A comparative cytotoxicity study of TiO2 nanoparticles under light and dark conditions at low exposure concentrations†
Swayamprava
Dalai
,
Sunandan
Pakrashi
,
R. S. Suresh
Kumar
,
N.
Chandrasekaran
and
Amitava
Mukherjee
*
Centre for Nanobiotechnology, VIT-University, Vellore-632014, India. E-mail: amit.mookerjea@gmail.com
First published on 16th May 2012
Abstract
The aim of the present study was to explore the difference in toxicity mechanism of TiO2 nanoparticles (NPs) at low concentrations (≤1 μg mL−1), in a freshwater bacterial isolate, Bacillus licheniformis, under light (UV-illuminated) and dark (non-illuminated) conditions. Standard plate count and MTT assays showed the dose dependent decrease in the bacterial cell viability. The difference in reduction of cell viability under light (20.7%) and dark conditions (21.3%) was statistically non-significant at 1 μg mL−1 concentration and 2 h interaction period. The fluorescence microscopy of the NP interacted cells (1.0 μg mL−1, 2 h) under light and dark conditions showed the mixture of live and dead cells. A significant dose dependent increase in intracellular ROS generation compared to control was noted. The ROS level after 2 h of interaction was significantly higher under light conditions (7.4 ± 0.13%) as compared to dark conditions (4.35 ± 0.12%). The LDH analyses confirmed a statistically significant increase in membrane permeability under dark conditions compared to the light conditions. The NPs were stable against aggregation in sterilized lake water matrix for a period of 24 h, under both light and dark conditions. However, in the presence of bacterial cells an elevated rate of sedimentation was noted under dark conditions. The electron microscopic (SEM, TEM) observations suggested the concentration buildup of NPs near the plasma membrane leading to internalization. The zeta-potential analysis proved that NP attachment was not charge based. The FTIR studies demonstrated the possible involvement of surface functional groups in the attachment. The concentration of dissolved Ti4+ ions was found to be negligible during the test period. The dominant cytotoxicity mechanism under light conditions was found to be ROS generation, whereas, NP attachment to the cell membrane leading to membrane damage significantly contributed in dark conditions.
Introduction
Titanium dioxide (TiO2) nanoparticles (NPs) have various consumer applications in food, cosmetics, filters, cleaning agents, plastics, electronics, glass and ceramics, light bulbs, batteries, ink and in textiles.1–4 Its high refractive index makes it an ideal material for light scattering and thereby they are extensively applied in paints, polymers, coatings and sunscreen lotions.5 Moreover, the efficient photoreactivity exhibited by nano-sized TiO2 makes it an excellent catalyst for waste water treatment and for combating air pollution.6,7 According to the estimates made by the United States Environmental Protection Agency (USEPA) in 2005, the annual production of TiO2 NPs was 2000 metric tons, with 65% of the product usage in cosmetics (USEPA, 2009).8 Due to ever-increasing market demand, the annual production of TiO2 NPs is predicted to reach 2.5 million tons by 2025.9Owing to the increased industrial and consumer applications, there is growing concern regarding the possible risk associated with their environmental exposure. TiO2 NPs can enter the aquatic environment either through direct or indirect release from nano-paints, sunscreen lotions, food additives, medical use, dismantling of batteries, recycling of plastic/glass/metal with nano-coating, groundwater remediation etc.10 The effluents from wastewater treatment plants and waste incineration of products were reported to be the major entry points of TiO2 NPs to the aquatic environment.11–13 Kaegi et al. first documented a direct evidence for the leaching of synthetic TiO2 NPs into the surface water (Ti concentration of 16 μg L−1) from facade paints.14 The predicted environmental concentration (PEC) for released TiO2 NPs in surface water was modeled to be below 1 μg mL−1.15 The National Institute for Occupational Health and Safety (NIOSH) has proposed a draft permissible exposure level (PEL) of 1.5 mg m−3 and a recommended exposure level (REL) of 0.1 mg m−3 for TiO2 nanomaterials.16 Hence the possible risks to aquatic organisms due to the remarkably high annual release of TiO2 NPs to the surface water need to be assessed.17
Understanding the fate and behaviour of NPs released into aquatic ecosystems has become a prerequisite for the assessment of their environmental as well as human health risks. The reactivity of TiO2 NPs in the environment largely depends on their colloidal stability against aggregation.18,19 Our previous ecotoxicity study on Al2O3 NPs using lake water medium suggested that NP stability plays a key role in assessing ecotoxicity.20 The nature of the experimental matrix plays a crucial role in deciding the stability and hence reactivity of NPs. Keller et al. reported the diverse aggregation tendency of TiO2 NPs in different water matrices indicating the impact of the environmental matrix on the stability of the NPs contained in it.21 The role of pH, ionic strength and natural organic matter (NOM) including humic or fulvic acid in stabilizing TiO2 NPs in the aquatic environment has also been stressed.21,22
Microorganisms form the base of the food web and are indispensable agents in maintaining biogeochemical cycles. Since commercial NPs eventually enter the water-bodies, their interaction with aquatic microorganisms is of primary consideration. Microbial metabolism is vital in maintaining equilibrium of the major elements like C, N, S and P.23,24 The microbial processes also influence the distribution of metals in a water body.25 Any damage to the microbes may disturb the geochemical cycles. Hence, the negative impact of NPs, released to the environment, on microbes needs to be assessed. Adams et al. reported 72% reduction in growth of Escherichia coli at 5 g L−1 TiO2 NPs whereas Bacillus subtilis was found to be slightly more sensitive with a similar reduction in growth at 1 g L−1.26 In another experiment, upon exposure to TiO2 NPs (80 μg mL−1), significant reduction in viability (19%) of E. coli was observed.27 Acute toxicity of TiO2 NPs was assessed by Jiang et al. in E. coli labeled with green fluorescent protein and a 50% reduction in cell viability was observed after 135 min of exposure.28
From a detailed literature review, the interaction between the NPs and the bacterial cells leading to cytotoxicity can be hypothesized to involve two steps. The first one involves oxidative damage by the NPs attached to the cell membrane resulting in membrane permeabilization without significant reduction in cell viability. The second step includes leaking out of intracellular components, which is the leading cause of decreased viability, and internalization of NPs damaging cell organelles including the nucleus.29 The observed cytotoxic effects of TiO2 NPs under UV-illumination (light) have largely been related to the involvement of free radicals. Recent studies on the mechanistic aspect of NP toxicity indicated that the photocatalytic effect of TiO2 NPs occurred via the extracellular generation of ROS, membrane damage and subsequently cell death.30 Kumar et al. suggested a more detailed mechanism which involves ROS generation, glutathione depletion, lipid peroxidation, membrane permeability, and subsequently DNA damage leading to cell death in E. coli at 80 μg mL−1 NP concentrations.27 Burello and Worth emphasized that, TiO2 NP is capable of inducing oxidative stress without any irradiation.31 Another mechanistic study showed adsorption of NPs onto the cell wall, the interruption of transmembrane electron transfer, modification of the membrane potential, physical damage resulting in leakage of the cell contents and/or the production of reactive oxygen species.29,32 In a study on E. coli, Maness et al. reported lipid peroxidation as the underlying killing mechanism of TiO2 in light condition.33 Most of these findings are based on the photocatalytic property of TiO2 NPs whereas a handful of reports are only available on toxicity under dark conditions.30,34 Fenoglio et al. reported non-UV-induced free radical production from anatase phase of TiO2 NPs, by means of a spin-trapping/ESR spectroscopy.35 Thill et al. proposed that the extracellular adsorption of NPs may induce intracellular ROS production which is a possible reason for toxicity under dark conditions.36
Most of the studies on the potential ecotoxicity of TiO2 NPs towards microorganisms are carried out at elevated exposure concentrations and the observed effects may undermine the lower dose consequences. In addition, nature of the experimental matrix plays a vital role in deciding the oxidative stress. Enriched media were reported to suppress the formation of radicals.37 Previous reports on TiO2 NP toxicity are mostly based on effect of irradiation and there is a lack of insight into the definite mechanisms by which microbial species respond to NP toxicity under dark conditions. Hence the underlying toxicity mechanisms at low exposure concentrations of TiO2 NPs in a nutrient minimal media towards a bacterial species under both light and dark conditions need to be explored. Hence, the aim of the current study was elucidating the different mechanistic modes of TiO2 NP toxicity at low exposure concentrations (≤1 μg mL−1) towards a freshwater bacterial isolate, Bacillus licheniformis, in both the presence and absence of UV-illumination. All the experiments were carried out using lake water matrix. Mechanistic endpoints included oxidative stress analysis, membrane permeability assessment and sedimentation studies.
Materials and methods
Materials
Dry titanium(IV) oxide nanopowder (TiO2, 99.7% anatase, particle size: <25 nm, CAS No.: 637254) and DCFH-DA (2′,7′-dichlorofluorescin diacetate) was procured from Sigma Aldrich (St. Louis, MO, USA). MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) and Acridine Orange (AO) was purchased from Hi-Media Pvt. Ltd (Mumbai, India). Ethidium bromide (EtBr) was obtained from Medox Biotech India Pvt. Ltd. All other chemicals were of analytical reagent grade.Fresh water, collected from VIT Lake, Vellore, India, without additional nutrients, was used for the study (conductance: 4.3 ± 0.13 mS cm−1; pH: 7.8; DO: 7.2 ± 0.46 μg mL−1; TDS: 800 ± 74 μg mL−1). Quantification of metal ions and other inorganic ions was carried out by ICP-OES (Perkin Elmer Optima 5300 DV, USA) by titrimetric methods respectively (details in our previous report, Pakrashi et al., 2011).20 Primary filtration through Whatman No. 1 filter paper was done, following sterilization to remove suspended dust particles, keeping the chemical composition of the water intact. This water was used as the experimental matrix throughout the study. A secondary filtration, through 0.22 μm membrane filter, was done to avoid the interference of large colloidal particles prior to hydrodynamic size measurement.
Characterization of as-received TiO2 NPs
Stability of the dispersion was examined with respect to time by analyzing the variation in particle size distribution at the top layer. Concentration of TiO2 NP at the top layer of dispersion was analyzed by measuring its UV absorbance at 336 nm (UV-Vis spectra of TiO2 NP dispersion in Millipore water and Lake water in ESI Fig. S1†).
Toxicity assessment
Bacteria were interacted with three different concentrations (0.05 μg mL−1, 0.5 μg mL−1, 1 μg mL−1) of TiO2 NP dispersion. All the experiments were carried out in parallel in two sets, under light and dark conditions. For light induced studies, test beakers were exposed to UV irradiation through UV lamps (Philips, 15 W) and for supporting non-illuminated studies, complete darkness was maintained. Control beakers without NP addition were kept for both light and dark conditions. Samples were analyzed after an interaction period of 2 h, 6 h and 24 h respectively.
Oxidative stress assessment
A negative control of NPs without cells was also analyzed to find out the autofluorescence of TiO2 NPs that may interfere with the DCFDA dye.
Membrane permeability assessment
Electron microscopic analyses
Surface interactions with NPs
Sedimentation analysis
The concentration of NPs at these three layers was determined by the UV-Vis spectroscopic method (absorbance in terms of concentration).24 TiO2 NP dispersed in lake water gave an absorbance peak at 336 nm. We validated this method by comparing the absorbance data with Ti4+ metal ion concentration data obtained by conventional inductively coupled plasma optical emission spectrometry (ICP-OES) analysis.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 20 min) and filtered through a 0.1 μm membrane filter. A particle size analyzer ensured complete removal of nanoparticles from the filtrate. Concentration of Ti4+ ions was quantified using ICP-OES at 2nd, 6th and 24th h of incubation. Particle free suspension was interacted with bacterial culture to detect toxicity of released ions.
000g, 20 min) and filtered through a 0.1 μm membrane filter. A particle size analyzer ensured complete removal of nanoparticles from the filtrate. Concentration of Ti4+ ions was quantified using ICP-OES at 2nd, 6th and 24th h of incubation. Particle free suspension was interacted with bacterial culture to detect toxicity of released ions.
Results
Characterization of as-received TiO2 NPs
Transmission electron micrograph of as-received NPs (Fig. 1) demonstrated the particles to be within 20–100 nm size. The particles were nearly spherical and cubical in shape. The observed aggregates were composed of small cubical and spherical shaped particles of size between 10 and 20 nm. The large sized (50–100 nm) particles were observed to be mostly spherical in shape. The X-ray diffraction pattern of TiO2 NPs confirmed the predominance of the anatase phase (details in ESI, Fig. S3†). TiO2 NP dispersion in Millipore water, at a concentration of 1 μg mL−1, showed the presence of more particles in the size range of 0–50 and 200–300 nm at 0 h (Fig. 2A). Within 2 h, the particles started aggregating, and the lower size range (0–50 nm) almost disappeared. With time, the aggregation tendency increased, and particle size moved towards micrometer size range, suggesting instability of NPs in Millipore water. | ||
| Fig. 1 Transmission electron micrograph of as-received TiO2 NPs. | ||
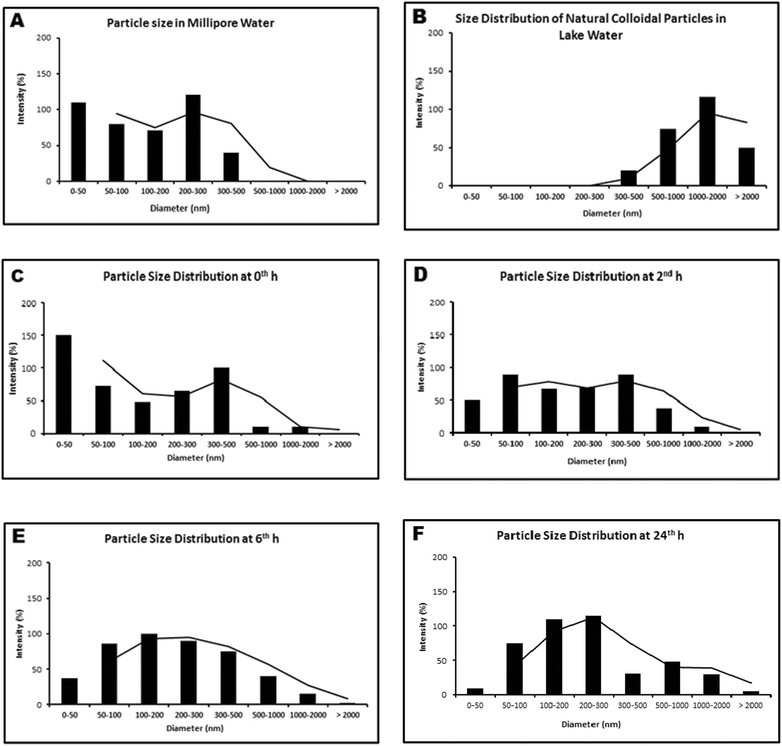 | ||
| Fig. 2 Hydrodynamic size distribution of (A): TiO2 NPs in Millipore water at 0 h; (B): Natural colloidal particles present in lake water; (C–F): TiO2 NPs in lake water (0 h–24 h). | ||
TiO2 nanoparticle stability in lake water
The hydrodynamic size distribution of particles in lake water (devoid of NPs) suggested the presence of natural colloids mostly in the size range of 0.5–2 μm (Fig. 2B). NPs dispersed in lake water at a concentration of 1μg mL−1 were analyzed at 0th, 2nd, 6th and 24th h for particle size distribution and stability studies (Fig. 2C–F). The three size ranges: 50–100 nm, 100–200 nm and 200–300 nm remained stable till 24 h of the experimental period. The additional size range of 0.5–1 μm observed could be attributed to the existence of natural colloids in the lake water. The variation in particle size distribution under both light and dark conditions was found to be insignificant (at p > 0.05) in the experimental period.To support the DLS data we studied the NP size distribution in lake water (after 2 h) through scanning electron microscopy. The SEM micrograph of TiO2 NP dispersion showed uniform distribution of 50–200 nm diameter particles (Fig. 3A). The nanoparticles appeared nearly spherical in shape. The SEM micrograph of the lake water suggested the presence of spherical natural colloids with 1–2 μm diameter (Fig. 3B). Another characterization process of TiO2 NPs is UV-Vis absorbance measurement. TiO2 NP dispersed in lake water gave a near-UV absorbance peak at 336 nm.
 | ||
| Fig. 3 Scanning electron micrograph of (A): TiO2 NPs dispersed in lake water; (B): Natural colloids present in lake water. | ||
The variation in particle size distribution in an abiotic environment (Fig. 4A, B) under both light and dark conditions was found to be insignificant till 24 h at a size range of 200–300 nm (Fig. 4C). Absorbance at 336 nm suggested steady concentration of NPs at the top layer (Fig. 4D) for both light and dark conditions. This result explains a stable dispersion of TiO2 NPs in lake water matrix, with a remarkably low rate of sedimentation.
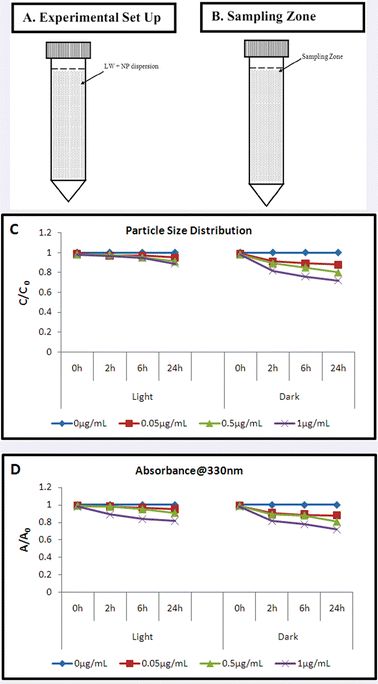 | ||
| Fig. 4 TiO2 nanoparticle stability in lake water (A): Experimental set up; (B): Sample collecting zone; (C): Particle size distribution (C/C0) at the top layer with respect to time; (D): Concentration at the top layer (A/A0) with respect to time. | ||
Cell viability assessment
A concentration dependent reduction in cell viability of B. licheniformis upon NP interaction was observed through plate count assay and MTT assay. A plate count assay showed a significant decline in viability of NP interacted cells (21.3 ± 1.7% and 23.4 ± 1.2% under light and dark conditions respectively, 2 h), as compared to control, at 1 μg mL−1 concentration (Fig. 5A). The cytotoxicity of NPs measured in terms of the viability of cells was found to decrease with time. The difference in toxicity under both test conditions (light and dark) was detected to be insignificant at all the three concentrations (0.05, 0.5 and 1.0 μg mL−1). The MTT assay also corroborated with the plate count results showing similar dose dependent and time dependent response (Fig. 5B). Percentage reduction in viability at 1 μg mL−1 NP concentration, calculated under light and dark conditions (20.7 ± 1.5% and 22.3 ± 1.1% respectively, 2 h) was statistically significant (p < 0.05) as compared to the control. At lower concentrations, the difference in cell viability under light and dark conditions was found to be non-significant. One-way ANOVA followed by Dunnett's post-hoc test was performed to calculate the statistical significance in values. Interestingly, with an increase in exposure concentration (from 1 μg mL−1 to 20 μg mL−1), significant deviation in cell viability was noted between light and dark conditions. EC50 value calculated (EPA Probit Analysis Program, Version 1.5) by plate count assay and MTT assay varied significantly for light (5.228 μg mL−1, 4.983 μg mL−1) and dark (19.568 μg mL−1, 17.664 μg mL−1) conditions (ESI†).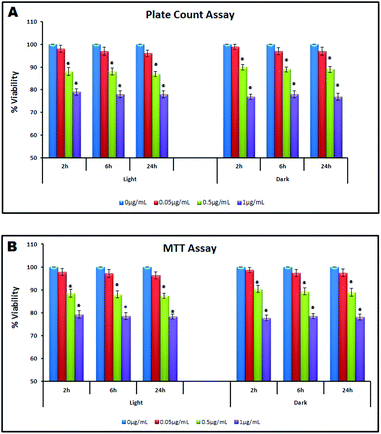 | ||
| Fig. 5 Cell viability assessment under light and dark conditions (0 h–24 h) (A): Plate count assay; (B): MTT assay. | ||
Live–dead discrimination through fluorescence microscopy
To visualize the live/dead cells in the sample, we performed AO-EtBr fluorescence staining. Control and NP interacted cells (1 μg mL−1, 2 h) stained with AO and EtBr were visualized under a fluorescence microscope. Live cells appeared green (Fig. 6A), and cells with compromised membrane, were observed to be bright red in colour. Interacted samples were found to be a mixture of green and bright red cells for both light (Fig. 6B, C) and dark conditions (Fig. 6D, E). Some intermediate or injured cells having motility (orange red) were also observed in dark treated samples.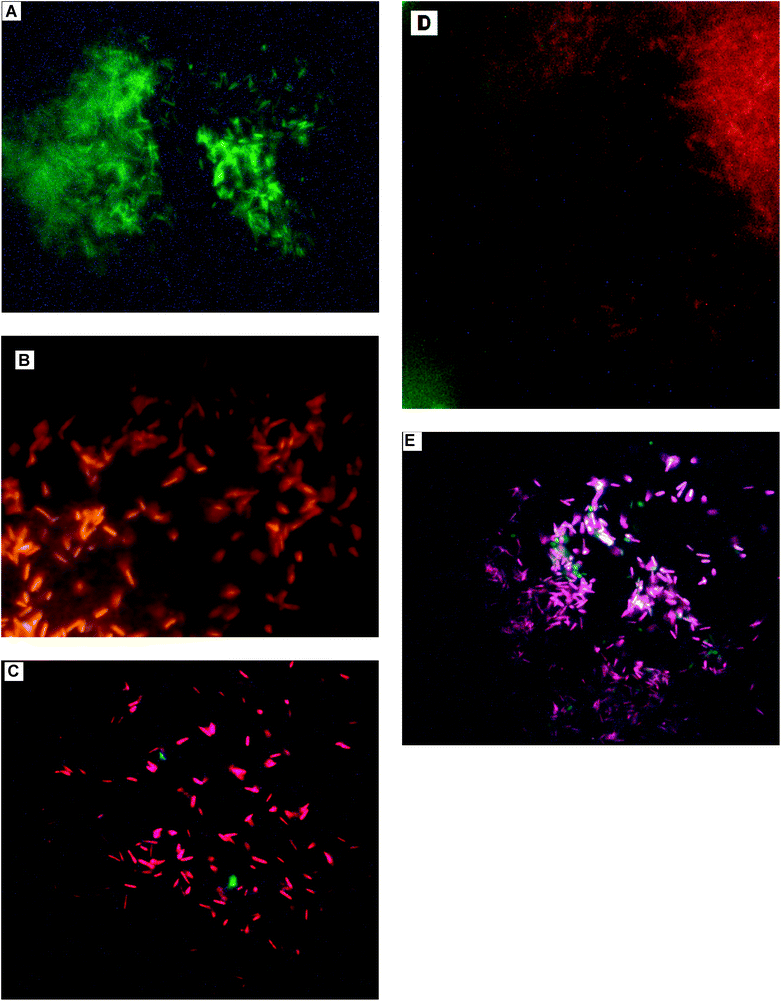 | ||
| Fig. 6 Fluorescence microscopic images of (A): untreated (control) cells showing no damage (green); (B, C): Treated cells under light conditions showing the presence of damaged (bright red) as well as undamaged cells (green); (D, E): Cells treated under dark conditions showing the presence of damaged (bright red), undamaged cells (green) and injured cells (orange red). | ||
Oxidative stress assessment
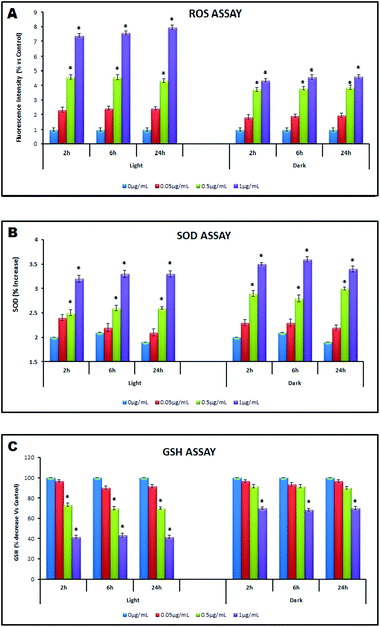 | ||
| Fig. 7 Oxidative stress assessment under light and dark conditions (0 h–24 h) by (A): ROS Assay; (B): SOD Assay; (C): GSH Assay. | ||
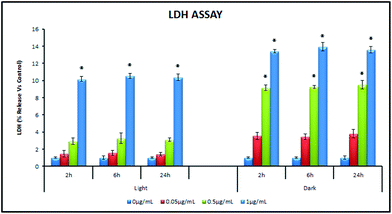 | ||
| Fig. 8 Assessment of membrane damage under light and dark conditions (0 h–24 h) by LDH Assay. | ||
The NP interacted (1 μg mL−1, 2 h) and control cells were analyzed for membrane permeability using spectrofluorimetry. The intensity of EtBr, staining both non-viable and damaged cells was measured compared to control. A higher emission peak intensity was noted for dark treated samples compared to light treated ones. The fluorescence spectrometric analysis suggested higher total cell membrane damage (owing to both the damaged cells as well as the dead cells) under dark conditions.
 | ||
| Fig. 9 Scanning electron micrograph of (A): untreated bacteria (control) showing intact cell; (B): NP attachment on bacterial cell surface; (C): Flocculation of NPs with the cell; (D): Roughening of cell surface and signs of membrane damage; (E): Bending of cells upon NP treatment. | ||
![Transmission electron micrograph (A): showing the typical appearance of B. licheniformis before NP treatment; (B): interacted cells showing disrupted morphology under light conditions; (C): appearance of small vacuole like structures in light treated samples; (D): internalization of NPs under light conditions; (E): accumulation of NPs on the outer and inner side of plasma membrane in dark treated samples; (F): dividing cell with NP accumulated near nuclear material. [Notations used: Blue arrow indicates membrane damage; red arrow indicates cell distortion; yellow arrow marks internalization of NPs into cytoplasm; red box demarcates localization of NPs onto cell membrane; blue box demarcates localization of NPs in nucleus; Yellow box demarcates vacuole formation].](/image/article/2012/TX/c2tx00012a/c2tx00012a-f10.gif) | ||
| Fig. 10 Transmission electron micrograph (A): showing the typical appearance of B. licheniformis before NP treatment; (B): interacted cells showing disrupted morphology under light conditions; (C): appearance of small vacuole like structures in light treated samples; (D): internalization of NPs under light conditions; (E): accumulation of NPs on the outer and inner side of plasma membrane in dark treated samples; (F): dividing cell with NP accumulated near nuclear material. [Notations used: Blue arrow indicates membrane damage; red arrow indicates cell distortion; yellow arrow marks internalization of NPs into cytoplasm; red box demarcates localization of NPs onto cell membrane; blue box demarcates localization of NPs in nucleus; Yellow box demarcates vacuole formation]. | ||
Based on the investigation of SEM and TEM, it can be concluded that TiO2 NPs adsorbed on the surface of the bacteria, damaged the cell membrane under both light and dark conditions and subsequently resulted in the leakage of the intracellular substances, thus, causing the cell death.
Surface interactions with NPs
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or C–O bonds were noted. The peak corresponding to COO− in hydrophobic glycerol was observed at ∼1365 cm−1.45 A characteristic peak attributed to PO2− vibrations present exclusively in nucleic acids with a little contribution from phospholipids of cell wall was evident at 1230 cm−1.45 A broad peak corresponding to glycogen units and polysaccharides in the cell wall was noted at the finger print region with peak intensity at 709 cm−1.46
O or C–O bonds were noted. The peak corresponding to COO− in hydrophobic glycerol was observed at ∼1365 cm−1.45 A characteristic peak attributed to PO2− vibrations present exclusively in nucleic acids with a little contribution from phospholipids of cell wall was evident at 1230 cm−1.45 A broad peak corresponding to glycogen units and polysaccharides in the cell wall was noted at the finger print region with peak intensity at 709 cm−1.46
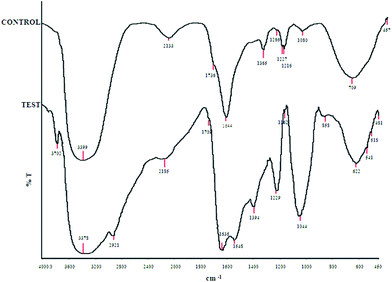 | ||
| Fig. 11 Comparative FTIR spectra for untreated (control) and NP treated (test) cells. | ||
After treatment of bacteria with nanoparticles, the bands at 3391, 1736, 1365, 1060 cm−1 were shifted to 3378, 1709, 1044 cm−1 respectively while a considerable decrease in peak intensity is observed at 1365 and 1227 cm−1. The band centered at about 3700 cm−1 shifted to lower frequencies (by 15–20 cm−1). A considerable increase in peak intensity at 1543 cm−1 suggested the involvement of amide II (protein N–H bending and C–N stretching) in the cell wall.47 A broad stretching peak noted around 2930 cm−1 was characteristic of weak C–H stretching band from alkyl groups. The characteristic Ti–O–Ti band appeared in the range of 500–900 cm−1 suggesting nanoparticles attachment to the cell wall.48
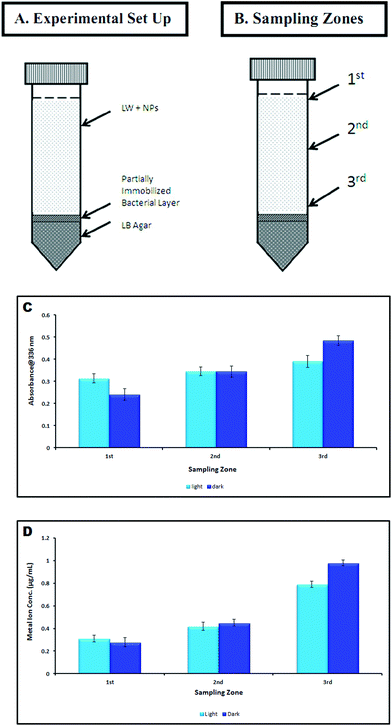 | ||
| Fig. 12 Sedimentation analysis (A): experimental set up; (B): sampling zones; (C): absorbance measurement at different sampling zones; (D): metal ion concentration at different sampling zones. | ||
Toxicity due to Ti4+ ion dissolution was detected to be negligible. Concentration of dissolved Ti4+ ion in NP dispersion at 2nd, 6th and 24th h was measured to be below detection limit of the instrument (ICP-OES) under both the conditions.
Discussion
The anatase phase TiO2 NPs were selected for this study owing to their greater relevance to toxicity assessment.49 These NPs were found to be stable against aggregation at concentrations employed (0.05, 0.5 and 1 μg mL−1) in lake water matrix as observed through dynamic light scattering analysis (Fig. 2C–F). The presence of natural colloids (1–2 μm) and uniform distribution of TiO2 NPs (50–200 nm) was confirmed through scanning electron microscopic analysis. This suggests the involvement of natural organic matter (NOM) in stabilizing the NPs.20,21,50 The previous reports strongly suggest that the colloidal stability of NPs plays an essential role in the interaction with cells leading towards adverse effects like membrane damage and cytotoxicity.51The dose and exposure dependent reduction in the cell viability was observed under both light and dark conditions (Fig. 5A, B). At low concentrations, the difference in cell viability reduction under light and dark conditions, was found to be non-significant. On the contrary, the EC50 value for the light reaction was found to be nearly 4 times less than the dark reaction (5.228 μg mL−1, 19.568 μg mL−1 respectively) confirming the concentration dependence of the photocatalytic effect of TiO2 NPs. The previous reports on photocatalytic toxicity of the TiO2 NP showed 19% reduction in E. coli cell viability at 8 μg mL−1 TiO2 NP concentration (experimental period: 60 min), using phosphate buffered saline as the experimental matrix.28 In another study, the nano-TiO2 suspension at 20 μg mL−1 was found to be not acutely toxic to V. fischeri.52 Brunet et al. also observed phototoxicity of 100 μg mL−1 TiO2 with 75% reduction in E. coli viability whereas no effect could be observed under dark conditions.37 In contrast, aggregation of E. coli cells and suppressed cell division induced by TiO2 NPs was reported at pH 4.0–4.5 and in the absence of any UV irradiation, demonstrating non photocatalytic mode of toxicity.34 Fenoglio et al. also reported that radicals generated from TiO2 NPs (anatase) under non-UV conditions may trigger cytotoxicity.35 Through fluorescence microscopy, live (green) and dead (bright red) cells were observed under both light and dark conditions. In dark treated samples, some intermediate/injured cells (orange red/pale red) were observed having motility which suggests membrane damaging property of TiO2 NPs. Battin et al. reported that, under dark conditions, nucleic acid stains and epifluorescence microscopy showed substantially damaged cell membranes after 24 h of exposure to nano-TiO2 (initial concentration, 5.3 μg mL−1).30
The oxidative stress induced by TiO2 NPs can be determined via production of reactive oxygen species (ROS), level of superoxide dismutase (SOD) and level of reduced glutathione (GSH).28 The cellular damage by ROS is a major contributor to its functional decline. Though the ROS level was found to be significantly higher under light conditions as compared to dark conditions (1 μg mL−1; 7.4 ± 0.13% and 4.35 ± 0.12%; 2 h), importantly a considerable amount of ROS was also generated even under dark conditions (Fig. 7A). The UV-excited TiO2 has long been known to generate ROS leading to cytotoxic effects.49,53 Another report by Burello and Worth suggests TiO2 is capable of inducing cytotoxicity due to oxidative stress irrespective of UV irradiation as the position of its conduction band lies in the range of biological redox potentials.31 A thorough literature survey reveals that the mechanism (s) explaining TiO2 NP toxicity under dark conditions is not well explored. The non-photocatalytic toxic effect may be attributed to (a) the extracellular adsorption of NPs inducing intracellular ROS generation36 (b) reactivity of TiO2 NPs (predominantly anatase) toward biomolecules producing carbon centered free radicals which exacerbate the oxidative damage of the bio-organics on the cell wall35 and (c) production of free radical species by TiO2 NPs even without irradiation.35 Several reports showing cytotoxicity and DNA damage (in prokaryotic cells) by TiO2 NPs irrespective of illumination conditions also support our finding.30,34 The observed ROS generation under dark conditions was supportive of such non photo-induced effects. The antioxidant enzyme superoxide dismutase (SOD) acts as a protective agent against increased oxidative stress due to ROS generation. The present study (Fig. 7B) shows an elevated level of SOD under dark conditions as compared to light conditions, suggesting strong resistance from bacteria against oxidative stress in the absence of light. Possibly, the dark treated cells were under enhanced stress conditions suggesting mechanistic modes other than ROS generation playing a significant role as oxidative stress triggers. Higher SOD levels indicate that bacteria could defend themselves against ˙OH attack to a greater extent.54,55 Reduced glutathione (GSH) is also engaged in cellular protection mechanism through chemical detoxification and antioxidant defence.56 According to a prior report, the depletion of GSH causes an imbalance of pro-oxidants (e.g. ROS) and antioxidants (e.g. GSH) leading to increased oxidative stress.57 In the present study the decreased GSH depletion (Fig. 7C) under dark conditions agrees well with the ROS data but, the rationale for higher SOD levels in dark treated samples need to be explained further. Thus in both light exposed and dark conditions, membrane permeability assessment was carried out to explore the possible toxicity mechanism.
The membrane permeability assessment through LDH release detection showed more membrane damage under dark conditions compared to light (Fig. 8). TiO2 NPs have been shown to cause membrane disorganization, increased membrane permeability as a result of perforation and finally leading to cell death.26,58 Previous study by our group on E. coli, B. subtilis and P. aeruginosa reported that the antibacterial mechanism of TiO2 NPs included the inner membrane permeabilization, destruction of cellular integrity and involvement of surface functional groups.59 The antibacterial effects of the NPs differed between the microbial strains due to the outer membrane structural differences. A recent study by Fang et al. also suggested that the TiO2 NPs attachment on Nitrosomonas europea cells caused apparent morphological damage making the cells heavier.60
The changes in surface morphology of the bacterial cells when exposed to the TiO2 NPs were studied through scanning electron microscopy. The smoothness of the membrane disappeared with NP interaction and the cells were found to form agglomerates along with NPs (Fig. 9) A previous report showed the membrane localization of TiO2 NPs on bacteria which was responsible for the cellular stress.61 The physiological adaptation of bacteria to stress conditions includes alteration of the membrane structure as a defence mechanism.62 The morphological changes of cells and the internalization and bio-distribution of NPs were further confirmed through transmission electron microscopy (Fig. 10). Recently, Pigeot-Rémy et al. examined the morphological damage caused by irradiated TiO2 to E. coli, using TEM.63 Bacteria were found to be non-viable after 1.5 h and membrane structure was observed to be disorganized. The membrane localization of TiO2 NPs was also observed in bacterial cells.32 A flow cytometric method was developed to quantify the amount of NP internalized by live bacterial cells.64 Ma et al. also observed time dependent adsorption of TiO2–Ag (2.0%) aggregates on E. coli cells through TEM imaging.65
The zeta potential measurement suggested no involvement of charge based interaction between NP and bacteria. Similar findings were reported by Jiang et al.66 The contribution of surface chemical groups in bacteria–NP interaction was investigated through FTIR analysis. The shifting of the band at about 3700 cm−1 to lower frequencies suggested OH stretching vibrations (due to water) and possible metal binding on amide groups in proteins.67 The TiO2 NPs attachment to the cell wall is evident from the characteristic Ti–O–Ti band appeared in the range of 500–900 cm−1.48 Our previous findings on TiO2 NP toxicity also suggested involvement of surface functional groups in cellular membrane damage upon NP interaction.59,68 The differences in NP attachment to the cell wall under light and dark conditions was also confirmed through a sedimentation experiment and quantified through ICP-OES analysis (Fig. 12). The faster sedimentation of TiO2 NPs in dark compared to light conditions related to enhanced bacteria-NP attachment and leading to cell damage/death.
To explain the differences in cytotoxicity mechanisms observed in this study under light and dark conditions, we propose (Fig. 13) that bacteria–NP interaction proceeded through different stages. For the light reaction, the sequence of events can be summarized as: sedimentation of nanoparticles to the bacterial zone, ROS generation and concentration buildup of NPs near the plasma membrane leading to membrane damage, internalization of NPs, leaking out of intracellular components leading to cell death. Under dark conditions, the cytotoxicity mechanism undergoes the following stages: sedimentation of NPs, agglomeration of NPs with bacteria, localization of NPs on the cell membrane, induction of the oxidative stress, increase in membrane permeability, internalization and bio-distribution of the NPs. While the ROS generated was the principal contributor for cytotoxicity in light condition, NP attachment to the cell membrane leading to membrane damage was the predominant effect in dark. A report by Maness et al. supported our findings, suggesting the likely reason to be the aggregation of TiO2 NPs with cells in the dark.33 Another study by Malato et al. reported a slight reduction in the concentration of bacterial colonies in the presence of TiO2 NPs in the dark, possibly due to agglomeration of TiO2 with the bacterial cells and subsequent sedimentation.69
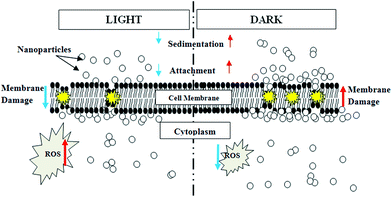 | ||
| Fig. 13 Schematic illustration of proposed TiO2 NP induced toxicity mechanisms under light and dark conditions. | ||
Several physico-chemical parameters like size, shape, crystallinity, stabilizing agents, agglomeration and/or aggregation in the medium, play a key role in determining toxicity of NPs towards bacteria.51 Environmental factors i.e. the characteristics of the exposure media (pH, composition, ionic strength) also greatly influence the behaviour of NPs and their interaction with cells.12 There is a possibility that other toxicants can be associated with TiO2 nanoparticles and therefore likely synergistic effects must be examined for additional toxicological concerns.70
Conclusion
The present study explores the mechanistic aspects of cytotoxicity of photo catalytic TiO2 NP towards bacterial systems at low exposure conditions under non-irradiated or dark conditions, and compares with those at irradiated or light conditions. The findings from this study can be extrapolated to study the environmental risk of these nanoparticles given their increasing usage in consumer products. In this context understanding the underlying fundamental chemistry of the TiO2 NP–cell interaction in dark conditions will be helpful for future studies. Notwithstanding the implied scientific merit in the present study, and related other studies on ecotoxicity of NPs, which are slowly growing in number, (i) a lack of standardized characterization protocol to study physico-chemical characteristics of the NPs, which influence their reactivity as well as toxicity, in differing real systems, (ii) the scattered data on different microbial systems employed in absence of a common test strategy, (iii) an acute lack of emphasis on the environmental parameters prevent a meaningful comparative assessment from the hitherto available nanotoxicity data with the bacterial systems. In this context, concerted research efforts in near future should concentrate on correlating NP characterization and behaviour in environmental matrices with its toxicological endpoints, and evolving a common test strategy for ecotoxicity studies of NPs taking care of various confounding factors relating to the particles, bacterial systems, and the test environment.Acknowledgements
We acknowledge Sophisticated Analytical Instrumentation Facility (SAIF), Department of Science & Technology (DST) at Indian Institute of Technology, Madras, for ICP-OES analysis facility and Christian Medical College, Vellore, India, for the Transmission Electron Microscopy facilities used for our study. We profoundly thank Ministry of Environment and Forests, Govt. of India, for funding this research. We are grateful to both anonymous reviewers for helping us in improving the quality of the manuscript.References
- B. Q. Wang, L. Q. Jing, Y. C. Qu, S. D. Li, B. J. Jiang, L. B. Yang, B. F. Xin and H. G. Fu, Appl. Surf. Sci., 2006, 252, 2817 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS.
- B. D. More, Indian J. Dermatol., Venereol. Leprol., 2007, 73, 80–85 CrossRef.
- N. C. Mueller and B. Nowack, Environ. Sci. Technol., 2008, 42, 4447–4453 CrossRef CAS.
- D. Chen, F. Huang, Y. B. Cheng and R. A. Caruso, Adv. Mater., 2009, 21, 2206–2210 CrossRef CAS.
- M. A. Ferguson, M. R. Hoffmann and J. G. Hering, Environ. Sci. Technol., 2005, 39, 1880–1886 CrossRef CAS.
- C. R. Esterkin, A. C. Negro, O. M. Alfano and A. E. Cassano, AIChE J., 2005, 51, 2298–2310 CrossRef CAS.
- USEPA, EPA/600/R-09/057, United States Environmental Protection Agency, Washington, DC, 2009 Search PubMed.
- C. O. Robichaud, A. E. Uyar, M. R. Darby, L. G. Zucker and M. R. Wiesner, Environ. Sci. Technol., 2009, 43, 4227–4233 CrossRef CAS.
- N. O'Brien and E. Cummins, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2010, 45, 992–1007 CrossRef CAS.
- M. A. Kiser, P. Westerhoff, T. Benn, Y. Wang, J. Perez-rivera and K. Hristovski, Environ. Sci. Technol., 2009, 43, 6757–6763 CrossRef CAS.
- T. M. Scown, R. van Aerle and C. R. Tylre, Crit. Rev. Toxicol., 2010, 40, 653–670 CrossRef CAS.
- N. Musee, M. Thwala and N. Nota, J. Environ. Monit., 2011, 13, 1164–1183 RSC.
- R. Kaegi, A. Ulrich, B. Sinnet, R. Vonbank, A. Wichser, S. Zuleeg, H. Simmler, S. Brunner, H. Vonmont, M. Burkhardt and M. Boller, Environ. Pollut., 2008, 156, 233–239 CrossRef CAS.
- F. Gottschalk, T. Sonderer, R. W. Scholz and B. Nowack, Environ. Sci. Technol., 2009, 43, 9216–9222 CrossRef CAS.
- R. Landsiedel, L. Ma-Hock, A. Kroll, D. Hahn, J. Schnekenburger, K. Wiench and W. Wohlleben, Adv. Mater., 2010, 22, 2601–2627 CrossRef CAS.
- F. Gottschalk, R. W. Scholz and B. Nowack, Environ. Modell. Software, 2010, 25, 320–332 CrossRef.
- Y. Zhang, Y. Chen, P. Westerhoff, K. Hristovski and J. C. Crittenden, Water Res., 2008, 42, 2204–2212 CrossRef CAS.
- V. K. Sharma, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2009, 44, 1485–1495 CrossRef CAS.
- S. Pakrashi, S. Dalai, D. Sabat, S. Singh, N. Chandrasekaran and A. Mukherjee, Chem. Res. Toxicol., 2011, 24, 1899–1904 CrossRef CAS.
- A. A. Keller, H. Wang, D. Zhou, H. S. Lenihan, G. Cherr, B. J. Cardinal, R. Miller and Z. Ji, Environ. Sci. Technol., 2010, 44, 1962–1967 CrossRef CAS.
- R. F. Domingos, N. Tufenkji and K. I. Wilkinson, Environ. Sci. Technol., 2009, 43, 1282–1286 CrossRef CAS.
- M. Schaechter, J. L. Ingraham and F. C. Neidhardt, Microbe., ASM Press, Washington, DC, 2006 Search PubMed.
- G. H. R. Osler and M. Sommerkorn, Ecology, 2007, 88, 1611–1621 CrossRef.
- T. Ford and D. Ryan, Environ. Health Perspect., 1995, 103, 25–28 CAS.
- L. K. Adams, D. Y. Lyon and P. J. Alvarez, Water Res., 2006, 40, 3527–3532 CrossRef CAS.
- A. Kumar, A. K. Pandey, S. S. Singh, R. Shanker and A. Dhawan, Free Radical Biol. Med., 2011, 51, 1872–1881 CrossRef CAS.
- G. Jiang, Z. Shen, J. Niu, Y. Bao, J. Chen and T. He, J. Environ. Monit., 2011, 13, 42–48 RSC.
- S. J. Klaine, P. J. Alvarez, G. E. Batley, T. F. Fernandes, R. D. Handy, D. Y. Lyon, S. Mahendra, M. J. McLaughlin and J. R. Lead, Environ. Toxicol. Chem., 2008, 27, 1825–1851 CrossRef CAS.
- T. J. Battin, F. V. D. Kammer, A. Weilhartner, O. Ottofuelling and T. Hofmann, Environ. Sci. Technol., 2009, 43, 8098–8104 CrossRef CAS.
- E. Burello and A. P. Worth, Nanotoxicology, 2011, 5, 228–235 CrossRef CAS.
- Y. Li, M. Ma, X. Wang and X. Wang, J. Environ. Sci., 2008, 20, 1527–1533 CrossRef CAS.
- P. C. Maness, S. Smolinski, D. M. Blake, Z. Huang, E. J. Wolfrum and W. A. Jacoby, Appl. Environ. Microbiol., 1999, 65, 4094–4098 CAS.
- L. V. Zhukova, J. Kiwi and V. V. Nikandrov, Dokl. Chem., 2010, 435, 279–282 CrossRef CAS.
- I. Fenoglio, G. Greco, S. Livraghi and B. Fubini, Chem.–Eur. J., 2009, 15, 4614–4621 CrossRef CAS.
- A. Thill, O. Zeyons, O. Spalla, F. Chauvat, J. Rose, M. Auffan and A. M. Flank, Environ. Sci. Technol., 2006, 40, 6151–6156 CrossRef CAS.
- L. Brunet, D. Y. Lyon, E. M. Hotze, P. J. Alvarez and M. R. Wiesner, Environ. Sci. Technol., 2009, 43, 4355–4360 CrossRef CAS.
- T. Mossman, J. Immunol. Methods, 1983, 65, 55–63 CrossRef.
- H. Wang, H. Cheng, D. Wei and F. Wang, J. Microbiol. Methods, 2011, 84, 140–143 CrossRef.
- S. Jakopec, K. Dubravcic, S. Polanc, J. Kosmrlj and M. Osmak, Toxicol. in Vitro, 2006, 20, 17–226 CrossRef.
- H. Wang and J. A. Joseph, Free Radical Biol. Med., 1999, 27, 612–616 CrossRef CAS.
- C. Wintherbourn, R. Hawkins, M. Brian and R. Carrell, J. Lab. Clin. Med., 1975, 85, 337–341 Search PubMed.
- D. M. Brown, M. R. Wilson, W. MacNee, V. Stone and K. Donaldson, Toxicol. Appl. Pharmacol., 2001, 175, 191–199 CrossRef CAS.
- A. A. Kamnev, Spectroscopy (Amsterdam), 2008, 22, 83–95 CAS.
- Z. Liu, S. Yang, Y. Bai, J. Xiu, H. Yan, J. Huang, L. Wang, H. Zhang and Y. Liu, Miner. Eng., 2011, 24, 839–944 CrossRef CAS.
- K. Nakamoto, Infrared spectra of inorganic and co-ordination compounds, John Wiley and Sons, New York, 1963, p. 107 Search PubMed.
- S. K. Das and A. K. Guha, Colloids Surf., B, 2007, 60, 46–54 CrossRef CAS.
- R. R. Pandey, K. K. Saini and M. Dhayal, J. Biosens. Bioelectron., 2010, 1, 101, DOI:10.4172/2155-6210.1000101.
- C. Jin, Y. Tang, F. G. Yang, X. L. Li, S. Xu, X. Y. Fan, Y. Huang and Y. J. Yang, Biol. Trace Elem. Res., 2011, 141, 3–15 CrossRef CAS.
- J. T. K. Quik, I. Lynch, K. V. Hoecke, C. J. H. Miermans, K. A. C. De Schamphelaere, C. R. Janssen, K. A. Dawson, M. A. C. Stuart and D. V. D. Meent, Chemosphere, 2010, 81, 711–715 CrossRef CAS.
- E. Navarro, A. Baun, R. Behra, N. B. Hartmann, J. Filser, A. J. Miao, A. Quigg, P. H. Santschi and L. Sigg, Ecotoxicology, 2008, 17, 372–386 CrossRef CAS.
- M. Heinlaan, A. Ivask, I. Blinova, H. C. Dubourguier and A. Kahru, Chemosphere, 2008, 71, 1308–1316 CrossRef CAS.
- N. Singh, B. Manshian, G. J. S. Jenkins, S. M. Griffiths, P. M. Williams, T. G. G. Maffeis, C. J. Wright and S. H. Doak, Biomaterials, 2009, 30, 3891–3914 CrossRef CAS.
- T. Y. Leung, C. Y. Chan, C. Hu, J. C. Yu and P. K. Wong, Water Res., 2008, 42, 4827–4837 CrossRef CAS.
- P. K. J. Robertson, J. M. C. Robertson and D. W. Bahnemann, J. Hazard. Mater., 2012, 211–212, 161–171 CrossRef CAS.
- R. M. Green, M. Graham, M. R. O'Donovan, J. K. Chipman and N. J. Hodges, Mutagenesis, 2006, 21, 383–390 CrossRef CAS.
- H. Sies, Klin. Wochenschr., 1991, 69, 965–968 CrossRef CAS.
- Y. H. Tsuang, J. S. Sun, Y. C. Huang, C. H. Lu, W. H. Chang and C. C. Wang, Artif. Organs, 2008, 32, 167–174 CrossRef CAS.
- M. Sadiq, N. Chandrasekaran and A. Mukherjee, Curr. Nanosci., 2010, 6, 381–387 CrossRef.
- X. Fang, R. Yu, B. Li, P. Somasundaran and K. Chandran, J. Colloid Interface Sci., 2010, 348, 329–334 CrossRef CAS.
- R. Morones, J. L. Elechiguerra, A. Camacho, K. Holt, J. B. Kouri, J. T. Ramirez and M. J. Yacaman, Nanotechnology, 2005, 16, 2346–2353 CrossRef.
- Y. Xie, H. Yiping, L. I. Peter, J. Tony and S. Xianming, Appl. Environ. Microbiol., 2011, 77, 2325–2331 CrossRef CAS.
- S. Pigeot-Rémy, F. Simonet, E. Errazuriz-Cerda, J. C. Lazzaroni, D. Atlan and C. Guillard, Appl. Catal., B, 2011, 104, 390–398 CrossRef.
- A. Kumar, A. K. Pandey, S. S. Singh, R. Shankar and A. Dhawan, Cytometry, Part A, 2011, 79, 707–712 Search PubMed.
- J. Ma, Z. Xiong, T. D. Waite, W. J. Ng and X. S. Zhao, Microporous Mesoporous Mater., 2011, 144, 97–104 CrossRef CAS.
- W. Jiang, H. Mashayekhi and B. Xing, Environ. Pollut., 2009, 157, 619–625 Search PubMed.
- J. Guo and X. Zhang, Carbohydr. Res., 2004, 339, 1421–26 CrossRef CAS.
- M. Sadiq, S. Dalai, N. Chandrasekaran and A. Mukherjee, Ecotoxicol. Environ. Saf., 2011, 74, 1180–1187 CrossRef.
- S. Malato, P. Fernández-Ibáñez, M. I. Maldonado, J. Blanco and W. Gernjak, Catal. Today, 2009, 147, 1–59 CrossRef CAS.
- D. Wang, H. Ji and R. David, Sci. Total Environ., 2011, 409, 1351–1356 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2tx00012a |
| This journal is © The Royal Society of Chemistry 2012 |