Naproxen causes cytotoxicity and induces changes in polyamine metabolism independent of cyclo-oxygenase expression
Alun
Hughes
,
Fiona R.
Saunders
and
Heather M.
Wallace
*
Division of Applied Medicine, College of Life Sciences & Medicine, University of Aberdeen, Polwarth Building, Foresterhill, Aberdeen. E-mail: h.m.wallace@abdn.ac.uk; Tel: +01224 437956
First published on 1st June 2012
Abstract
The ability of the non-steroidal anti-inflammatory drugs (NSAIDs) to prevent colorectal cancer is well established and has been assumed to be mediated through a cyclo-oxygenase (COX) dependent pathway. In this study we demonstrate that naproxen induces cytotoxicity in a COX-2 null human colorectal cancer cell line (HCT-15) and was equipotent in COX expressing cells (DLD-1). Significant decreases in polyamine content (60–45% of control) were observed in both cell lines, with a corresponding increase in the activity of the two major polyamine catabolic enzymes, spermine/spermidine-N1-acetyltransferase (SSAT) and acetylpolyamine oxidase (APAO). Quantitative PCR confirmed a third catabolic enzyme, spermine oxidase (SMO), was also upregulated in both cell lines following exposure to naproxen. These findings indicate that naproxen is capable of inducing changes in polyamine metabolism that could account for its cytotoxicity and possibly also the chemopreventative actions of the NSAIDs, and further confirms a toxicological effect of the NSAIDs independent of COX inhibition.
Introduction
The chemopreventative effects of the non-steroidal anti-inflammatory drugs (NSAIDs) were first discovered by Kune et al.1 who noted that frequent users of NSAIDs had a 50% reduced risk of developing colorectal cancer. Since this initial discovery, the NSAIDs, mainly aspirin, have been shown to be effective chemopreventative agents against other types of cancer, including breast,2 bladder3 and lung.4 Use of NSAIDs as chemopreventative agents would seem promising, but the gastrointestinal side effects associated with long term use limits their utility.5 Pharmacologically, the NSAIDs act through inhibition of cyclo-oxygenase (COX) of which there are two isoforms; one is expressed constitutively (COX-1), whilst the second is inducible (COX-2). Both produce prostaglandins, which are signalling molecules involved in both normal cellular homeostasis and in the immune response (for review see ref. 6). Classic NSAIDs (such as aspirin, sulindac and naproxen) inhibit both isoforms of COX and are associated with gastrointestinal side effects following prolonged use due to inhibition of COX-1.6 Highly selective inhibitors of COX-2 have been developed which are considerably more potent than classic NSAIDs and there is some evidence to suggest that this new class of drugs is also effective in chemoprevention.7,8 However, they can display an increased incidence of cardiovascular events (reviewed in ref. 9).The majority of colorectal tumours over-express COX-2 compared to normal mucosa.10,11 COX-2 overexpression is sufficient for transformation both in vitro (reviewed in ref. 12), and in vivo models of spontaneous polyp generation in rat,13,14 demonstrating a causative role for COX-2 in tumour development. It has been suggested that inhibition of COX-2 by NSAIDs prevents the increase in prostaglandin concentrations responsible for cellular transformation into a tumour phenotype (reviewed in ref. 15). There are a number of studies that call into question the necessity of COX inhibition for the chemopreventative or toxic effects of the NSAIDs. Several studies have demonstrated that the toxicity of the NSAIDs in vitro cannot be reversed by addition of prostaglandins or their stable analogues.16 Furthermore, sulindac sulfone (a metabolite of sulindac with no COX-2 activity) is equally as potent in vitro as the COX-2 active metabolites of the parent compound,17 and is also capable of preventing azoxymethane-induced colorectal tumours in rats.18 This suggests that an alternative pathway of NSAID-induced chemoprevention must exist.
We have previously suggested that a viable alternative pathway could be through modulation of polyamine biosynthesis.19 Like the prostaglandins, the polyamines are multi-factorial in their regulation of both cell growth and cell death (for review see ref. 20). Additionally, the polyamines are significantly elevated in all tumour types compared to normal tissue.21 The first step in polyamine biosynthesis is the conversion of ornithine to putrescine by ornithine decarboxylase (ODC). In tumourigenesis, ODC activity and polyamine concentrations are increased within the cell and this transforms cells, maintains the altered phenotype, and supports the increased growth rate of tumour cells (reviewed in ref. 22). We, and others, have reported significant changes in polyamine metabolism in vitro in response to NSAID treatment.19,23,24 The addition of exogenous polyamines can be sufficient to prevent the induction of apoptosis and the loss of viable cell number.19,24 This evidence shows that the polyamine metabolic pathway plays an important role in the cytotoxicity of the NSAIDs in vitro, which in turn suggests that modulation of the polyamine pathway in vivo could be a crucial step in the chemopreventative effects of the NSAIDs. The three main polyamine catabolic enzymes, spermine/spermidine-N1-acetyltransferase (SSAT), spermine oxidase (SMO) and acetylpolyamine oxidase (APAO), can be upregulated by cytotoxic stimuli, demonstrating that both the synthesis and catabolism of the polyamines hold potential as therapeutic targets for intervention.25,26
The aim of this study was to examine the influence of COX expression on the effects of naproxen on cytotoxicity and upon polyamine metabolism in human colorectal cancer cells in vitro. Naproxen was chosen as an alternative to aspirin which has been used in most studies so far as naproxen is a reversible competitive inhibitor of COX-1 and 2, while aspirin irreversibly inactivates the enzyme. We show that naproxen is equipotent in both COX−/− HCT-15 cells and COX+/+ DLD-1 cells. We demonstrate that polyamine depletion and export are both caused by naproxen independently of COX expression and this seems to be due to the increased activity of the polyamine catabolic enzymes SSAT and APAO, and the increased mRNA expression of SMO. This evidence indicates that the modulation of polyamine catabolism by the NSAIDs is independent of COX-2 expression and therefore the polyamine pathway is an alternative pathway for the chemopreventative effects of both the NSAIDs and selective COX inhibitors. This is the first study comparing directly the effect of an NSAID on all of the major enzymes and transport of polyamines.
Experimental
Reagents
Naproxen, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Sigma-Aldrich Co. (Poole, Dorset, UK). SC-236 was purchased from Cayman Chemical Company (Estonia, Europe). Cell culture plastics, DMEM growth media, penicillin–streptomycin, foetal bovine serum and trypsin were from Life Technologies (Paisley, Scotland, UK). The DLD-1 and HCT-15 cell lines were obtained from the European Collection of Cell Cultures (Salisbury, Wiltshire, UK).Radioisotopes
3H acetyl coenzyme A (37.0–370.0 GBq mmol−1). DuPont NEN, Boston, MA, USA.14C ornithine dyhydrochloride (1.85–2.22 GBq mmol−1) was from American Radiolabeled Chemicals Inc., St Louis, MO, USA. 1,4(n)-3H putrescine dihydrochloride (0.18–1.48 TBq mmol−1) was from Amersham Life Sciences, Bucks, UK.
Cell culture
Human colon cancer lines (DLD-1 and HCT-15) were grown in DMEM medium, supplemented with 10% (v/v) foetal bovine serum (FBS), 50 units per ml penicillin and 50 μg ml−1 streptomycin. Cultures were maintained in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. A seeding density of 2.0 × 104 cells per cm2 was used for all experiments, and cells were allowed at attach for 4 h, grown for 24 h after which the appropriate treatments were started.Cytotoxicity assay
Cytotoxicity was quantified by the method of Mosmann27 as modified by Denizot and Lang.28 Briefly, cells were grown in the presence of drug and at the desired time, 10 μl of a 5 mg ml−1 sterile solution of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) in DMEM was added to the cells and incubated for 4 h at 37 °C. The formazan salt formed by actively respiring mitochondria was dissolved in dimethyl sulfoxide and detected at 570 and 690 nm.Cell growth determination
Determination of cell number and viability was assessed by the exclusion of Trypan blue. Cells were grown in the presence of drug on 50 mm diameter plates and harvested using trypsin/EDTA solution, then counted on an improved Neubauer haemocytometer. Cells stained blue were counted as non-viable, while those excluding the dye were viable.Protein determination
Total cellular protein content was determined using a modification of the method described by Lowry et al.29 Standards were prepared from a stock solution of BSA (500 μg ml−1) by serial dilution with 0.3 M NaOH to give standards within the range 0–250 μg ml−1 BSA.Polyamine analysis
Cells for polyamine analysis were harvested in perchloric acid as described by Wallace et al.30 and the acid fraction containing the polyamines was stored at −20 °C until analysis by HPLC. Polyamines and their monoacetyl derivatives were separated and quantified by a modification of the HPLC method of Seiler and Knodgen31 as modified by Wallace et al.30Ornithine decarboxylase enzyme activity
ODC activity was measured by a modification of the method of Coleman and Pegg.32 Briefly, the amount of radiolabelled CO2 released from 14C-ornithine in one hour by cleavage from ornithine to putrescine by ODC was measured by collection in benzethonium hydroxide.Polyamine oxidase and spermidine oxidase activity
APAO or SMO activity was determined by the amount of hydrogen peroxide generated in 30 min caused by the oxidation of N1-acetylspermine or spermine respectively. The method is described in full elsewhere.19Spermidine/spermine acetyltransferase activity
SSAT activity was measured according to the method of Wallace and Evans.33 Cells were lysed in Tris buffer (10 mM Tris HCl pH 7.5 @ 4 °C with 1 mM EDTA and 2.5 mM dithiothreitol) then ultracentrifuged at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 gav for 1 h 10 min at 4 °C. The supernatant was transferred to eppendorf tubes with 10 μl 30 mM spermidine and 10 μl 1 M Tris HCl (pH 7.8 at 37 °C), 10 μl 250 μM acetyl-CoA and 10 μl 0.33 μCi 3H-acetyl-CoA. The reaction was terminated after 10 min with 20 μl 1 M hydroxylamine. 30 μl of each supernatant were dried on Whatman P81 paper discs, counted in a scintillation counter and calculated as pmol N-acetylspermidine formed per min per mg of protein.
000 gav for 1 h 10 min at 4 °C. The supernatant was transferred to eppendorf tubes with 10 μl 30 mM spermidine and 10 μl 1 M Tris HCl (pH 7.8 at 37 °C), 10 μl 250 μM acetyl-CoA and 10 μl 0.33 μCi 3H-acetyl-CoA. The reaction was terminated after 10 min with 20 μl 1 M hydroxylamine. 30 μl of each supernatant were dried on Whatman P81 paper discs, counted in a scintillation counter and calculated as pmol N-acetylspermidine formed per min per mg of protein.
Morphological characterisation of cell death
Cells detached into the medium were carefully retained, and those attached to the plates were harvested with trypsin/EDTA solution. Cells were washed with PBS and pellets resuspended in 4% (v/v) formaldehyde in PBS. Cytospins of total cells (cells attached pooled with cells in medium) were prepared on a Shandon Cytospin at 28 gav for 5 min. Cell nuclei were stained with 1 μg ml−1 DAPI (4′,6-diamidino-2-phenylindole) in complete PBS. Samples were counted blind with 100 cells counted in five different fields per slide. Percentage apoptosis was scored on the morphological features of apoptosis, including chromatin condensation and cell shrinkage, and expressed as a percentage.Quantitative PCR
Total RNA was extracted using Trizol (Invitrogen), as per the manufacturer's protocol and quantified on a Nanodrop spectrophotometer (Thermo Scientific). Two micrograms of total RNA was reverse-transcribed to cDNA using 2.5 μM random hexamer primers, 500 μM dNTPs and 200 U SuperScript II (Life Technologies) and adjusted to a final volume of 100 μl with molecular biology grade water. Intron-spanning FAM-labelled hydrolysis probe assays for SSAT, ODC, SMOX and APAO were designed using the Universal Probe Library (see Table 1; Sigma-Aldrich), and a pre-validated VIC-labelled hydrolysis probe assay for GAPD was purchased from Applied Biosystems. Quantitative PCR was performed on a Roche LightCycler480 using 20 μl reaction volumes consisting of 10 μl of Roche LightCycler480 Probes Master enzyme mix, 400 nM forward and reverse primers and 200 nM of each relevant Universal Probe Library probe, or 1 μl of the Applied Biosystems GAPD endogenous reference gene assay. Amplification was performed for 45 cycles of 10 s at 95 °C and 20 s at 60 °C, after an initial 5 min enzyme activation step at 95 °C. Relative quantification of target genes was normalised to reference gene expression using curves of serially diluted cDNA to generate estimates of qPCR efficiency.| Gene | Primer sequence | UPL probe number |
|---|---|---|
| ODC | Left: aaaacatgggcgcttacact | 34 |
| Right: tggaattgctgcatgagttg | ||
| SSAT | Left: gactgttcaagatcgacaagga | 82 |
| Right: tgtcatctacagcagcactcc | ||
| AZ1 | Left: gaggggagcaaggacagc | 2 |
| Right: cggttcttgtggaagcaaat | ||
| APAO | Left: gtcaccgtgcccttaggtt | 29 |
| Right: tcccaaagcctatcttcctg | ||
| SMOX | Left: tcacggatgtcactgtgctt | 5 |
| Right: ctcaaaggtggcgtgtcc |
Results
HCT-15 cells have been characterized previously as lacking both isoforms of COX16 and we confirmed the absence of COX-2 by immunohistochemistry (Fig. 1b). DLD-1 cells stained positive for COX-2 protein (Fig. 1a), but less intensely than a third colorectal cancer cell line, HT-115 (data not shown), indicating that whilst DLD-1 cells express COX-2, they do not appear to substantially overexpress the protein. As also observed by Hanif et al.16 HCT-15 cells did not express mRNA transcripts for COX-1 or COX-2 as confirmed by RT-qPCR, whilst transcripts for both were detected in DLD-1 cells (data not shown).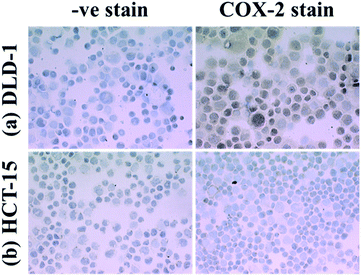 | ||
| Fig. 1 Immunohistochemical staining for COX-2 in DLD-1 and HCT-15 cells. | ||
Having confirmed the expression of COX was absent in HCT-15, we compared the toxicity of naproxen in both cell lines. IC50 values were calculated from MTT cytotoxicity assays following 48 h and 96 h exposure. As can be seen in Fig. 1, naproxen reached IC50 values of 2.5 mM and 1 mM in both the COX-2 expressing DLD-1 cells and the COX-2 null HCT-15 cells respectively. Naproxen reached 100% toxicity by 5 mM in HCT-15 cells, and 10 mM in DLD-1 cells, confirming that the effect is, at high concentrations at least, cytotoxic and more evident in the HCT-15 cells (Fig. 2). The duration of exposure was chosen based on previously published results,19 which indicated toxicity from a single dose of naproxen takes between 48–96 h to have significant effects on cell growth and toxicity.
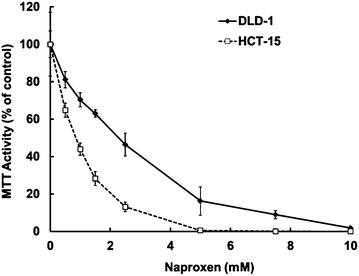 | ||
| Fig. 2 Effects of naproxen on the growth of DLD-1 and HCT-15 cells. | ||
As MTT assays are a surrogate measure of viable cell number and may often underestimate the effect of cytotoxic agents, we performed growth curve and fixed time-point analysis of cell number, cell viability and apoptosis using IC50 and IC90 doses of naproxen calculated from the 96 h MTT assays (Fig. 3). Naproxen appeared to induce a growth inhibitory or cytostatic response at IC50 concentrations in time course experiments (data not shown). In accord with this observation, no decrease in cell viability or increase in the morphological signs of apoptosis was observed following 96 h exposure to IC50 concentrations of naproxen in either DLD-1 or HCT-15 cells (Fig. 3b and c). Increased concentrations of naproxen (equivalent to IC90 values obtained from MTT assays) decreased viable cell number further (Fig. 3a) but did not alter the viability of adhered cells, suggesting a lack of necrotic cell death (Fig. 3b). However, naproxen caused significant levels of apoptosis in both DLD-1 and HCT-15 cells at the elevated IC90 concentrations, as measured by DAPI staining (Fig. 3c) and confirmed by caspase-3 activation and DNA fragmentation ELISAs (data not shown).
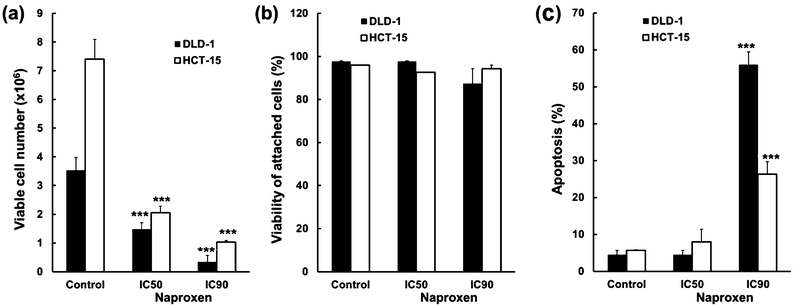 | ||
| Fig. 3 Cell number, viability and apoptosis following naproxen treatment; ***P < 0.001. | ||
We have previously reported that NSAIDs decrease ODC activity and intracellular polyamine content and upregulate polyamine export.20 We examined the effect of naproxen treatment on COX-null HCT-15 cells and observed that both polyamine content decreased and polyamine export increased as we had previously observed in the DLD-1 cells (Table 2). However, these changes were associated with an approximate doubling of ODC activity in the HCT-15 cells (Table 2) where we had previously observed a 50% decrease in activity in the DLD-1 cells.19
| ODC activity (pmol CO2 h−1 mg−1) | Putrescine (nmol mg−1) | Spermidine (nmol mg−1) | Spermine (nmol mg−1) | Total polyamine content (nmol mg−1) | Polyamine export (% exported to medium) | |
|---|---|---|---|---|---|---|
| ***P < 0.001. | ||||||
| Control | 139.5 ± 16.0 | 1.1 ± 0.3 | 6.2 ± 0.9 | 16.5 ± 4.3 | 23.8 ± 3.1 | 18.7 ± 1.1 |
| Naproxen | 236.7 ± 38.4 | 1.4 ± 0.5 | 1.8 ± 1.0 | 7.6 ± 1.9 | 10.8 ± 1.4*** | 33.9 ± 3.4*** |
As polyamine content decreased despite the differential effects of naproxen on ODC activity, we postulated that increased catabolic enzyme activity was responsible for the polyamine depletion observed. Changes in polyamine content and ODC activity were observed after 48 h (Table 2), suggesting that modulation of the polyamine pathway may be occurring by alterations in gene expression of the regulatory enzymes. By using RT-qPCR, we examined expression of ODC, SMOX, APAO and SSAT in response to naproxen in both HCT-15 and DLD-1 cells (Fig. 4). With the exception of 48 h treatment in DLD-1 cells, no significant alteration in ODC expression was observed in response to naproxen in either cell line. An increase in ODC expression was observed at 48 h in naproxen treated DLD-1 cells but did not correlate to the decreased enzyme activity we previously observed at the same timepoint,19 suggesting a post-translational mechanism was regulating ODC activity. As antizyme regulates ODC activity, we measured expression of AZ in both DLD-1 and HCT-15 cells and found no difference in expression at the mRNA level in response to naproxen (data not shown). Expression of APAO was variable, with a trend to decreased expression in response to naproxen in HCT-15 cells, whilst SSAT expression tended to increase in response to naproxen in both cell lines, reaching significance at a few timepoints (Fig. 4). The strongest response at the mRNA level was from SMOX expression, which was significantly elevated several fold in response to naproxen in DLD-1 cells at all time points measured, with similar levels of increase observed in HCT-15 cells between 8–24 h (Fig. 4). Taken together, this data indicates that the largest and most reproducible changes in polyamine metabolism at the mRNA level due to naproxen are via increased SSAT and SMOX expression.
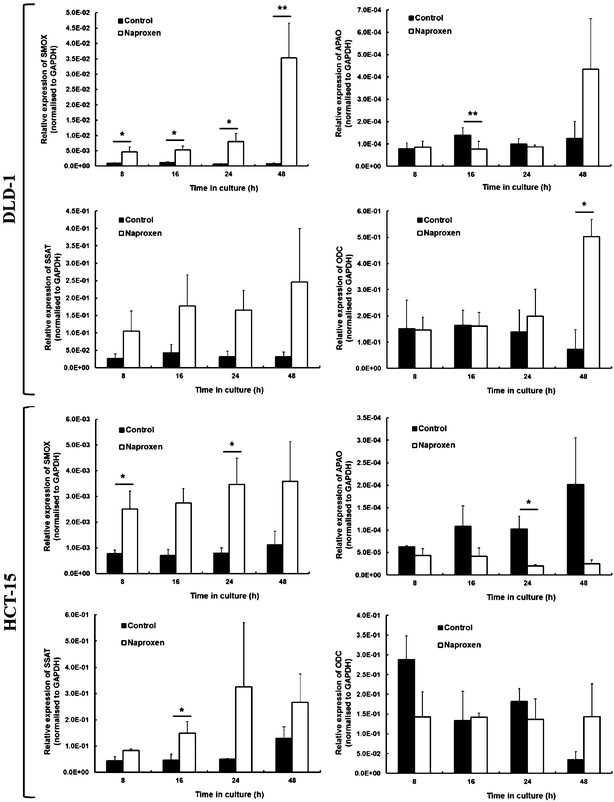 | ||
| Fig. 4 Expression of ODC, SSAT, APAO and SMOX in DLD-1 and HCT-15 cells following treatment with naproxen; *P < 0.05, **P < 0.01. | ||
In accordance with the RT-qPCR results, SSAT activity was significantly increased in both HCT-15 and DLD-1 cells 24 h following naproxen exposure (Table 3). Whilst there was a trend for continued elevated activity in HCT-15 cells, SSAT activity was still significantly 3.5 fold higher in treated DLD-1 cells at 48 h (Table 3). APAO activity did not significantly increase in DLD-1 cells at either timepoint examined, but did significantly double in activity at 24 h in response to naproxen, which appeared to remain elevated by 48 h but did not reach statistical significance (Table 3). Basal levels of SSAT and APAO were markedly lower in DLD-1 cells than measured in the HCT-15 cells: Basal SSAT activity in DLD-1 cells was 110 times lower than the HCT-15 cells at 24 h, whilst basal APAO activity was 42 times lower in DLD-1 cells at the same time point. Despite the differences in basal activity, naproxen caused significant increases in SSAT activity at the IC50 doses tested, albeit peaking earlier in the HCT-15 cells than in the DLD-1 cells. The increase in SSAT activity observed in the HCT-15 cells was transient with a peak at 24 h, as levels had returned to basal levels by 48 h. The overall net effect of the NSAID-induced changes in polyamine metabolism favours retro-conversion and excretion of the polyamines from within the cell, as can be observed in the up-regulation of SSAT and APAO in conjunction with the decrease in intracellular polyamines and an increase in the export of 3H-labelled polyamines from both DLD-1 and HCT-15 cells (Table 2). SMO activity in both DLD-1 and HCT-15 cells was low and highly variable when detectable (Table 3), suggesting that the levels of mRNA detected does not relate proportionally to the generated protein or its subsequent activity. SMO activity is measured by a simple substitution of the substrate used in the APAO enzyme assay, suggesting the assay system is working but SMO activity is too low in these cells to reliably measure.
| SSAT activity (pmol N-acetylspermidine min−1 mg−1) | APAO activity (pmol H2O2 min−1 mg−1) | SMO activity (pmol H2O2 min−1 mg−1) | ||||
|---|---|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | |
| *P < 0.05, ***P < 0.001, n.d. = not determined as below limit of detection. | ||||||
| DLD-1 | ||||||
| Control | 0.08 ± 0.01 | 0.69 ± 0.01 | 1.2 ± 0.3 | 2.9 ± 1.3 | n.d. | 2.5 ± 2.0 |
| Naproxen | 0.42 ± 0.11* | 2.42 ± 0.29*** | 1.4 ± 0.6 | 4.2 ± 0.7 | n.d. | 4.0 ± 2.0 |
| HCT-15 | ||||||
| Control | 8.87 ± 1.28 | 2.88 ± 0.49 | 50.5 ± 4.2 | 84.1 ± 12.1 | n.d. | 3.6 ± 3.4 |
| Naproxen | 30.09 ± 4.24* | 5.04 ± 1.01 | 111.4 ± 12.2*** | 179.7 ± 22.4 | n.d. | 4.3 ± 2.8 |
Discussion
Hanif et al. provided compelling evidence for a COX-independent mechanism of NSAID action by utilizing the HCT-15 cell line: a completely COX-1−/− and COX-2−/− null human colorectal cancer cell line that produced no prostaglandins.16 This cell line was shown to be as equally sensitive to the effects of sulindac and piroxicam in vitro as several COX-2 expressing colorectal cancer cell lines, and our study extends this work to confirm there is no selectivity between COX-2 expressing or non-expressing cells with a third NSAID, naproxen. The ability of naproxen to induce toxicity independently of COX expression indicates that alternative cellular targets for NSAID binding may exist. Recent studies have revealed the existence of one such target, the so-called ‘NSAID activated gene’ product (NAG-1), which has been shown to be over-expressed in prostate cancer and colorectal cancer34,35 and is inducible by chemopreventative agents in a p53 dependent manner.36,37 NAG-1 is known to have anti-tumourigenic properties, thought to be linked to the down-regulation of COX-2,35 but as both DLD-1 and HCT-15 cells express mutant forms of p53 it is unlikely that naproxen is inducing changes in polyamine pathway enzymes via this mechanism. There is much potential for exploration in this area, but it demonstrates that NSAIDs can induce key changes within the cell that may instigate anti-tumour effects independent of their established direct actions upon COX. It is unknown as of yet if components of the polyamine pathway are downstream of NAG-1 or if selective COX-2 inhibitors are more potent inducers of NAG-1 than the NSAIDs, but this will make for interesting future investigation.Our study extends the understanding of the effects of NSAIDs upon polyamine metabolism in the absence of COX-2 expression, by characterising the full catabolic and anabolic pathway of polyamine metabolism at both the expression and activity level. In this study partial depletion of polyamines by naproxen correlated with growth inhibitory and cytotoxic effects, and it is well established that depletion of intracellular polyamine content can prevent the proliferation of cells, particularly cancer cells.38 Both cell lines displayed increased catabolic activity following NSAID treatment, whilst differential regulation of ODC activity was observed between the two lines. In conjunction with the observed increase in polyamine export and the effects on gene expression of polyamine metabolic enzymes, this suggests that the depletion observed was more likely due to increased catabolism than decreased biosynthesis.
We demonstrate that the two main polyamine catabolic enzymes, APAO and SSAT, are substantially elevated in enzyme activity in response to naproxen treatment. Although we did observe substantial and significant increases in the gene expression of SMOX, this did not translate favourably to activity. SMO activity was low and highly variable, unlike the activity of APAO, suggesting either a less important role for SMO in the polyamine catabolic response to naproxen treatment or a need for a more sensitive method of measuring SMO activity. Both SMO and APAO generate H2O2 as a by-product of breakdown of their polyamine substrates,39,40 so persistent increases in catabolic activity would both deplete intracellular polyamines and may also generate significant quantities of H2O2. SSAT activity is in turn stimulated by H2O2 and this may provide a critical cell-death signal-generating loop.40
Many anti-cancer agents have been shown to substantially induce SSAT activity41 and it has previously been speculated that the ability of an agent to induce SSAT is related to its effectiveness as a cytotoxic agent.42 The ability of the NSAIDs to stimulate polyamine catabolism may provide an explanation as to the effectiveness of the drugs as chemopreventative agents. Inhibition of ODC activity would prevent the signals associated with transformation to a tumour phenotype whilst stimulation of catabolism could initiate cell death and remove pre-cancerous cells before a tumour can be established. Our study would suggest, however, that polyamine catabolism (mediated in particular by SSAT and APAO) plays a larger role in NSAID-induced polyamine depletion in colorectal cancer cells in vitro, due to the differential effects observed on ODC activity in the DLD-1 and HCT-15 cells.
The concept of modulating polyamine metabolism as a chemopreventative strategy is not new and has previously centered on the ability of many preventative agents to inhibit ODC and prevent the increase of intracellular polyamine levels.38 Indeed, recent work has suggested that polymorphisms in the ODC gene may influence an individual's response to NSAID-induced chemoprevention, plus it is well known that increased ODC expression and activity is sufficient to cause cellular transformation.43,44 This strategy means that the classical ODC inhibitor DFMO may yet see resurgence as a potent chemopreventative agent despite its previous failings as a chemotherapeutic treatment for colon cancer.45 A recent trial examining the chemopreventative potential of DFMO in combination with the COX-2 selective inhibitor Celecoxib was stopped early due to high levels of efficacy.45,46 This study suggests that inhibition of polyamine biosynthesis sensitizes cells to the effects of a selective COX-2 inhibitor, although it is unknown as of yet if this would be as effective in combination with the classic NSAIDs. If so, our work would suggest this treatment would be effective due to the combination of inhibition of polyamine biosynthesis by DFMO and the stimulation of catabolism by the NSAIDs.
Conclusions
Naproxen can induce toxicity in vitro independently of COX-2 expression. Polyamine depletion is induced regardless and appears to be facilitated mainly by increased polyamine catabolism. This decrease in polyamine content appears to be causative to the apoptosis induced by the NSAIDs, suggesting that the NSAIDs may exert their effects in part through a COX-independent mechanism dependent on polyamine depletion.Acknowledgements
We would like to thank the European Social Fund, TENOVUS, Sir Samuel Scott of Yews Trust, University of Aberdeen and the Grampian University Hospital NHS Trust for the financial support of this work.References
- G. A. Kune, S. Kune and L. F. Watson, Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne colorectal cancer study, Cancer Res., 1988, 48, 4399–404 CAS.
- S. A. Khuder and A. B. Mutgi, Breast cancer and NSAID use: a meta-analysis, Br. J. Cancer, 2001, 84(9), 1188–92 CrossRef CAS.
- J. E. Castelao, J. M. Yuan, M. Gago-Domingue, M. C. Yu and R. K. Ross, Non-steroidal anti-inflammatory drugs and bladder cancer prevention, Br. J. Cancer, 2000, 82, 1364–1369 CrossRef CAS.
- S. A. Khuder, N. A. Herial, A. B. Mutgi and D. J. Federman, Nonsteroidal anti-inflammatory drug use and lung cancer: a meta analysis, Chest, 2005, 127, 748–54 CrossRef.
- D. M. McCarthy, Comparative toxicity of nonsteroidal anti-inflammatory drugs, Am. J. Med., 1999, 107, 37–47S CrossRef.
- A. Lupulescu, Prostaglandins, their inhibitors and cancer, Prostaglandins, Leukotrienes Essent. Fatty Acids, 1996, 54, 83–94 CrossRef CAS.
- B. S. Reddy, C. V. Rao and K. Seibert, Evaluation of cyclooxygenase-2 inhibitor for potential chemopreventive properties in colon carcinogenesis, Cancer Res., 1996, 56, 4566–9 CAS.
- B. S. Reddy, Y. Hirose, R. Lubet, V. Steele, G. Kelloff, S. Paulson, K. Seibert and C. V. Rao, Chemoprevention of colon cancer by specific cyclooxygenase-2 inhibitor, celecoxib, administered during different stages of carcinogenesis, Cancer Res., 2000, 60, 293–7 CAS.
- M. M. Bertagnolli, Chemoprevention of colorectal cancer with cyclooxygenase-2 inhibitors: two steps forward, one step back, Lancet Oncol., 2007, 8, 439–43 CrossRef CAS.
- O. C. Trifan and T. Hla, Cyclooxygenase-2 modulates cellular growth and promotes tumorigenesis, J. Cell. Mol. Med., 2003, 7, 207–22 CrossRef CAS.
- S. L. Kargman, G. P. O'Neill, P. J. Vickers, J. F. Evans, J. A. Mancini and S. Jothy, Expression of prostaglandin G/H synthase-1 and -2 protein in human colon cancer, Cancer Res., 1995, 55, 2556–9 CAS.
- F. A. Sinicrope, Targeting cyclooxygenase-2 for prevention and therapy of colorectal cancer, Mol. Carcinog., 2006, 45, 447–454 CrossRef CAS.
- M. Oshima, J. E. Dinchuk, S. L. Kargman, H. Oshima, B. Hancock, E. Wong, J. M. Trzaskos, J. F. Evans and M. M. Taketo, Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2), Cell, 1996, 87, 803–809 CrossRef CAS.
- R. N. DuBois, A. Radhika, B. S. Reddy and A. J. Entingh, Increased cyclooxygenase-2 levels in carcinogen-induced rat colonic tumors, Gastroenterology, 1996, 110, 1259–1262 CrossRef CAS.
- J. B. Tuynman, M. P. Peppelenbosch and D. J. Richel, COX-2 inhibition as a tool to treat and prevent colorectal cancer, Crit. Rev. Oncol. Hematol., 2004, 52, 81–101 CAS.
- R. Hanif, A. Pittas, Y. Feng, M. I. Koutsos, L. Qiao, L. Staiano-Coico, S. I. Shiff and B. Rigas, Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway, Biochem. Pharmacol., 1996, 52(2), 237–45 CrossRef CAS.
- M. Richter, M. Weiss, I. Weinberger, G. Fürstenberger and B. Marian, Growth inhibition and induction of apoptosis in colorectal tumor cells by cyclooxygenase inhibitors, Carcinogenesis, 2001, 22, 17–25 CrossRef CAS.
- G. A. Piazza, D. S. Alberts, L. J. Hixson, N. S. Paranka, H. Li, T. Finn, C. Bogert, J. M. Guillen, K. Brendel, P. H. Gross, G. Sperl, J. Ritchie, R. W. Burt, L. Ellsworth, D. J. Ahnen and R. Pamukcu, Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in Rats without reducing prostaglandin levels, Cancer Res., 1997, 57, 2909–2915 CAS.
- A. Hughes, N. I. Smith and H. M. Wallace, Polyamines reverse non-steroidal anti-inflammatory drug-induced toxicity in human colorectal cancer cells, Biochem. J., 2003, 374, 481–8 CrossRef CAS.
- H. M. Wallace, A. V. Fraser and A. Hughes, A perspective of polyamine metabolism, Biochem. J., 2003, 376, 1–14 CrossRef CAS.
- A. N. Kingsnorth, A. B. Lumsden and H. M. Wallace, Polyamines in colorectal cancer, Br. J. Surg., 1984, 71, 791–4 CrossRef CAS.
- M. Auvinen, Cell transformation, invasion, and angiogenesis: a regulatory role for ornithine decarboxylase and polyamines?, J. Natl. Cancer Inst., 1997, 89(8), 533–7 CrossRef CAS.
- L. Turchanowa, N. Dauletbaev, V. Milovic and J. Stein, Nonsteroidal anti-inflammatory drugs stimulate spermidine/spermine acetyltransferase and deplete polyamine content in colon cancer cells, Eur. J. Clin. Invest., 2001, 31, 887–93 CrossRef CAS.
- N. Babbar, E. W. Gerner and R. A. Casero Jr., Induction of spermidine/spermine N1-acetyltransferase (SSAT) by aspirin in Caco-2 colon cancer cells, Biochem. J., 2006, 394, 317–24 CrossRef CAS.
- Y. Wang and R. A. Casero Jr., Mammalian polyamine catabolism: a therapeutic target, a pathological problem or both?, J. Biochem., 2006, 139, 17–25 CrossRef CAS.
- G. S. Lindsay and H. M. Wallace, Changes in polyamine catabolism in HL-60 human promyelogenous leukaemic cells in response to etoposide-induced apoptosis, Biochem. J., 1999, 337, 83–87 CrossRef CAS.
- T. Mosman, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63 CrossRef.
- F. Denizot and R. Lang, Rapid colorimetric assay for cell growth and survival, J. Immunol. Methods, 1986, 89, 271–277 CrossRef CAS.
- O. H. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, Protein measurement with the phenol reagent, J. Biol. Chem., 1951, 193, 265–275 CAS.
- H. M. Wallace, M. E. Nuttall and F. C. Robinson, Acetylation of spermidine and methylglyoxal bis(guanylhydrazone) in baby-hamster kidney cells (BHK-21/C13), Biochem. J., 1988, 253, 223–7 CAS.
- N. Seiler and B. Knodgen, High-performance liquid chromatographic procedure for the simultaneous determination of the natural polyamines and their monoacetyl derivatives, J. Chromatogr., Biomed. Appl., 1980, 221, 227–35 CrossRef CAS.
- C. S. Coleman and A. E. Pegg, Assay of mammalian ornithine decarboxylase activity using 14C ornithine. in Methods in Molecular Biology: Polyamine Protocols, ed. D. M. L. Morgan, Humana Press Inc., New Jersey, USA, 1998, pp. 41–44 Search PubMed.
- H. M. Wallace and D. M. Evans, Measurment of spermidine/spermine N1-acetyltransferase. in Methods in Molecular Biology: Polyamine Protocols, ed. D. M. L. Morgan, Humana Press Inc., New Jersey, USA, 1998, pp. 59–68 Search PubMed.
- K. A. Iczkowski and C. G. Pantazis, Overexpression of NSAID activated gene product in prostate cancer, Int. J. Surg. Pathol., 2003, 11(3), 159–166 CrossRef CAS.
- G. Iguchi, K. Chrysovergis, L. Seong-Ho, J. B. Seung, R. Langenbach and T. E. Eling, A reciprocal relationship exists between non-steroidal anti-inflammatory drug-activated gene-1 (NAG-1) and cyclooxygenase-2, Cancer Lett., 2009, 282, 152–158 CrossRef CAS.
- L. C. Wilson, S. J. Baek, A. Call and T. E. Eling, Nonsteroidal anti-inflammatory drug-activated gene (NAG-1) is induced by genistein through the expression of p53 in colorectal cancer cells, Int. J. Cancer, 2003, 105(6), 747–753 CrossRef CAS.
- S. J. Baek, L. C. Wilson, C. H. Lee and T. E. Eling, Dual function of nonteroidal anti-inflammatory drugs (NSAIDs): inhibition of cyclooxygenase and induction of NSAID-activated gene, J. Pharmacol. Exp. Ther., 2002, 301(3), 1126–1131 CrossRef CAS.
- T. Nitta, K. K. Igarashi and N. Yamamoto, Polyamine depletion induces apoptosis through mitochondria-mediated pathway, Exp. Cell Res., 2002, 276(1), 120–8 CrossRef CAS.
- N. Babbar, T. Murray-Stewart and R. A. Casero Jr., Inflammation and polyamine catabolism: the good, the bad and the ugly, Biochem. Soc. Trans., 2007, 35, 300–304 CrossRef CAS.
- S. Chopra and H. M. Wallace, Induction of spermidine/spermine N1-acetyltransferase in human cancer cells in response to increased production of reactive oxygen species, Biochem. Pharmacol., 1998, 55(7), 1119–1123 CrossRef CAS.
- H. M. Wallace and A. V. Fraser, Inhibitors of polyamine metabolism: review article, Amino Acids, 2004, 26, 353–365 CrossRef CAS.
- W. L. Allen, E. G. McLean, J. Boyer, A. McCulla, P. M. Wilson, V. Coyle, D. B. Longley, R. A. Casero Jr. and P. G. Johnston, The role of spermidine/spermine N1-acetyl-transferase in determine response to chemotherapeutic agents in colorectal cancer cells, Mol. Cancer Ther., 2007, 6(1), 128–137 CrossRef CAS.
- J. T. Cross, E. M. Poole and C. M. Ulrich, A review of gene-drug interactions for non-steroidal anti-inflammatory drug use in preventing colorectal neoplasia, Pharmacogenomics J., 2008, 8(4), 237–247 CrossRef CAS.
- M. Auvinen, A. Passinen, L. C. Andersson and E. Holtta, Ornithine decarboxylase activity is critical for cell transformation, Nature, 1992, 360(6402), 355–358 CrossRef CAS.
- E. W. Gerner and F. L. Meyskens Jr., Combination chemoprevention for colon cancer targeting polyamine synthesis and inflammation, Clin. Cancer Res., 2009, 15(3), 758–761 CrossRef CAS.
- F. L. Meyskens Jr., C. E. Mclaren, D. Pelot, S. Fujikawa-Brooks, P. M. Carpenter, E. Hawk, G. Kelloff, M. J. Lawson, J. Kidao, J. Mccracken, C. G. Albers, D. J. Ahnen, D. K. Turgeon, S. Goldschmid, P. Lance, C. H. Hagedorn, D. L. Gille and E. W. Gerner, Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomised placebo-controlled, double-blind trial, Cancer Prev. Res., 2008, 1, 32–38 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |