chemical and mechanical properties.1 Particularly, polymer gel networks generated from derivatives of PEG, namely hydrogels, have been examined and used for drug delivery and tissue engineering based on their ability to imbibe a large amount of water and their soft and rubbery consistency.2–5 Besides these beneficial properties, PEG-based gels are known to be nontoxic, non-immunogenic and possess anti-fouling properties.6 Due to the unique ability of PEG to block serum protein adsorption as well as the ability to prevent prokaryotic and eukaryotic cell adhesion, PEG can be further used as a surface coating material for the passivation of biomaterial surfaces.7–9 The non-fouling character of such coatings relies on steric repulsion effects between the protein and the hydrated polymer chains at the surface as well as on parameters such as chain length and surface density.10 Further studies proved the prevention of unspecific protein adsorption using an ultrathin star-shaped PEG layer.11Surface wettability or hydrophilicity of the surface and the resulting protein adsorption and cell adhesion onto PEG-based surfaces can be affected by changing the ratio of PEG and poly(propylene glycol) (PPG) block lengths in tri-block copolymer systems PEG-b-PPG-b-PEG.12,13 These tri-block copolymers, namely Pluronics®, are used for drug delivery applications and as emulsion stabilizers in pharmaceutical formulations.14 In this study, we report on the use of polymeric gels that are cross-linked from such tri-block copolymer precursors bearing telechelic acrylate groups.
In order to use polymeric (hydro)gels in biomedical applications, intense nutrient transport to cells as well as free diffusion of waste products are required.15 In this context, metabolomics is a valuable tool to investigate biochemical changes in the surrounding medium and to characterize small molecules.
A variety of different analytical techniques, e.g. nuclear magnetic resonance (NMR) and mass spectrometry coupled to chromatographic techniques such as gas chromatography (GC) and liquid chromatography (LC), can be used to measure the metabolic status of cells.16 Among these, GC-MS is one of the most common analytical techniques to analyze changes within primary metabolism, e.g. low-molecular-weight compounds such as amino acids, lipids and organic acids, since this technique offers a high separation efficiency, high chromatographic resolution and is further a relatively low-cost method.17
So far, metabolomics has been a useful tool for analyzing biofluids, for drug development and for functional genomics of plants.18–20
In the context of biomaterials research, metabolomics has only been used to study cellular responses to smooth and patterned titanium substrates.21 Nevertheless, there are no studies concerning global metabolic changes of cells on synthetic gels such as PEG-based substrates. Thus, we performed untargeted metabolite profiling experiments of murine fibroblasts (L-929) on selected PEG-based substrates and on tissue culture polystyrene using gas chromatography coupled to mass spectrometry (GC-MS) in order to define extracellular and intracellular metabolite changes between the different substrates particularly with regard to cell phenotype in response to nutritional, toxicological and environmental (e.g. surface chemistry) changes.
Since our PEG-based gel compositions have not been reported in the literature yet, cytocompatibility, wettability, plasma protein adsorption and cellular behavior on these substrates were investigated as well.
Experimental section
Acrylation of PEG monomers with hydroxyl end groups
PEG diol (3400 Da) and PEG-b-PPG-b-PEG diol (4400 Da) were purchased from Sigma-Aldrich and 8-arm PEG octaol (15 kDa) from Jenkem Technology USA. Potassium carbonate (K2CO3), sodium chloride (NaCl) and magnesium sulfate (MgSO4) as well as dry dichloromethane (CH2Cl2), petroleum ether and acryloyl chloride were obtained from Sigma-Aldrich. All solvents were of analytical grade.PEG-b-PPG-b-PEG diol (4400 Da) was functionalized to yield the acrylated derivative 3BC as previously described elsewhere.22 Briefly, PEG-b-PPG-b-PEG diol and potassium carbonate were mixed in dry dichloromethane under a nitrogen-based atmosphere. After cooling the solution to 0 °C, acryloyl chloride was added and the mixture stirred at 50 °C for 2 days. Subsequently, the solution was filtered and poured into cold petroleum ether. Following stirring for 10 min, the petroleum ether was decanted. Finally, the crude product was dissolved in dichloromethane and washed with saturated sodium chloride solution. The organic layer was collected and dried with magnesium sulfate overnight. After filtration, the solvent was removed under reduced pressure resulting in a colourless liquid with a moderate yield (64%). 1H NMR (CDCl3) of 3BC: OCH2CHCH3O 1.12 ppm; OCH2CHCH3O 3.38 ppm; OCH2CHCH3O 3.52 ppm; OCH2CH2O 3.63 ppm; (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OCH2 4.30 ppm; C–Htrans 5.83 ppm; CHC 6.15 ppm; C–Hcis 6.42 ppm.
O)OCH2 4.30 ppm; C–Htrans 5.83 ppm; CHC 6.15 ppm; C–Hcis 6.42 ppm.
The acrylation of PEG diol (3400 Da) and 8-arm PEG octaol (15 kDa) was conducted as outlined above for PEG-b-PPG-b-PEG diol (4400 Da) to yield the new acrylated PEG and 8-PEG derivatives. In these cases, the mixtures were stirred at 60 °C for 4 days. A white solid was obtained for PEG and 8-PEG with a yield of 70% and 72%, respectively. NMR analysis revealed complete conversion of hydroxyl-groups into acrylate functions. 1H NMR (CDCl3) of PEG: OCH2CH2O 3.64 ppm; (CO)OCH2 4.31 ppm; C–Htrans 5.83 ppm; CHC 6.15 ppm; C–Hcis 6.42 ppm. 1H NMR (CDCl3) of 8-PEG: OCH2CH2O 3.64 ppm; (CO)OCH2 4.31 ppm; C–Htrans 5.83 ppm; CHC 6.15 ppm; C–Hcis 6.42 ppm.
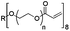
Physicochemical properties of PEG-based precursors are shown in Table 1.
| Material | PEG diacrylate (PEG) | PEG-b-PPG-b-PEG diacrylate (3BC) | 8-arm PEG acrylate (8-PEG) |
|---|---|---|---|
| Structure |
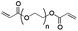
|
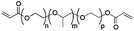
|
|
| Mw [kDa] | 3.4 | 4.4 | 15 |
| Chain length | n ∼ 72 | n + p ∼ 12; m ∼ 57 | n ∼ 40 |
| PEG [%] | 100 | ∼ 30, (∼ 70% PPG) | 100 |
| State at r.t. | Solid | Liquid | Solid |
Gel formation
The liquid, acrylated precursor PEG-b-PPG-b-PEG (3BC) was mixed with 1% of the photoinitiator (PI) Irgacure 2959 (Sigma-Aldrich) in acetone. Subsequently, a mild stream of nitrogen was applied to evaporate the solvent. In order to mix the PI with the solid precursors, i.e. linear PEG (PEG) and 8-arm PEG (8-PEG), aqueous solutions (33 wt%) containing 1% of PI (1 wt% with respect to the amount of the precursor) were prepared. For surface analysis, the solid precursors PEG and 8-PEG were transformed into a melt using an oven.Silicon wafers were cleaned with acetone, activated using hydrogen peroxide–sulphuric acid (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7, v/v) and fluorinated with trichloro(1H, 1H, 2H, 2H-perfluorooctyl)silane (Sigma-Aldrich) prior to use. Subsequently, 50 μl of the selected PEG precursor mixtures were dispensed on fluorinated silicon wafers (CrysTec GmbH), capped with a cover glass (18 mm × 18 mm; Carl Roth GmbH & Co. KG) and exposed to UV light (λ = 365 nm, Vilber Lourmat GmbH) for 30 min using a working distance of 10 cm, in a nitrogen-filled glovebox. Finally, the cured transparent gels were peeled off with tweezers and kept in clean petri dishes (VWR International GmbH) until further use.
:7, v/v) and fluorinated with trichloro(1H, 1H, 2H, 2H-perfluorooctyl)silane (Sigma-Aldrich) prior to use. Subsequently, 50 μl of the selected PEG precursor mixtures were dispensed on fluorinated silicon wafers (CrysTec GmbH), capped with a cover glass (18 mm × 18 mm; Carl Roth GmbH & Co. KG) and exposed to UV light (λ = 365 nm, Vilber Lourmat GmbH) for 30 min using a working distance of 10 cm, in a nitrogen-filled glovebox. Finally, the cured transparent gels were peeled off with tweezers and kept in clean petri dishes (VWR International GmbH) until further use.
Swelling tests
After polymerization, gels were washed in deionised water to remove unreacted precursors and dried in a vacuum oven for 48 h. Subsequently, the dry gels of known weight (Wd) were immersed in a cell culture medium at 37 °C. After appropriate time points, swollen gels were taken out from the medium, blotted dry with a tissue paper and weighed again (Ws) immediately. The swelling degree (SD) was determined according to eqn (1), where Ws is the gel mass after swelling and Wd is the dry gel mass.22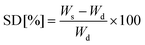 | (1) |
Surface analysis
Water contact angles were measured by the sessile drop method using a goniometer (Data Physics) equipped with a video camera. Two μl of deionised water were deposited onto the material surfaces using the goniometer syringe. Images of droplets were taken at 21 °C. Contact angle values were recorded and averaged from five independent samples.Protein adsorption
In water swollen PEG-based gels PEG, 3BC and 8-PEG (1 cm × 1 cm) were placed in μ-slides (Ibidi GmbH) and incubated with 0.3 ml of a solution of fluorescently labelled albumin (50 μg ml−1 fluorescein isothiocyanate conjugated albumin in phosphate buffered saline, Sigma-Aldrich) for 1 h in a humidified and thermostated incubator (37 °C and 5% of CO2). After the incubation period, samples were washed twice with phosphate buffered saline (PBS) and analysed by fluorescence microscopy using an inverted microscope (Axio Observer.Z1, Carl Zeiss) equipped with a light emitting diode source (Colibri, Carl Zeiss). Pictures were analysed using the Axio Vision software (V 4.8.2, Carl Zeiss).Cell culture
Mouse connective tissue fibroblasts (L-929) were grown in 75 cm2 cell culture flasks (Greiner Bio-One) in a RPMI 1640 medium containing 10% fetal bovine serum and 1% penicillin/streptomycin (all PAA Laboratories GmbH). The cultures were incubated in a humidified incubator (CB 150, Binder GmbH) at 37 °C and 5% CO2 atmosphere as previously described elsewhere.22 For the following experiments L-929 cells were used between passages 9 and 15. The cell culture medium was refreshed every second day.Cell viability
000 cells were seeded into each well and incubated at 37 °C, 5% CO2 atmosphere and 100% humidity. The viability of cells on the three gels PEG, 3BC, and 8-PEG was estimated after 24 h, 48 h and 72 h of incubation. Following incubation, cells were stained with 100 μl of a vitality staining solution containing fluorescein diacetate (stock solution 0.5 mg ml−1 in acetone, Sigma-Aldrich) and propidium iodide (stock solution 0.5 mg ml−1 in Ringer's solution, Fluka). Viable and dead cells were quantified by fluorescence microscopy.
The cell culture medium was not exchanged during the examined period due to the fact that particularly cells seeded on PEG or 8-PEG do not adhere to the polymer surface and would be sucked away. The number of viable and dead cells was counted; values were determined in triplicate.
5000 L-929 cells per 100 μl were seeded into each well of a 96-microwell plate. The plate was pre-incubated for 24 h at 37 °C and 5% CO2. Next, the cell culture medium was aspirated through a pipette. 100 μl of the medium that had been on top of the gels for 24 h, 48 h or 72 h were added on top of the newly seeded cells. After an incubation period of 24 h, 10 μl of Cell Counting Kit-8 (Sigma-Aldrich) containing the water soluble WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] were added to each well. The 96-microwell plate was incubated for 1 h in an incubator and the absorbance of the formazan product (λ450 nm) was measured using a microplate reader (infinite® 200, Tecan). Wells containing medium without cells were used as background control. The number of viable cells was expressed as the relative percentage of the control; values were determined in triplicate. Cells cultured on tissue culture polystyrene (TCPS) were used as control.
Optical and scanning electron microscopy
300 μl medium containing 50000 cells were seeded onto every material. After 24 h, 48 h and 72 h optical images were taken with the Axio Oberserver.Z1 (Carl Zeiss). The diameter of cells on PEG, 3BC, 8-PEG and on tissue culture polystyrene (TCPS) was measured using the Axio Vision software (V4.8.2 Carl Zeiss).
For scanning electron microscopy (SEM) of L-929 cells on 3BC, cells were rinsed with PBS and fixed with formaldehyde (4%, Carl Roth GmbH & Co. KG) for 30 min. Subsequently, the samples were dehydrated in a graded acetone series, dried with critical point drying (CPD 030, Baltec) as previously described and sputtered with gold using a sputter coater (SCD 030, Balzers).23 Scanning electron images were taken with a Hitachi S-520 using an acceleration voltage of 20 kV and a working distance of 10 mm. Pictures were taken using the Digital Image Processing System (2.6.20.1, Point Electronic). Since cell adhesion on the pure PEG-based gels PEG and 8-PEG was minimal, hardly any cells would withstand the rinsing and fixation procedure and therefore these samples were not investigated by SEM.
Metabolite profiling via GC-MS
000 cells per ml) were seeded onto each sample and cultured for a defined time period (24–72 h) in a humidified incubator with 5% CO2 and 37 °C. TCPS was used as reference material.
In the case of the reference material TCPS and the PEG-based gel 3BC, on which cells could adhere, the supernatant was removed and stored at −80 °C until further analysis. The adherent cells were washed with DPBS to remove extracellular metabolites and treated with Trypsin-EDTA (PAA Laboratories GmbH) to detach the cells from the polymer surface. Subsequently, detached cells were resuspended in a RPMI 1640 cell culture medium. The solution was transferred into a falcon tube (VWR International GmbH) and centrifuged at 1300 rpm for 5 min at 4 °C (Eppendorf 5810R). Finally, the cell pellet was washed twice with DPBS. Cells were counted with a haemocytometer (Paul Marienfeld GmbH & Co. KG). Cells from three wells were combined into one Eppendorf-tube (aliquot approx. 1 × 106 cells). The cell pellet was immediately frozen in liquid nitrogen after removing DPBS and stored at −80 °C until further analysis. Experiments were performed in triplicate.
The cell culture medium along with the non-adherent cells on PEG and 8-PEG was collected and centrifuged after 24 h, 48 h and 72 h, respectively. The supernatant was stored at −80 °C until further use, while the cell pellet was washed twice with DPBS. Cells were counted according to the procedure described for 3BC and TCPS. The main preparation steps for the GC-MS based analysis of extracellular and intracellular metabolites are illustrated in Fig. 1.
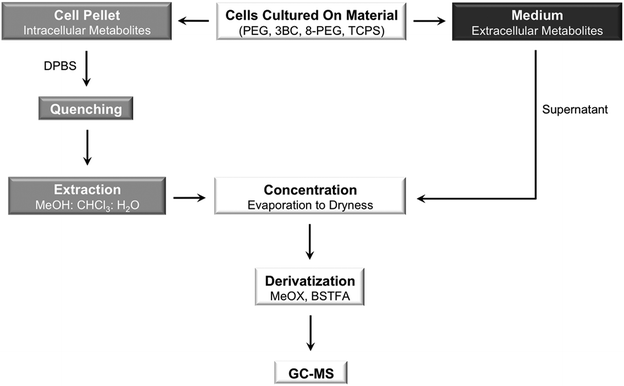 | ||
| Fig. 1 Schematic illustration of the main preparation steps for the analysis of intracellular (cell pellet; depicted in grey) and extracellular (medium; depicted in black) metabolites. Methoxyamine (MeOX) hydrochloride in pyridine and N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) were used for derivatization. | ||
:1:0.1, v/v/v), were added to the frozen cell pellet. Subsequently, the mixture was vortexed, thawed in an ice bath for 10 min and transferred to liquid nitrogen for an additional 10 min. The thawing and freezing were repeated twice. After centrifugation (5 min at 4 °C and with 1350 rpm), the supernatant was transferred into a new Eppendorf-tube. The cell pellet was re-extracted with 400 μl of ice-cold extraction solvent, treated at 70 °C for 15 min and centrifuged.
Prior to derivatisation, 800 μl of the cell pellet extract containing intracellular metabolites were evaporated to dryness in a speed-vac concentrator (RVC 2-25CD plis, CHRIST).
000 rpm for 1 min (Eppendorf 5415R) and the supernatant transferred into a glass vial (Chromatographiezubehör Trott).
Extracellular metabolites were derivatised using the MPS 2XL-Twister Autosampler (Gerstel GmbH & Co. KG). Prior to derivatisation, 10 μl medium were evaporated until dryness in a speed-vac concentrator (RVC 2-25CD plis, CHRIST). 20 μl of methoxyamine hydrochloride in pyridine (20 mg ml−1), 40 μl of BSTFA and 5 μl of the alkane mixture were used. All other conditions were maintained as described for the derivatisation of intracellular metabolites.
Data analysis and statistics
Data analysis was performed with the help of the MetAlign software (version 10/2007) and the TagFinder software (version 4.1), in succession.25 In short, peak intensities above 1000 arbitrary ion current units were imported, aligned and grouped according to their common retention time and mass spectral features. The relevant mass spectral features were identified with the help of the Golm Metabolome Database (gmd.mpimp-golm.mpg.de; version June 2011).For statistics, peak intensities were baseline-corrected and normalized to cell number and abundance of the alkanes; medium samples were only normalized to alkanes. Then, the data were mean-centred per analyte, log 2-transformed and submitted to statistical analysis (two-way analysis of variance, ANOVA: time × material). This was conducted with the help of the Multi Experiment Viewer software (MeV, version 4.7.2).26
Results and discussion
Surface analysis, protein adsorption and cell morphology
PEG-based gels with different swelling properties were fabricated by photoinitiated crosslinking using acrylated linear and multi-arm PEG-based precursors as well as poly(propylene glycol)-b-poly(ethylene glycol) precursors. The crosslinked gels prepared with PEG and 8-PEG can take up more water than the crosslinked gel prepared with 3BC.22 The swelling degree of the gels after 24 h is shown in the ESI (Table S3†).The cellular response and protein adsorption of L-929 cells to the materials PEG, 3BC, 8-PEG and TCPS were investigated (using optical-, scanning electron- and accordingly fluorescence microscopy) and correlated to their wettability, since cell adhesion and protein adsorption onto biomaterial surfaces are known to be influenced amongst others by the wettability of the surface.27 An optimum in protein adsorption and cell adhesion occurs on surfaces with an intermediate wettability ranging from 60 to 80°.28 Consequently, too hydrophilic (water contact angle below 60°) and too hydrophobic substrates (water contact angle above 80°) support protein adsorption and cell adhesion to a lesser extent.
According to our expectation due to the hydrophilic surface, smooth PEG and 8-PEG were anti-adhesive (Fig. 2A), showing a round-shaped cell morphology (17 ± 1 μm in diameter) indicative of the absence of spreading, and cell clustering, Fig. 2B. Furthermore, no protein adsorption was observed on the hydrophilic gels PEG and 8-PEG as expected, Fig. S1.† In contrast, the more hydrophobic substrates TCPS and 3BC did induce protein adsorption, cell adhesion and spreading, resulting in a cell diameter of 63 ± 15 μm and 57 ± 15 μm, respectively.
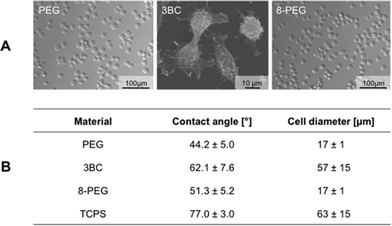 | ||
| Fig. 2 (A) Representative microscopy images showing the morphology of L-929 cells on PEG, 3BC and 8-PEG. (B) Contact angles onto various materials were measured with the help of the sessile drop method. Cell diameter of L-929 cells on PEG, 3BC, 8-PEG and TCPS was obtained by the Axio Vision software. The cell diameter of cells cultured on 3BC and TCPS was measured using the long axis of the cells, while the cell diameter of cells cultured on PEG and 8-PEG was obtained by measuring the cross-section dimension. | ||
In fact, PEG surfaces are very well known to prevent protein adsorption and cell adhesion.29 Recently, we have demonstrated the cell-repellent effect towards a hydrogel prepared from a star-shaped PEG with acrylate end groups due to its hydrophilic surface (static contact angle 43° ± 1°).30 A likewise, relatively low contact angle (52°) was detected for a gel prepared from a linear PEG macromolecule with diacrylate end-groups.31
Changing the ratio of PEG in tri-block copolymer systems increases the hydrophobicity of the surface, and thereby enables more protein adsorption and cell adhesion.32 Thus, the surface of 3BC was indeed found to be more hydrophobic in comparison to PEG and 8-PEG, since the gel prepared from 3BC contains a small PEG (30%) and a large PPG (70%) content in this gel formulation.
Cell viability
For the prospective use of PEG-based gels in biomedical applications, gels require excellent cytocompatibility. Nevertheless, unreacted monomers and photoinitiator fragments can be physically trapped in UV-cured gels. These compounds can be released into the cell culture medium, most likely leading to in vitro toxicity, which can result in cell death, loss of membrane integrity, altered cell morphology and reduced biosynthetic activity.33 Thus, cytocompatibility of PEG, 3BC and 8-PEG was verified after direct cell contact using a live–dead assay and indirectly by a water soluble tetrazolium salt based assay after 24 h, 48 h and 72 h of incubation.The live–dead assay stains viable and non-viable cells by means of fluorescein diacetate and propidium iodide, respectively. Viable cells express green fluorescence under UV illumination, representing transformation of fluorescein diacetate into fluorescein by the action of esterases inside the living cells. Propidium iodide passes through damaged plasma membranes and binds to nucleic acids resulting in red fluorescence.34 Our live–dead staining results indicate that cells remain viable after 24 h, 48 h as well as after 72 h (cell viability is as follows [%]: PEG 98.5 ± 1.3, 97.7 ± 3.3, 98.1 ± 2.7; 3BC 98.9 ± 1.7, 97.9 ± 1.2, 97.4 ± 0.4 and 8-PEG 98.6 ± 2.5, 98.9 ± 1.9, 97.9 ± 3.6), Fig. 3A. Thus, loss of membrane integrity of L-929 cells on PEG, 3BC and 8-PEG did not occur within the time frame of 72 h.
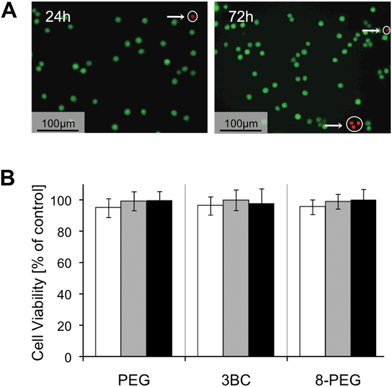 | ||
| Fig. 3 (A) Live–dead assay indicating viable (green) and dead (red) L-929 cells on 8-PEG after 24 h and 72 h. Dead cells are encircled. (B) WST assay illustrating the cell viability of L-929 cells which were incubated with cell culture medium that had been on top of PEG, 3BC and 8-PEG for 24 h (white bar), 48 h (grey bar) and 72 h (black bar). | ||
In a second step, we quantified the number of viable cells via a nonradioactive colorimetric assay. In the course of this assay, a tetrazolium salt is converted by a mitochondrial succinate dehydrogenase into a yellow-coloured soluble formazan. The cell viability of cells incubated with the medium that had been on top of the PEG-based gels was not significantly different from those cultured on the reference material TCPS, Fig. 3B. As expected, L-929 cells showed more than 90% viability after 24 h, 48 h and 72 h, respectively.
Thus, the direct and indirect contact of the PEG-based gels (PEG, 3BC and 8-PEG) with L-929 cells indicated no in vitro cytotoxicity. Any remaining toxic components on the gel surfaces such as photoinitiator fragments and unreacted monomers were most likely washed away with ethanol and Dulbecco's PBS before cell contact. The non-toxic potential of our PEG-based gels corresponds to results obtained for UV-curable gels prepared from linear PEG with diacrylate end groups reported in the literature and star-shaped PEG precursors investigated by our group.35,36 Both groups report high levels of cell viability by live–dead assay after an incubation period of 24 h to 72 h.
Metabolite profiling via GC-MS
Next, we performed untargeted metabolite profiling experiments of murine fibroblasts on smooth PEG-based gels PEG, 3BC and 8-PEG using gas chromatography coupled to mass spectrometry to examine global intra- and extracellular metabolic responses of L-929 cells on distinct substrates at three default time points (24, 48 and 72 h). TCPS was used as reference material. During these studies we identified 16 extracellular metabolites and 15 intracellular metabolites changing in the course of the predefined time frame in at least one of the materials using two-way analysis of variance (two-way ANOVA).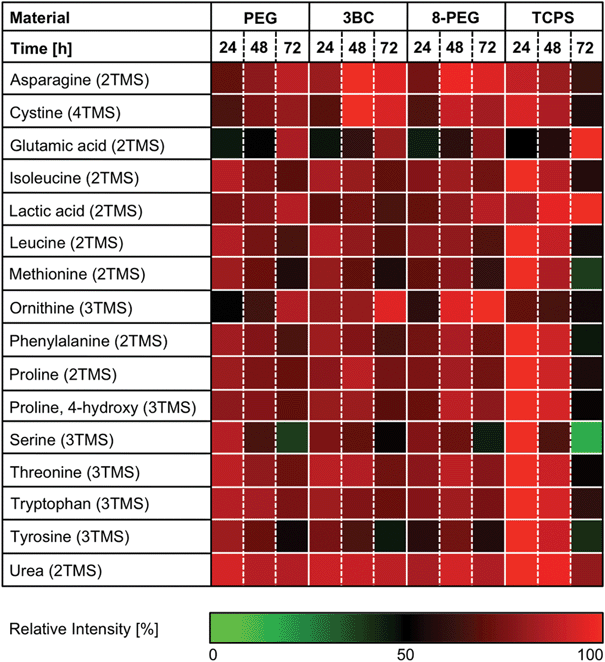 | ||
| Fig. 4 Relative changes of ANOVA positive extracellular analytes. L-929 cells were incubated with PEG, 3BC and 8-PEG for 24, 48 and 72 h. TCPS was used as reference material. The number of derivatised functional groups is given in brackets (TMS: trimethylsilyl). ANOVA positive analytes were maximum normalized. | ||
The analysed medium of cells cultured on PEG, 3BC and 8-PEG exhibited similar metabolic responses in comparison to the medium of cells cultured on TCPS. Consequently, these studies provide valuable information on the overall physiological state of the cells.
As can be seen in Fig. 4, cells incubated on materials prepared with precursor PEG, 3BC, or 8-PEG as well as cells on TCPS released lactic acid continuously into the cell culture medium. This corresponds to previous studies reported in the literature, in which the release of metabolites by L-929 mouse fibroblasts cultured on polystyrene tissue culture flasks into the cell culture medium was determined.37 Furthermore, an incorporation of 13C labelled carbon indicated that lactic acid was derived from glucose. Since a high amount of glucose (2000 mg l−1) is supplemented in our cell culture medium as well, L-929 cells can utilize glucose as an energy source for cellular maintenance and cellular growth.
In contrast, the amino acids isoleucine, leucine, methionine, phenylalanine, threonine and tryptophan decreased gradually in the course of the examined period in all cases. These amino acids are supplied among others in the cell culture medium, since they cannot be synthesized de novo by eukaryotic cells.38 The consumption of essential amino acids by L-929 fibroblasts is consistent with a previous study revealing that the essential amino acids isoleucine, leucine, methionine, phenylalanine, threonine and tryptophan are required for cell metabolism.39,40 Furthermore, these amino acids were proven to be essential for cell growth in tissue culture.41
A similar trend was observed for the non-essential amino acids proline, hydroxyproline, serine and tyrosine. Interestingly, asparagine, cystine, glutamic acid and ornithine increased gradually in the medium for cells cultured on PEG, 3BC and 8-PEG, whereas, surprisingly, the medium of cells cultured on TCPS exhibited a slight decrease of asparagine, cystine and ornithine.
The increasing utilization of cystine, proline and tyrosine as well as the increase of glutamic acid in the cell culture medium has been described for L-929 fibroblasts in chemically defined media.37,39 The accumulation of glutamic acid in the cell culture medium was expected, since glutamic acid is derived from glutamine, which is a component in our cell culture medium.42
High levels of urea were obtained in media of cells cultured on any material, while no clear temporal difference was observed. The secretion of urea into the cell culture media with distinct kinetics is described in the literature, indicating a slight increase during 48 h.43
GC-MS based metabolite profiling led to the identification of 15 analytes belonging to diverse biochemical classes such as amino acids (aspartic acid, glutamic acid, glycine and tyrosine), fatty acids (arachidic acid, myristic acid, palmitic acid, stearic acid), monosaccharides (fructose and glucose 6-phosphate), nucleotides (adenosine monophosphate (AMP)), polyamines (putrescine), polyols (glucitol, myo-inositol), and steroids (cholesterol), Fig. 5 and Table S1.†
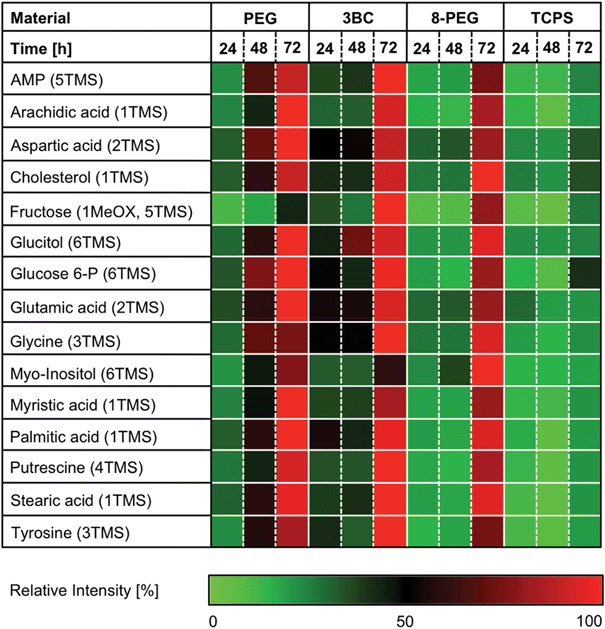 | ||
| Fig. 5 Relative amount of ANOVA positive intracellular analytes. L-929 cells were cultured on PEG, 3BC and 8-PEG for 24, 48 and 72 h. TCPS was used as reference material. The number of derivatised functional groups is given in brackets (TMS: trimethylsilyl, MeOX: methoxyaminated). ANOVA positive analytes were maximum normalized. | ||
The temporal metabolite profile of cells cultured on PEG, 3BC and 8-PEG was similar to the metabolite profile of cells cultured on TCPS, although higher metabolic activity was observed for cells cultured on PEG, 3BC and 8-PEG when compared to cells cultured on TCPS. This observation is surprising, since the cell phenotype of non-adherent cells on PEG and 8-PEG reflects a similar metabolic activity in comparison to adherent and spread cells on 3BC. Presumably, the higher metabolic activity of cells cultured on PEG-based gels is attributed to the more efficient nutrient exchange, by reason that our PEG-based gels swell in the presence of water and the cell culture medium, respectively.
A number of amino acids (i.e. aspartic acid, glutamic acid, glycine and tyrosine) increased gradually in cells cultured on PEG, 3BC and 8-PEG, while only a moderate increase of amino acid intensities within time was observed for cells cultured on TCPS.
The increasing level of glycine and tyrosine in the cell pellet material might have resulted from the consumption of extracellular serine and phenylalanine, respectively. The corresponding, gradual decrease of these amino acids is depicted in Fig. 4. Moreover, accumulated aspartate and glutamate within time can arise from enhanced tricarboxylic acid cycle (TCA) activity, since aspartate and glutamate are derived from the TCA cycle intermediates oxaloacetate and glutamine, respectively.
Similar results concerning the accumulation of aspartate, glycine, glutamate and tyrosine had been observed for mesenchymal stem cells (MSC) cultured for 7 days on various titanium substrates, using liquid chromatography tandem mass spectrometry.21 An increased capacity for protein synthesis on the titanium substrates was proposed as explanation for the accumulation of amino acids. Concerning our materials, cells cultured on PEG-based gels (PEG, 3BC and 8-PEG) possibly increased their capacity for protein synthesis as well.
Amino acids with nitrogen-containing residues can break down into polyamines like putrescine. A cumulative response within time was observed for cells cultured on PEG, 3BC and 8-PEG. A slight increase was also observed for cells cultured on TCPS. Since the cell culture medium was not exchanged at all during the examined period due to non-adherent cells on PEG and 8-PEG, an accumulation of putrescine within time is plausible. However, putrescine is toxic and can inhibit cell proliferation even at low concentration.44 Nevertheless, direct and indirect cell contact with PEG, 3BC, 8-PEG and TCPS indicated no toxicity over the examined period.
Besides the temporal increase of amino acids and polyamines, elevated monosaccharide (fructose, glucose 6-phosphate) and polyol (glucitol, myo-inositol) abundances were observed for cells cultured on PEG, 3BC and 8-PEG, while these metabolites were only slightly increased with respect to cells cultured on TCPS. Elevated intracellular fructose and glucitol had been observed for Chinese hamster ovary (CHO) cells, arising from glucitol pathway, an alternate pathway for glucose.45 Myo-inositol derives from glucose 6-phosphate, which is an intermediate in glycolysis. Interestingly, both analytes accumulate during the examined period.
The relative intensity of adenosine monophosphate (AMP) is gradually increased for cells cultured on PEG, 3BC and 8-PEG as well as slightly enhanced for cells cultured on TCPS. This could be attributed to the labile adenosine triphosphate (ATP), which is an important energy transfer compound within cells. Due to the high reactivity of ATP, we could only detect the breakdown product AMP after derivatisation.46
The cell membrane of eukaryotic cells consists of phospholipids and cholesterol, among others, while fatty acids are part of phospholipids. Relative cholesterol and fatty acid (e.g. arachidic acid, myristic acid, palmitic acid and stearic acid) levels increased within time independent of the material. Since cholesterol is an essential structural component for cell membranes and is further required to ensure membrane stability, permeability and fluidity, spread cells cultured on 3BC and TCPS were expected to exhibit a higher relative cholesterol level in comparison to round cells cultured on PEG and 8-PEG.47 Interestingly, an intense cholesterol accumulation was observed for round cells cultured on PEG and 8-PEG as well as for spread cells cultured on 3BC in comparison to spread cells cultured on TCPS.
Concerning fatty acid accumulation, an elevated phospholipid breakdown resulting in the build-up of fatty acids under conditions of oxygen deficiency has been reported.48 Since oxygen levels in cell cultures are cell density dependent, an increase of fatty acids, as our results show, is comprehensible.49
Conclusions
In conclusion, PEG and 8-PEG containing pure poly(ethylene glycol) are protein and cell repellent due to the hydrophilic surface, while protein adsorption and cell adhesion occur on the more hydrophobic 3BC surface containing a relatively small PEG content. Thus, protein adsorption and cell adhesion can be consequently influenced by changing the ratio of cell repellent material, namely PEG. Our results also indicated that all the studied PEG-based gels are cytocompatible.Our investigations concerning metabolism reveal that GC-MS analysis can be used to evaluate cell responses to biomaterial surfaces. Indeed, our untargeted metabolite profiling results are not coincident with cell phenotypes. In addition, untargeted metabolite profiling revealed similar trends of extracellular metabolites obtained from the medium of cells cultured on PEG, 3BC, 8-PEG and TCPS, respectively. In contrast, relative intensities of intracellular metabolites obtained from round L-929 cells on PEG and 8-PEG as well as spread cells on 3BC differed from those on TCPS. This leads to the conclusion that cells cultured on PEG-based gels have an increased intracellular metabolic activity in comparison to cells cultured on TCPS. In the case of PEG, 3BC and 8-PEG, which swell in the presence of water or cell culture medium, a better diffusion of nutrients and an eventually increased metabolism can be held responsible for this observation.
Acknowledgements
The authors thank Dr J. Lehmann, Fraunhofer Institute for Cell Therapy and Immunology Leipzig, for kindly providing mouse connective tissue fibroblasts (L-929) and greatly acknowledge funding in the form of a Sofja Kovalevskaja Award granted to M. C. Lensen by the Alexander von Humboldt Foundation and funded by the Federal Ministry for Education and Research (BMBF). The authors also thank the Deutsche Forschungsgemeinschaft (DFG) for financial support within the framework of the German Initiative for Excellence the Cluster of Excellence “Unifying Concepts in Catalysis” (EXC 314) coordinated by the Technische Universität Berlin.References
- E. S. Place, J. H. George, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139–1151 RSC.
- N. B. Graham and M. E. McNeill, Biomaterials, 1984, 5, 27–36 CrossRef CAS.
- J. K. Tessmar and A. M. Göpferich, Macromol. Biosci., 2007, 7, 23–39 CrossRef CAS.
- H. Park and K. Park, in Hydrogels and Biodegradable Polymers for Bioapplications, ed. R. M. Ottenbrite, S. J. Huang and K. Park, American Chemical Society, Washington DC, 1996, p. 2 Search PubMed.
- K. B. Keys, F. M. Andreopoulos and N. A. Peppas, Macromolecules, 1998, 31(23), 8149–8156 CrossRef CAS.
- S Zalipsky and J. M. Harris, in Poly(ethylene glycol): Chemistry and Biological Applications, ed. J. M. Harris and S. Zalipsky, American Chemical Society, Washington DC, 1997, p. 1 Search PubMed.
- N. P. Desai, S. F. A. Hossainy and J. A. Hubbell, Biomaterials, 1992, 13(7), 417–420 CrossRef CAS.
- P. D. Drumheller and J. A. Hubbell, J. Biomed. Mater. Res., 1995, 29, 207–215 CrossRef CAS.
- E. W. Merrill and E. W. Salzman, ASAIO J, 1982, 6, 60 Search PubMed.
- S. I. Jeon and J. D. Andrade, J. Colloid Interface Sci., 1991, 142(1), 159–166 CrossRef CAS.
- J. Groll, Z. Ademovic, T. Ameringer, D. Klee and M. Moeller, Biomacromolecules, 2005, 6(2), 956–962 CrossRef CAS.
- M. Amiji and K. Park, Biomaterials, 1992, 13(10), 682–692 CrossRef CAS.
- S. M. O'Connor, A. P. DeAnglis, S. H. Gehrke and G. S. Retzinger, Biotechnol. Appl. Biochem., 2000, 31, 185–196 CrossRef CAS.
- V. Y. Alakhov, E. Y. Moskaleva, E. V. Batrakova and A. V. Kabanov, Bioconjugate Chem., 1996, 7(2), 209–216 CrossRef CAS.
- C.-C. Lin and K. S. Anseth, Pharm. Res., 2009, 26(3), 631–643 CrossRef CAS.
- S. H. G. Khoo and M. Al-Rubeai, Biotechnol. Appl. Biochem., 2007, 47, 71–84 CrossRef CAS.
- S. G. Villas-Bôas, S. Mas, M. Åkesson, J. Smedsgaard and J. Nielsen, Mass Spectrom. Rev., 2005, 24, 613–646 CrossRef.
- O. Beckonert, H. C. Keun, T. M. D. Ebbels, J. Bundy, E. Holmes, J. C. Lindon and J. K. Nicholson, Nat. Protoc., 2007, 2(11), 2692–2703 CrossRef CAS.
- G. Schlotterbeck, A. Ross, F. Dieterle and H. Senn, Pharmacogenomics, 2006, 7, 1055–1075 CrossRef CAS.
- O. Fiehn, J. Kopka, P. Dörmann, T. Altmann, R. N. Trethewey and L. Willmitzer, Nat. Biotechnol., 2000, 18, 1157–1161 CrossRef CAS.
- L. E. McNamara, T. Sjöström, K. E. V. Burgess, J. J. W. Kim, E. Liu, S. Gordonov, P. V. Moghe, R. M. D. Meek, R. O. C. Oreffo, B. Su and M. Dalby, Biomaterials, 2011, 32, 7403–7410 CrossRef CAS.
- S. Kelleher, A. Jongerius, A. Loebus, C. Strehmel, Z. Zhang and M. C. Lensen, Adv. Eng. Mater., 2012, 14(3), B56–B66 CrossRef.
- M. E. Smith and E. H. Finke, Invest. Ophthalmol., 1972, 11(3), 127–132 CAS.
- K. Dettmer, N. Nürnberger, H. Kaspar, M. A. Gruber, M. F. Almstetter and P. J. Oefner, Anal. Bioanal. Chem., 2011, 399, 1127–1139 CrossRef CAS.
- A. Luedemann, K. Strassburg, A. Erban and J. Kopka, Bioinformatics, 2008, 24(5), 732–737 CrossRef CAS.
- A. Saeed, V. Sharov, J. White, J. Li, W. Liang, N. Bhagabati, J. Braisted, M. Klapa, T. Currier, M. Thiagarajan, A. Sturn, M. Snuffin, A. Rezantsev, D. Popov, A. Ryltsov, E. Kostukovich, I. Borisovsky, Z. Liu, A. Vinsavich, V. Trush and J. Quackenbush, BioTechniques, 2003, 34(2), 374–378 CAS.
- P. B. van Wachem, T. Beugeling, J. Feijen, A. Bantjes, J. P. Detmers and W. G. van Aken, Biomaterials, 1985, 6, 403–408 CrossRef CAS.
- Y. Tamada and Y. Ikada, J. Biomed. Mater. Res., 1994, 28, 783–789 CrossRef CAS.
- F. Zhang, E. T. Kang, K. G. Neoh, P. Wang and K. L. Tan, Biomaterials, 2001, 22, 1541–1548 CrossRef CAS.
- M. C. Lensen, V. A. Schulte, J. Salber, M. Diez, F. Menges and M. Möller, Pure Appl. Chem., 2008, 80(11), 2479–2487 CrossRef CAS.
- G. Tan, Y. Wang, J. Li and S. Zhang, Polym. Bull., 2008, 61, 91–98 CrossRef CAS.
- Y. Chang, W.-L. Chu, W.-Y. Chen, J. Zheng, L. Liu, R.-C. Ruaan and A. Higuchi, J. Biomed. Mater. Res., Part A, 2010, 93(1), 400–408 Search PubMed.
- C. J. Kirkpatrick and C. Mittermayer, J. Mater. Sci.: Mater. Med., 1990, 1(1), 9–13 CrossRef CAS.
- F. Dankberg and M. D. Persidsky, Cryobiology, 1976, 13, 430–432 CrossRef CAS.
- M. B. Browning and E. Cosgriff-Hernandez, Biomacromolecules, 2012, 13(3), 779–786 CrossRef CAS.
- V. A. Schulte, M. Diez, M. Möller and M. C. Lensen, Biomacromolecules, 2009, 10(10), 2795–2801 CrossRef CAS.
- K. W. Lanks, J. Biol. Chem., 1987, 262(21), 10093–10097 CAS.
- H. Eagle, Science, 1959, 130, 432–437 CAS.
- J. Mohberg and M. J. Johnson, J. Natl. Cancer Inst., 1963, 31(3), 611–625 CAS.
- J. B. Griffiths, J. Cell Sci., 1970, 6, 739–749 CAS.
- H. Eagle, J. Biol. Chem., 1955, 214(2), 839–852 CAS.
- L. Levintow, H. Eagle and K. A. Piez, J. Biol. Chem., 1957, 227(2), 929–941 CAS.
- R. J. X. Zawada, P. Kwan, K. L. Olszewski, M. Llinas and S. G. Huang, Biochem. Cell Biol., 2009, 87, 541–544 CrossRef CAS.
- G. Stabellini, G. Mariani, F. Pezzetti and C. Calastrini, Exp. Mol. Pathol., 1997, 64, 147–155 CrossRef CAS.
- J. Luo, N. Vijayasankaran, J. Autsen, R. Santuray, T. Hudson, A. Amanullah and F. Li, Biotechnol. Bioeng., 2012, 109(1), 146–156 CrossRef CAS.
- K. E. Weibel, J.-R. Mor and A. Fiechter, Anal. Biochem., 1974, 58, 208–216 CrossRef CAS.
- P. L. Yeagle, Biochim. Biophys. Acta, 1985, 822, 267–287 CrossRef CAS.
- D. A. Ford, Prog. Lipid Res., 2002, 41(1), 6–26 CrossRef CAS.
- S. P. Bruttig and W. L. Joyner, J. Cell. Physiol., 1983, 116, 173–180 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: In the ESI fluorescence micrographs of protein adsorption to pure PEG-gel and to the block copolymer are shown and mass spectral data of identified compounds as well as the swelling degree, bulk elasticity and surface roughness of the crosslinked PEG-gels are listed. See DOI: 10.1039/c3bm60055f |
| This journal is © The Royal Society of Chemistry 2013 |