Paper-based electroanalytical sensing platforms†
Jonathan P.
Metters
,
Said M.
Houssein
,
Dimitrious K.
Kampouris
and
Craig E.
Banks
*
Faculty of Science and Engineering, School of Chemistry and the Environment, Division of Chemistry and Environmental Science, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, Lancs, UK. E-mail: c.banks@mmu.ac.uk; Web: http://www.craigbanksresearch.com Fax: +44 (0)1612476831; Tel: +44 (0)1612471196
First published on 28th November 2012
Abstract
We report the fabrication of paper-based graphite screen printed electroanalytical sensors. Consideration is taken as to determine the most suitable paper-based material enabling reproducible, robust and low cost effective sensor development, after which the paper-based sensor is electrochemically characterised and utilised for the sensing of the important model analytes NADH and nitrite. The analytical performance of the sensors are compared and contrasted to that of commercially available graphite based screen printed sensors fabricated upon traditional polyester substrates. It is found that the paper-based screen printed sensors offers competitive analytical performance when compared with traditional screen printed sensors and other graphite based electrodes offering limits of detection of 1.8 μM and 15.1 μM for NADH and nitrite respectively. As such, these screen printed paper-based analytical devices offer a innovative alternative to the well-established screen printing history of electrodes for use in electroanalytical sensing; the ability to fabricate screen printed electroanalytical sensors which are highly flexible provides the potential for a new class of point-of-care diagnostic devices that are ultra-low-cost, easy to use, and can be designed specifically for use in developing countries.
1 Introduction
Society is in a constant state of growth and development and it is inevitable that demands for sensing devices related to clinical and industrial applications will increase. In order to achieve this, inexpensive and disposable, yet highly accurate and rapid devices are greatly sought. Additionally the portability of such devices is of fundamental importance, especially when the sensing in hard to reach locations is required. One of the fundamental developments which continues to successfully allow for the miniaturisation and low cost electrochemical systems is screen printing and associated fabrication techniques which allows simplification of electrochemical systems allowing the transition from the laboratory to the field to be realised.1–5 These inherent advantages of screen printing allowed the commercialisation of glucose sensors such that diabetics may routinely monitor their blood glucose levels on the spot within their homes.The ease of electrode modification and design through the use of screen printing makes it an exciting and ever evolving technique.5 Those skilled in the ‘art’ are able to fabricate sensors specific to particular requirements utilising the easily modified systems to affect parameters such as improvements in mass transport to impart low detection limits and greater sensitivities and the fabrication of multiple-sensors upon a single strip allows for a range of analytes to be screened within a single sample while still being economical in nature.
One such demographic for which low cost, rapid sensors such as screen printed sensors offer great promise is the developing countries. Typical diagnostic technologies that are successful in the economically developed world often are difficult to use in developing countries due to high costs, with those in developing countries struggling to afford even modestly expensive tests. Furthermore, basic infrastructures such as; reliable power, refrigeration, and trained personnel are often not available in such areas, compounding the case for the requirement of cheap, low cost sensors that can provide in-the-field diagnosis without the necessity for specialised personnel and high cost equipment.6–10 Such sentiments are outlined by the World Health Organisation using the acronym ‘ASSURED’: Affordable, Sensitive, Specific, User-friendly, Rapid and Robust, Equipment-free and Deliverable to end-users.11 The potential for the coupling of screen printing technologies and real-world applications for the detection of disease and illness is perfectly captured through fascinating work by Rusling et al.12 who utilise multielectrode screen printed sensors within an immunoassay system with the study holding great promise for accurate, sensitive multiplexed detection of diagnostic cancer biomarkers.
A further advantage of screen printed technology is the feasibility of printing upon many different substrate materials. Typically screen printed electrode are produced upon ceramics due to the high firing temperatures that are required with some inks but generally plastic substrates are more favoured due to its flexibility (depending on its thickness). Wang and co-workers13 recently reported of the elegant use of screen printing technologies within clothing, “biosensors in briefs”. The group developed durable biosensors that can be printed directly onto clothing, namely underwear, which could potentially enable continuous biomedical monitoring outside hospitals. The tight contact of the clothing and direct exposure to the skin is shown to allow the detection of hydrogen peroxide and the enzyme co-factor NADH; both of which are associated with numerous biomedical processes to be readily monitored.5,13,14
Inspired by this exciting work, we herein report the first study into the utilisation of different paper materials for the fabrication of electrochemical sensors. We note that other work has focussed on paper-based microfluidics15–19 and flow-injection analysis,20 where capillary wicking facilitates the microfluidic flow and recently, a potentiometric sensor has been reported,21 but to date, paper-based electroanalytical sensors produced via screen printing have not yet been reported which allow for highly bendable electroanalytical sensors based upon renewable substrate materials which are ultra-low cost. Consequently, these exciting paper-based electrochemical sensing platforms are physically and electrochemically characterised and explored towards the sensing of key model analytes in this paper.
2 Experimental
All chemicals used were of analytical grade and were used as received without any further purification and were obtained from Sigma-Aldrich. All solutions were prepared with deionised water of resistivity not less than 18.2 MΩ cm.Voltammetric measurements were carried out using a μ-Autolab III (ECO-Chemie, The Netherlands) potentiostat. All measurements were conducted using a screen-printed electrode configuration consisting of a disc-shaped working electrode with a geometric working electrode area of 3 mm diameter. Additionally a saturated calomel electrode (SCE) and platinum wire were externally utilised as the reference and counter electrodes respectively for all measurements.
Screen-printed graphite electrodes were fabricated in-house with appropriate stencil designs using a microDEK1760RS screen-printing machine (DEK, Weymouth, UK). A carbon–graphite ink formulation (Product Code: C2000802P2) (Gwent Electronic Materials Ltd, UK) previously utilised was first screen printed onto pre-selected substrates. For the fabrication of the standard SPE, a polyester flexible film (Autostat, 250 μm thickness) was utilised as the substrate. For the fabrication of the paper based electrodes the substrates utilised included: A4 text and graphic paper 160 g m−2, A4 lined refill pad paper 80 g m−2 and Filter Paper QL 100 for the IP-SPE, RP-SPE, and FP-SPE respectively. This layer was cured in a fan oven at 60° for 30 minutes. This layer produced the working electrodes, contacts and counter electrode. Last a dielectric paste ink (Product Code: D2070423D5) (Gwent Electronic Materials Ltd, UK) was printed to cover the connections and define the 3 mm diameter graphite working electrode. After curing at 60° for 30 minutes the fabricated screen printed electrodes are ready to use. Throughout all electrochemical measurements, an external SCE reference electrode was utilised.
Canal water was sampled at the edge of the canal bank (Rochdale Canal, Oxford Road, Manchester, UK) and collected in a polycarbonate bottle which was washed three times with canal water before being taken back to the laboratory. The sample was stored at room temperature and used within a day of sampling and was simply utilised in place of deionised water for the preparation of specific buffers before electroanalytical measurements were commenced.
Scanning electron microscope (SEM) images and surface element analysis were obtained with a JEOLJSM-5600LV model having an energy-dispersive X-ray microanalysis package.
3 Results and discussion
Substrate selection
Paper-based screen printed sensors were fabricated as described within the Experimental section. With a plethora of paper-based materials available, the decision was made to first test the viability of screen printing using various paper substrates. Of the paper-based substrates available, the four most practical and readily available were selected for printing upon. Those selected included inkjet, ruled pad paper and filter paper, denoted as IP-SPE, RP-SPE and FP-SPE throughout. Successful screen printing of the inks was possible at each of the substrates all allowing for highly reproducible and well defined electrode geometries. No substrate contraction was noted at any of the substrates as is highlighted through the successful screen printing of the subsequent non-conductive dielectric ink layer. Slight curling of the substrates was noted after curing which was remedied through the standard storage procedure of vacuum packing the electrodes to avoid contamination/fouling prior to use.Next, to further examine the feasibility of the paper-based substrates for use as electroanalytical sensors, cyclic voltammetric measurements were conducted in 1 mM ferrocyanide(II)/0.1 M KCl using each of the screen printed sensors, which are depicted in Fig. 1. Additionally comparisons were made using a previously characterised22 graphite based screen printed electrode printed upon a polyester substrate (see Experimental section). Note, cyclic voltammetry using the FP-SPE is not depicted in Fig. 1, as upon introduction to the solution, the highly absorbent paper-based substrate rapidly became saturated with water, with the crucial connections made between the sensor and the potentiostat being compromised due to rapid capillary wicking of the paper. A substrate with such high porosity and therefore rapid absorption of the solution thus deems the electrode unsuitable for use in this intended application area; note that such a material is reported to be useful where capillary wicking is required such as in the detection of glucose in urine where a flow of solution is continuously required.20
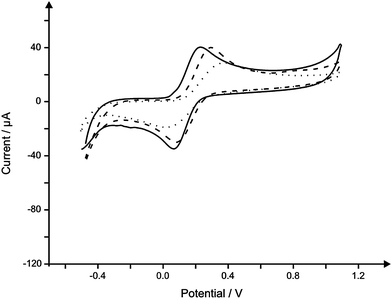 | ||
| Fig. 1 Typical cyclic voltammagrams comparing the response in 1 mM ferrocyanide(II) in 0.1 M KCl using a standard SPE (solid line), IP-SPE (dashed line) and RP-SPE (dotted line). Scan rate: 50 mV s−1. | ||
Characterisation of the screen printed sensors was sought using SEM analysis of the graphite based surface of the working electrode of each. Inspection via SEM analysis reveals a typical graphitic surface at the IP-SPE (see Fig. 2B–D), not dissimilar to that found at the standard SPE which has been fabricated upon polyester substrates.22 As is clear in Fig. 2B, a well-defined working electrode geometry is achieved when printing upon the paper-based substrate. ESI 1† also depicts typical SEM images of the RP-SPE and FP-SPE, initial inspection at a low magnification of ×35 which appear to indicate that although the substrate utilised is clearly much more porous and fibrous in appearance, successful screen printing of the graphite ink is achievable. Critically however, at an increased magnification of ×1000 it is evident that a badly formed working electrode surface had in fact been produced (viz ESI 1D†) far removed from the ideal geometry offered by the standard SPE and also the IP-SPE and RP-SPE. Such a deviation from the ideal geometry observed at the FP-SPE is attributed to the fibrous surface.
 | ||
| Fig. 2 An image demonstrating the ultra flexible and robust nature of the IP-SPE (A) and typical SEM images of the sensor at increasing magnifications; ×250 (B), ×2500 (C) and ×5000 (D). | ||
In comparing the IP-SPE and RP-SPE it is clear that IP-SPE exhibits voltammetric signatures which are most similar to that of the ideal and well characterised response of the standard SPE. As has been highlighted by Wang and co-workers,13 when fabricating screen printed sensors, additional parameters such as flexibility and durability are important factors for consideration. In the case of our paper-based sensors, using paper as a substrate offered excellent durability and flexibility allowing complete folding of the sensor without any detrimental effect upon the physical appearance of the sensor, and more importantly, its electroanalytical performance; see Fig. 2A which shows a complete paper-based sensor which is extremely light and flexible due to the paper as the electrode substrate.
In addition to the observed electrochemical responses (vizFig. 1) and due to the nature of paper-based materials, that is, capillary wicking, out of the three fabricated sensors, IP-SPE was selected for further use within the study due to its excellent electrochemical performance as demonstrated in Fig. 1 with the electrode being able to remain in solution for over 1 hour without electrical connection being compromised (see above). To further reduce capillary wicking and to examine the electrochemical performance of the sensors, the working electrode was isolated using Sellotape on both the back and front of the sensor; note that while this reduced capillary wicking, it will not completely eliminate it (see later, viz.Fig. 4) but allow for reproducible sensors to be obtained.
Characterisation of the paper-based sensor
Electrochemical characterisation was first carried out utilising well-established outer-sphere and inner-sphere redox probes, namely potassium ferrocyanide(II), hexaammine-ruthenium(III) chloride, potassium hexachloroiridate and N,N,N′,N′-tetramethyl p-phenylenediamine (TMPD) the responses of which are shown in Fig. 3. Observation of voltammetric peak height, plotted as peak current (IP) against square root of the applied scan rate (υ1/2) over the range 5–200 mV s−1 was found to be linear at each of the electrochemical probes utilised; potassium ferrocyanide(II) (IP (μA) = 192.6 μA/(V s−1)1/2 + 16.10 μA, R2 = 0.99), hexaammine-ruthenium(III) chloride (IP (μA) = −41.70 μA/(V s−1)1/2 + 2.133 μA, R2 = 0.99), potassium hexachloroiridate (IP (μA) = 80.93 μA/(V s−1)1/2 − 0.27 μA, R2 = 0.99) and TMPD (IP (μA) = 108.8 μA/(V s−1)1/2 + 13.96 μA, R2 = 0.96 at ∼0.25 V and IP (μA) = 125.0 μA/(V s−1)1/2 + 16.96 μA, R2 = 0.99 at ∼0.75 V) suggesting a diffusional process occurring at the electrode surface when using each electrochemical probe.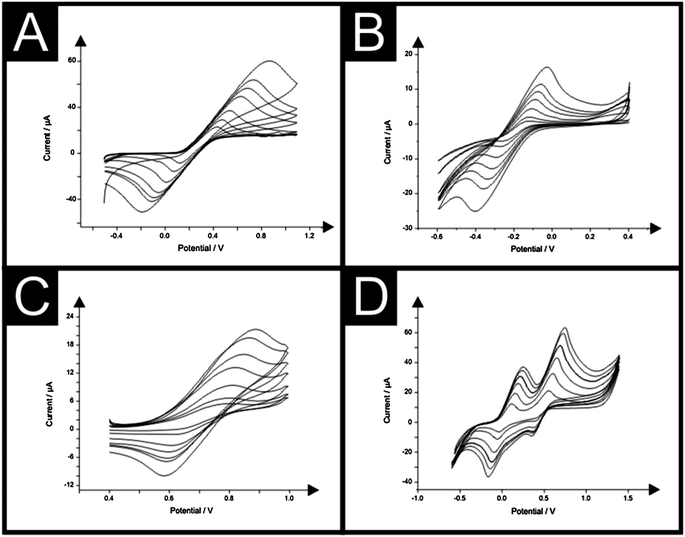 | ||
| Fig. 3 Typical cyclic voltammagrams resulting from increasing scan rates (mV s−1) at the IP-SPE in 1 mM potassium ferrocyanide(II) and 0.1 M KCl (A), 1 mM hexaammine-ruthenium(III) chloride in 0.1 M KCl (B), 1 mM potassium hexachloroiridate in 0.1 M KCl (C) and 1 mM TMPD in 0.1 M KCl (D). Scan rate range in all cases: 5–200 mV s−1. | ||
The heterogeneous electron transfer rate constant, k0, was estimated at the IP-SPE and standard SPE when studied using the outer-sphere electron transfer probe hexaammine-ruthenium(III) chloride. Visual inspection of the cyclic voltammograms depicted in Fig. 3 suggest a quasi-reversible response. The Nicholson method is routinely used to estimate the observed standard heterogeneous electron transfer rate for quasi-reversible systems using the following equation:23
| ψ = k0[πDnυF/(RT)]−1/2 | (1) |
| ψ = (−0.6288 + 0.021X)/(1 − 0.017X) | (2) |
Additionally, the working electrode area of the electrodes was experimentally determined using the Randles–Sevčík equation for an electrochemically quasi-reversible case:22
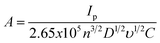 | (3) |
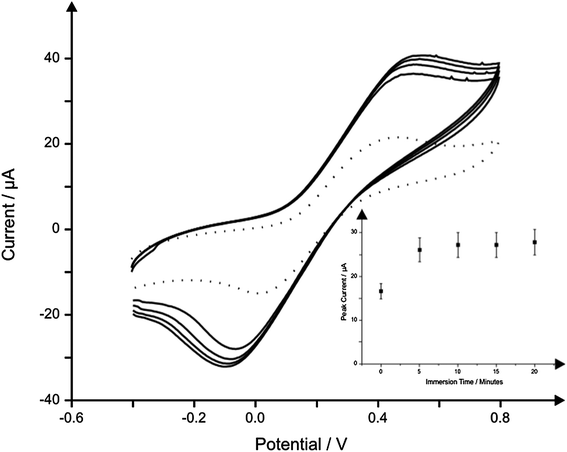 | ||
| Fig. 4 The effect of immersion time in solution upon the observed cyclic voltammetric response using a single IP-SPE in 1 mM potassium ferrocyanide/0.1 M KCl with scans carried out at 5 minutes intervals. The first scan at 0 minutes is depicted using a dotted line. Scan rate: 50 mV s−1. Inset: corresponding plot of peak height versus immersion time. | ||
Electroanalytical performance of the paper-based sensor
Following characterisation of the novel paper-based sensor the task of assessing the sensor's electroanalytical robustness was undertaken, first through determination of the biologically important molecule; NADH. The electrochemical oxidation of NADH (dihydronicotinamideadenine dinucleotide reduced form) to the corresponding oxidised form (NAD+) receives considerable attention owing to its very important role as a cofactor in many enzymatic reactions, and mainly because of the potential application in over 300 NAD+/NADH-dependent dehydrogenase based biosensors.25–30Cyclic voltammetric measurements were undertaken at increasing concentrations of NADH in a pH 7 phosphate buffer using the IP-SPE. Note, a buffer of pH 7 was selected as this pH is typical of biological samples, which would be of interest when monitoring NADH. As is shown in Fig. 5, a peak due to the oxidation of NADH is observed at ∼+0.6 V (vs. SCE), with the peak current found to increase upon additions of NADH (see Fig. 5B). A linear response over the entire analytical range (10–100 μM) was observed at the IP-SPE (I/μA = 1.5 × 10−2 μA μM−1 + 1.0 × 10−1 μA; R2 = 0.99, N = 10). For a comparison, cyclic voltammetric measurements were also carried out over the same concentration range using the standard SPE printing upon a traditional polyester substrate (I/μA = 1.7 × 10−2 μA μM−1 + 6.2 × 10−1 μA; R2 = 0.99, N = 10). Impressively, the IP-SPE was found to perform in accordance with the response observed at a standard SPE as shown in Fig. 5B, demonstrating the excellent performance of the IP-SPE yielding the equivalent electroanalytical performance of the standard SPE whilst offering the added attribute of being ultra-flexible, light weight and low cost. The limit of detection (3σ) for NADH using the IP-SPE was deduced to correspond to 1.8 μM. Such low levels of detection are comparable, not only to that obtained at the standard graphite based screen printed electrode, but also to other screen printed electrode configurations reported within the literature. A limit of detection of 2.5 μM was reported by Hart et al.,31 using a graphite based screen printed sensor which was coupled with meldola blue reagent during fabrication, which is reported to be electrocatalytic towards the NADH detection. Further studies using meldola blue as an electrocatalytic species within screen printed electrodes for the sensing of NADH by Marty and co-workers report a limit of detection of 2 μM at the optimised electrode configuration.32 It is important to note that the incorporation of the mediator meldola blue reagent does have the added attribute of facilitating the electrochemical oxidation of NADH at lower, more facile potentials.31,32 Clearly the limit of detection obtained using the IP-SPE is highly competitive, not only with the standard graphite screen printed electrode used herein, but also with existing literature.
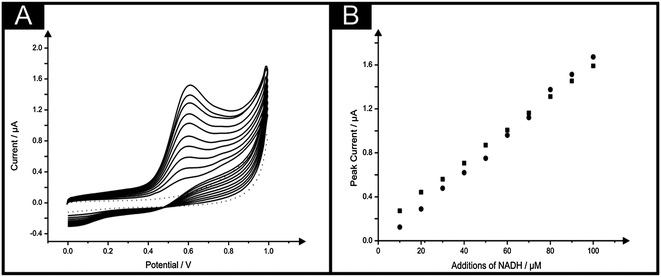 | ||
| Fig. 5 (A) Typical cyclic voltammagrams arising from additions of NADH in to a pH 7 buffer using the IP-SPE. Scan rate: 100 mV s−1. (B) Corresponding calibration plots over the range studied (10–100 μM) NADH using the IP-SPE (squares) and standard SPE (circles). | ||
Following confirmation that the IP-SPE offered comparable sensitivity to the standard SPE for the detection of NADH, the performance of the electrode was tested towards the monitoring of nitrite. Nitrite is reported to be a human health-hazard chemical the excess of which may cause poisoning and its derivatives are also major components in low-level radioactive waste solution.33,34 The excess uptake of nitrite can potentially cause gastric cancer35 and it is therefore necessary to develop a reliable and sensitive sensor to detect nitrite in food, drinking water and environmental samples.30
In the same manner as the IP-SPE was compared and contrasted with a standard SPE for the sensing of NADH, cyclic voltammetric measurements for the sensing of nitrite were carried out. A peak arising from the oxidation of nitrite in a pH 7 phosphate buffer solution was evident at ∼+1.0 V (vs. SCE) using the IP-SPE as seen in Fig. 6A; such a potential value is in agreement with the literature using carbon-based electrodes.36 The oxidation peak was found to increase upon the addition of nitrite over the concentration range 100–1000 μM with a linear response observed throughout. Fig. 6B shows the linear range studied using the IP-SPE with an additional plot demonstrating the response obtained over the same set concentrations of nitrite using a standard SPE. It is clear that the response obtained using the IP-SPE is comparable to that when using a standard SPE with both sensors demonstrating no deviation from linearity over the entire range studied (IP-SPE: I/μA = 4.4 × 10−2 μA μM−1 − 8.0 × 10−1 μA; R2 = 0.99, N = 10, standard SPE: I/μA = 5.3 × 10−2 μA μM−1 − 5.3 × 10−1 μA; R2 = 0.99, N = 10), though a slightly improved response in terms of peak current is seen using the standard SPE. The limit of detection (3σ) for nitrite using the IP-SPE was deduced to be 15.1 μM. Such a limit of detection demonstrates the great potential of the novel paper-based sensor with the fatal dose of nitrite ingestion reported as being between 8.7 μM and 28.3 μM.37–39 Clearly, although the paper-based sensor does not enable the monitoring of nitrite at the ultra-low concentrations of ∼8.7 μM, the limit of detection does allow for concentrations within the fatal range. It is however critical to note that the requirement of such relatively high oxidation potentials could give rise to interference by compounds such as ascorbic and uric acid.
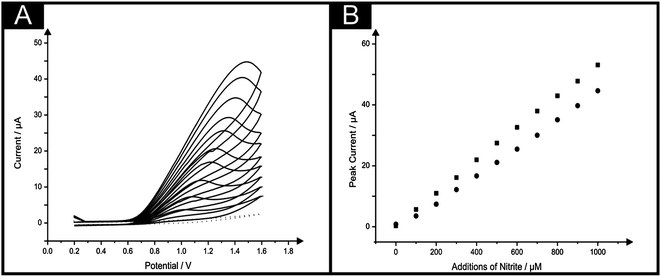 | ||
| Fig. 6 (A) Typical cyclic voltammagrams arising from additions of nitrite in to a pH 7 buffer using the IP-SPE. Scan rate: 100 mV s−1. (B) Corresponding calibration plots over the range studied (100–1000 μM) nitrite using the IP-SPE (circles) and standard SPE (squares). | ||
To gain further insights into the behaviour of the IP-SPE, particularly when used for analytical purposes, further information relating to the effect of the solution upon the electrode surface was sought. To do so, comparisons were made between the cyclic voltammetric measurement of increasing nitrite concentrations over the same range (100–1000 μM) using a single IP-SPE throughout and also a new IP-SPE for each addition of nitrite. Calibration plots corresponding to the peak currents observed through each method are shown in Fig. 7. It is apparent that a slightly greater peak current in observed when using the single IP-SPE over that of a new IP-SPE for each addition throughout the analysis, though this deviation is not sufficient enough to affect the sensitivity of the IP-SPE. Such responses indicate the excellent reproducibility of the paper-based sensors.
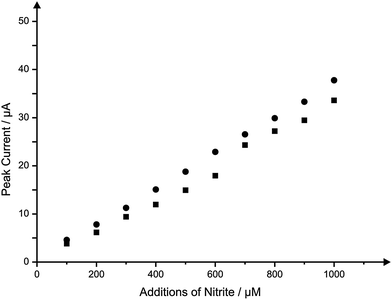 | ||
| Fig. 7 Typical calibration plots resulting from the addition of nitrite in to a pH 7 buffer using a single IP-SPE for the entire concentration range (circles) and a new IP-SPE (squares) for each concentration. Scan rate: 100 mV s−1. | ||
Exploring the applicability of the paper-based sensor towards ‘real-world’ samples
Finally, after concluding that the IP-SPE allowed for comparable analytical performance with that found using a standard SPE, attempts were made to determine whether the performance of the paper-based sensor was hampered in any way when utilised for the sensing of the model analyte within a sample that would typically be presented. Once more the model analyte nitrite was chosen for determination. The media selected was canal water (collected and pre-treated as described within the Experimental section), with the requirement for the monitoring of nitrite typically being within water sources.30Additions of nitrite over the same set range of concentrations (100–1000 μM) were made to a canal water buffer using the IP-SPE. The peak currents recorded upon each addition of nitrite in to the canal water sample are overlaid with a typical calibration plot constructed through measurement under ‘ideal’ buffer conditions using an IP-SPE; Fig. 8 depicts typical calibration plots obtained within the two buffer media resulting from cyclic voltammetric measurements. Clearly, no detrimental effect upon the sensitivity and performance of the electrode is found towards the detection of nitrite within a canal water sample with the resultant calibration plot providing an almost perfect overlay when compared with that arising from additions in to an ‘ideal’ buffer solution. Such findings highlight the robust nature of the sensor with the novel substrate utilised allowing for excellent sensitivity even in difficult sample media.
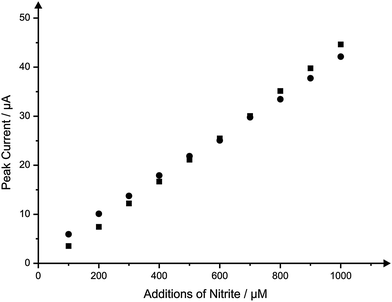 | ||
| Fig. 8 Typical calibration plots corresponding to additions of nitrite into a pH 7 buffer solution (squares) and canal water solution (circles) using the IP-SPE. Scan rate: 100 mV s−1. | ||
4 Conclusions
We have reported the characterisation and application of novel, disposable, single-shot paper-based screen printed sensors with their analytical performance towards the sensing of NADH and nitrite being compared and contrasted with well characterised commercially available screen printed sensors printed upon a traditional polyester substrate. The potential for paper-based substrates for use within screen printing was explored providing insights into the essential parameters requiring consideration when using such material, in particular, the porosity of the substrate material.The paper-based sensors were found to be robust in nature, whilst providing highly reproducible responses when utilised for the model analytes selected. Critically, when contrasted with the commercial available standard screen printed electrode, the paper-based sensors were found to exhibit almost identical electrochemical characteristics to their commercial counter-parts for the sensing of the model analytes NADH and nitrite. Furthermore the paper-based sensors were found to successfully maintain excellent levels of sensitivity even within samples of canal water.
References
- J. P. Hart and S. A. Wring, TrAC, Trends Anal. Chem., 1997, 16, 89 CrossRef CAS.
- J. P. Hart and S. A. Wring, Electroanalysis, 1994, 6, 617 CrossRef CAS.
- M. A. Sirvent, A. Merkoci and S. Alegret, Sens. Actuators, B, 2000, 69, 153 CrossRef.
- A. Morrin, A. J. Killard and M. R. Smyth, Anal. Lett., 2003, 36, 2021 CrossRef CAS.
- J. P. Metters, R. O. Kadara and C. E. Banks, Analyst, 2011, 136, 1067 RSC.
- C. D. Chin, V. Linder and S. K. Sia, Lab Chip, 2007, 7, 41 RSC.
- S. K. Sia, V. Linder, B. A. Parviz, A. Siegel and G. M. Whitesides, Angew. Chem., Int. Ed., 2004, 43, 498 CrossRef CAS.
- A. S. Daar, H. Thorsteindottir, D. K. Martin, A. C. Smith, S. Nast and P. A. Singer, Nat. Genet., 2002, 32, 229 CrossRef CAS.
- P. Yager, T. Edwards, E. Fu, K. Helton, K. Nelson, M. R. Tam and B. H. Weigl, Nature, 2006, 442, 412 CrossRef CAS.
- D. Mabey, R. W. Peeling, A. Ustianowski and M. D. Perkins, Nat. Rev. Microbiol., 2004, 2, 231 CrossRef CAS.
- R. W. Peeling, K. K. Holmes, D. Mabey and A. Ronald, Sex. Transm. Infect., 2006, 50, V1 CrossRef.
- B. V. Chikkaveeraiah, V. Mani, V. Patel, J. S. Gutkind and J. F. Rusling, Biosens. Bioelectron., 2011, 26, 4477 CrossRef CAS.
- Y.-L. Yang, M.-C. Chuang, S.-L. Lou and J. Wang, Analyst, 2010, 135, 1230 RSC.
- J. R. Windmiller and J. Wang, Electroanalysis, 2012 DOI:10.1002/elan.201200349.
- W. Dungchai, O. Chailapakul and C. S. Henry, Anal. Chem., 2009, 81, 5821 CrossRef CAS.
- Z. Nie, C. A. Nijhuis, J. Gong, X. Chen, A. Kumachev, A. W. Martinez, M. Narovlyansky and G. M. Whitesides, Lab Chip, 2009, 10, 477 RSC.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and C. Carrilho, Anal. Chem., 2010, 82, 3 CrossRef CAS.
- Z. H. Nie, C. A. Nijhuis, J. L. Gong, X. Chen, A. Kumachev, A. W. Martinez, M. Narovlyansky and G. M. Whitesides, Lab Chip, 2010, 10, 477 RSC.
- H. Liu and R. M. Crooks, Anal. Chem., 2012, 84, 2528 CrossRef CAS.
- J. Lankelma, Z. Nie, E. Carrilho and G. M. Whitesides, Anal. Chem., 2012, 84, 41 CrossRef.
- M. Novell, M. Parrilla, G. A. Crespo, F. X. Rius and F. J. Andrade, Anal. Chem., 2012, 84, 4695 CrossRef CAS.
- R. O. Kadara, N. Jenkinson and C. E. Banks, Sens. Actuators, B, 2009, 138, 556 CrossRef.
- R. S. Nicholson, Anal. Chem., 1965, 37, 1351–1355 CrossRef CAS.
- I. Lavagnini, R. Antiochia and F. Magno, Electroanalysis, 2004, 16, 505 CrossRef CAS.
- L. Gorton, Encyclopedia of Electrochemistry, Wiley-VCH, Weinhem, 2002, p. 67 Search PubMed.
- H. K. Chenault and G. M. Whitesides, Appl. Biochem. Biotechnol., 1987, 14, 147 CrossRef CAS.
- W. M. Clark, Oxidation–Reduction Potentials of Organic Systems, R. E. Krieger Publishing, Huntington, N.Y., 1972 Search PubMed.
- F. L. Rodkey, J. Biol. Chem., 1955, 213, 777 CAS.
- J. Wang, Electroanalysis, 2005, 17, 7 CrossRef CAS.
- J. P. Metters, R. O. Kadara and C. E. Banks, Sens. Actuators, B, 2012, 169, 136 CrossRef CAS.
- S. D. Sprules, J. P. Hart, S. A. Wring and R. Pittson, Analyst, 1994, 119, 253 RSC.
- A. Vasilescu, T. Noguer, S. Andreescu, C. Calas-Blanchard, C. Bala and J.-L. Marty, Talanta, 2003, 59, 751 CrossRef CAS.
- Drinking Water Standards, US Department of Health, Education and Welfare, Public Health Service, Washington DC, 1988, p. 1962 Search PubMed.
- J. M. Concon, Food Toxicology: Principles and Concepts: Part A, Marcel Dekker, New York, 1988 Search PubMed.
- W. Lijinsky and S. S. Epstein, Nature, 1970, 225, 21 CrossRef CAS.
- Y. Wang, E. Laborda and R. G. Compton, J. Electroanal. Chem., 2012, 670, 56 CrossRef CAS.
- J.-L. Chang and J.-M. Zen, Electroanalysis, 2006, 18, 941 CrossRef CAS.
- US Department of Health, Drinking Water Standards, Washington DC, 1962, p. 47 Search PubMed.
- M. Khairy, R. O. Kadara and C. E. Banks, Anal. Methods, 2010, 2, 851 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ay26396c |
| This journal is © The Royal Society of Chemistry 2013 |